
Abstract
Lentivirus (LV) vectors offer permanent delivery of therapeutic genes to the host through an RNA intermediate genome. They are one of the most commonly used vectors for clinical gene therapy of inherited disorders such as immune deficiencies and cancer immunotherapy. One of the most difficult challenges facing their widespread application to patients is the large-scale production of highly pure vector stocks. To improve vector production and downstream purification, there has been a recent investment in the United Kingdom to establish good manufacturing process (GMP)-licensed centers for manufacture and quality control. Other requirements for these vectors include their target cell specificity and tropism, how to regulate gene expression of the therapeutic payload and their potential side effects. Comprehensive detail on the full nucleic acid content of LV is unknown, even though they have entered clinical trials. With potential adverse effects in mind, it is important to identify these contents to assess their safety and purity. In this study, we used highly sensitive PacBio long-distance, next-generation sequencing of reverse-transcribed vector component RNA to investigate the nucleic acid composition of recombinant HIV-1 particles generated by human 293T packaging cells. In this article, we describe our findings of nucleic acids other than the recombinant vector genome that exist, which could potentially be delivered during gene transfer, and suggest that removal of these unwanted components be considered before clinical LV application.
INTRODUCTION
To date, more than 3,000 clinical trials have been initiated in the field of gene and cell therapy. 1,2 Many use retrovirus (RV) or lentivirus (LV)-based vectors due to their excellent gene transfer capabilities and long-term and persistent therapeutic gene expression. 3,4 To meet safety requirements and reach therapeutic potential for their use, significant modifications to these vectors have been made. As a result, RV produced by human embryonic kidney cells (HEK 293T) has been replaced by LV based on HIV-1, which unlike RV can infect nondividing cells. Essential to LV production from HEK293T is the isolation of pure particles using methodologies that isolate vector from supernatant impurities such as producer cell debris and media components that may elicit an immune response by the host. 5 In large-scale production, ultracentrifugation that is used for particle concentration is replaced by tangential flow filtration, benzoate treatment, and gradient purification or chromatography to remove cellular genomic DNA followed by elution and filtration. 6,7
Other impurities, important to vector safety, include virus components that may enable replication competence to support propagation and spread within the host. Replication competence was observed in early generation RVs, and consequently, the genes coding for virus packaging have been removed by recombinant vector design. This uses genome splitting onto discrete plasmid constructs with reduced homology to generate vector particles carrying cis genomes and trans-packaging genes to avoid generating recombination and replication of competent virus. Historically, first-generation LV was produced by splitting the HIV 1 genome into three plasmids for transfection of virus-producer cells. The first plasmid carrying the vector genome included the expression cassette that comprised an internal promoter to drive therapeutic gene expression, a Rev response element, and a genome psi (ψ) packaging signal recognized by the LV GAG nucleoprotein to ensure only the RNA genome is packaged with native long terminal repeats (LTR) needed for vector integration. A second plasmid was used that included HIV transgenes essential for genome packaging into virus particles, and the third plasmid provided an envelope gene to generate a surface glycoprotein (e.g., VSV-G) for virus attachment to the target cell for infection. 8 Importantly, as neither second nor third plasmids carry ψ packaging signals, their packaging is prevented to circumvent virus propagation. To further reduce the potential for recombination leading to replication competence, second-generation vectors were engineered with even less plasmid homology by removal of accessory genes (i.e., Vif, Vpu, Vpr, and Nef). 9 –11 Third-generation vectors include additional modifications by removal of Tat and the addition of Rev separately on a fourth plasmid to improve vector genome nuclear export and titer. Third-generation vectors also use self-inactivating design with a deletion in the 3′ LTR containing the TATA box to abolish LTR promoter activity and reduce the potential for oncogene activation near the vector integration site. 12,13
It has been shown that the packaging of vector genomes into RV and LV particles is enriched by the presence and specificity of the psi (ψ) packaging signal generally located near the 5′-end of the viral RNA recognized by the GAG protein. 14 It is unclear, however, to what extent nonviral RNA or DNA is packaged. These could include parts of the HEK293T genome, single genomes, human endogenous RV (HERV) such as sequences, spliced viral mRNAs, other cellular mRNAs, or the trans-plasmids used for vector production, 15 and these should be avoided in gene delivery. In addition, alternative contaminants for gene transfer could originate from ectosomes and exosomes that pinch off from HEK293 producer cells. Importantly, ectosomes and exosomes that are not usually removed in LV production are known to contain nucleic acids, lipids, metabolites, and proteins and have been reported to be associated with several cellular activities and cancer progression.
As the contents of packaged LV have not been fully characterized, we investigated in detail the nucleic acid composition of “full” genome and “empty” recombinant LV particles. For this study, particles not separated from ectosome and exosome vesicles were generated by three plasmid transfections of HEK293T cells and isolated for sensitive long-range PacBio RNA sequencing. PacBio sequencing, using single-molecule real-time (SMRT) sequencing, provides long-read sequences to identify complete genomes, through single-molecule sequencing. 16 This has been performed previously using the genomes packaged within adeno-associated viral vectors to identify chimeric genomes with the packaging of aberrant genomes. 17,18
We describe our observations of a range of nucleic acids not of HIV-1 origin, in addition to the vector genome present in LV particles. Our data suggest that further investigation of the origin of these nucleic acids and their potential side effects on target cells should be considered and propose that removal of these contaminants would be a valuable step in manufacturing safer recombinant LV particles.
METHODS
Vector production and titration
The production of HR’SIN-cPPT-SEW-eGFP-W LV (pHR, full) and no transgene equivalent (empty) LV was carried out as previously described.
19
pHR LV vector expresses eGFP under the internal promoter of SFFV. Infectious LV titer was calculated as previously reported.
19
Briefly, 2 × 105 HEK293T cells were seeded and incubated at 37°C, 5% CO2 overnight to adhere. Serial dilutions of the virus were prepared and incubated in a complete cell culture medium with 5 μg/mL polybrene (Sigma Aldrich), for 20 min at room temperature before addition to cells; 72 h post-transfection, cells were harvested for green flourescent protein (GFP) expression analysis via flow cytometry using ACEA Novocyte flow cytometer and NovoExpress software v1.2.5 (Agilent Technologies, Didcot, England). Dilutions expressing 1–30% GFP expression were analyzed as accurate representations of viral titer, calculated as shown in the following equation:
Empty LV, carrying no reporter transgene, were titrated using Lenti-X™ p24 Rapid Titer Kit (Takara Bio), according to manufacturer’s instructions. Absorbance was read on Elx808 absorbance reader at 450 nm and analyzed using Gen5 software (v2.06.10).
Nucleic acid harvesting
Viral RNA was harvested via QIAamp® viral RNA mini kit (Qiagen) according to the manufacturer’s instructions. The nucleic acid concentration was determined using Nanodrop 2000C UV-Vis (v1.2.1) (Fisher Scientific). RNA samples were treated to remove gDNA contamination using RNase-Free DNase set and RNase kit (Qiagen) according to the manufacturer’s instructions.
PacBio DNA sequencing
RNA samples were sequenced by the Center for Genomic Research, University of Liverpool. Sample preparation of involved RNA integrity using the Agilent 6000 Pico Bioanalyser and RNA pico chips; 1 μg of RNA per sample was depleted of rRNA using a Ribozero kit followed using RNA clean AMPure beads and repeated Bioanalyser testing for diminished rRNA profiles. Treated with Cell Script plus poly-A polymerase tailing kit, undergoing 15 cycles of cDNA synthesis being once again purified with the AMPure beads and reanalyzed this with a DNA HS Bioanalyser chip.
The library was produced using the Pac Bio DNA template kit 1.0 followed by a final AMPure bead cleaning and bioanalyser control step. Samples were mixed with polymerase and diffusion loaded into LR cells and into a PacBio Sequel 2.1 chemistry sequencing machine. Settings were 1,200 min movie times and 240 min of preextension with an on plate load of 8 pM. The data were processed using the Iso-Seq workflow to retrieve CCS.
DNA contaminants identification
Prior to sequencing, RNA samples were analyzed for gDNA contamination prior to and subsequent to conversion to cDNA using GoScript™ reverse transcriptase (Promega) according to manufacturer’s instructions. Samples were amplified via PCR against β-actin and the viral LTR, shown below.
LTR-F GAGCTCTCTGGCTAACTAGG
LTR-R GCTAGAGATTTTCCACACTG
SY100216195-080 AAGAGAGGCATCCTCACCCT
SY100216195-081 TACATGGCTGGGGTGTTGAA
Screening for HERVS, cloning vectors, and packaging sequences
For all searches, the same search keys were used to enable comparison, sequence coverage had to be greater or equal to 80 bp, had to be derived from CCS of 100 bp or greater, and alignment had to be 10% or greater than the CCS length. The single best result was determined for each CCS where applicable.
To identify the relative presence of HERV-K113 in the samples, the prevalence of human genomic reads was calculated by aligning all CCS to the human reference genome (GRCh38) and HERV-K113 genome and normalized per kilobase of genome per million reads.
RESULTS
Production of HIV-1 LV vectors
Replication defective second-generation SIN configuration HIV-1-based particles, HR’SIN-cPPT-SEW-eGFP-W (abbreviated to pHR LV in this article), or no vector genome (empty vector) (Fig. 1), were generated by three plasmid transient transfection of HEK293T virus-producer cells. 20 For this, packaging plasmids were used at identical concentrations plus or minus a third plasmid at the same concentration carrying the vector backbone. Viruses were harvested at 24, 48, and 72 h post-transfection and pooled for concentration. pHR LV particles were titrated via flow cytometry for GFP expression using infected HEK 293T cells. Vector titer for both pHR and empty LV was also determined using a p24 gag ELISA (Takara Bio). Each was diluted to 2.42 × 109 TU/mL to ensure that equal LV titers were used for subsequent investigation.
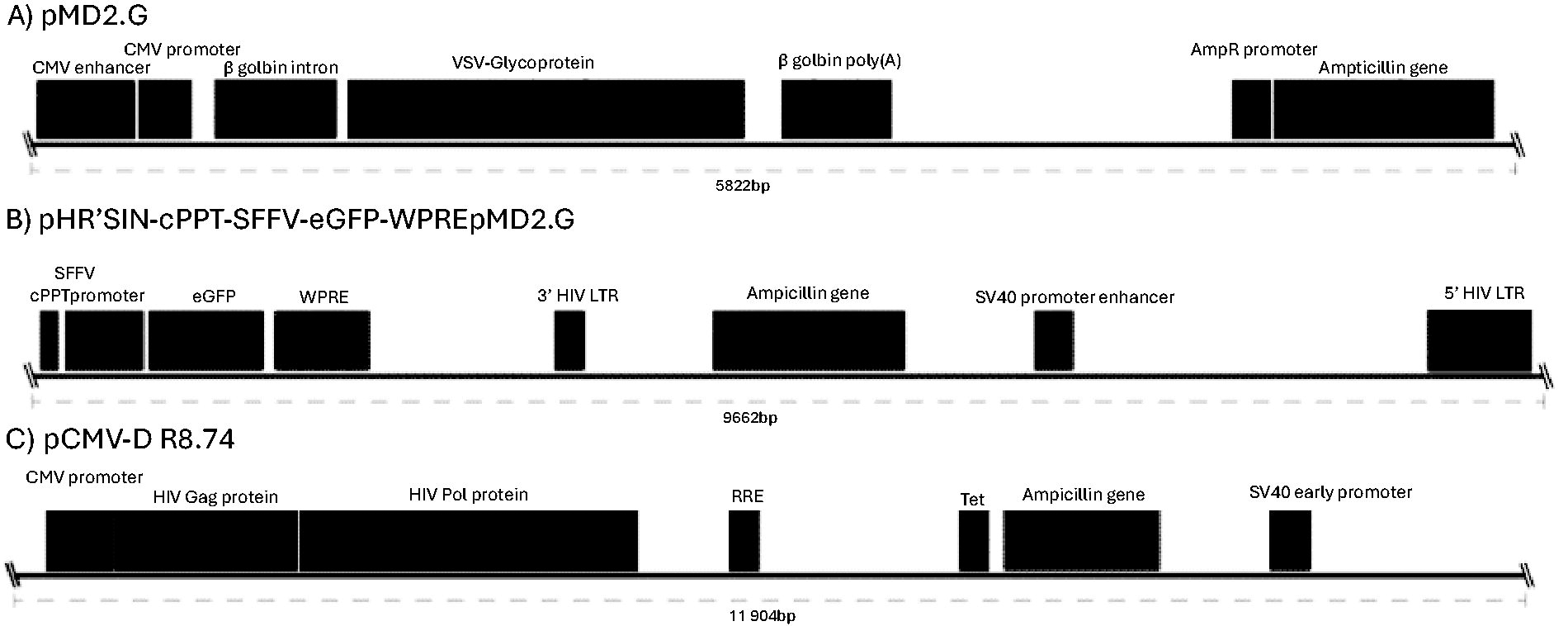
Vector used for LV production.
Collected particle supernatants were treated by DNAse I and RNAse digestion to remove external contaminants. Viral RNA, purified from lysed particles, was treated with and without reverse transcription and analyzed for the absence of HEK293T DNA by polymerase chain reaction (PCR) of the β-actin gene and vector LTRs (Fig. 2). The absence of bands for β-actin in viral samples but their presence in control cellular nucleic acid demonstrated the vector to be free of gDNA contamination at this level of detection. Batches of vectors were also subjected to more sensitive quantitative PCR that also showed no presence of contaminating HEK293T gDNA (data not shown). PCR of the HIV-1 LTR in samples without reverse transcription identified weak bands suggesting these could have arisen from reverse transcription within HIV-1 particles that can occur during maturation. Positive LTR bands in the empty virus particles suggest that the plasmid DNA carrying the HIV-1 trans-acting components used to generate virus particles was present. This corresponds with the data analyzed from PacBio sequencing of vector RNA (Fig. 2).
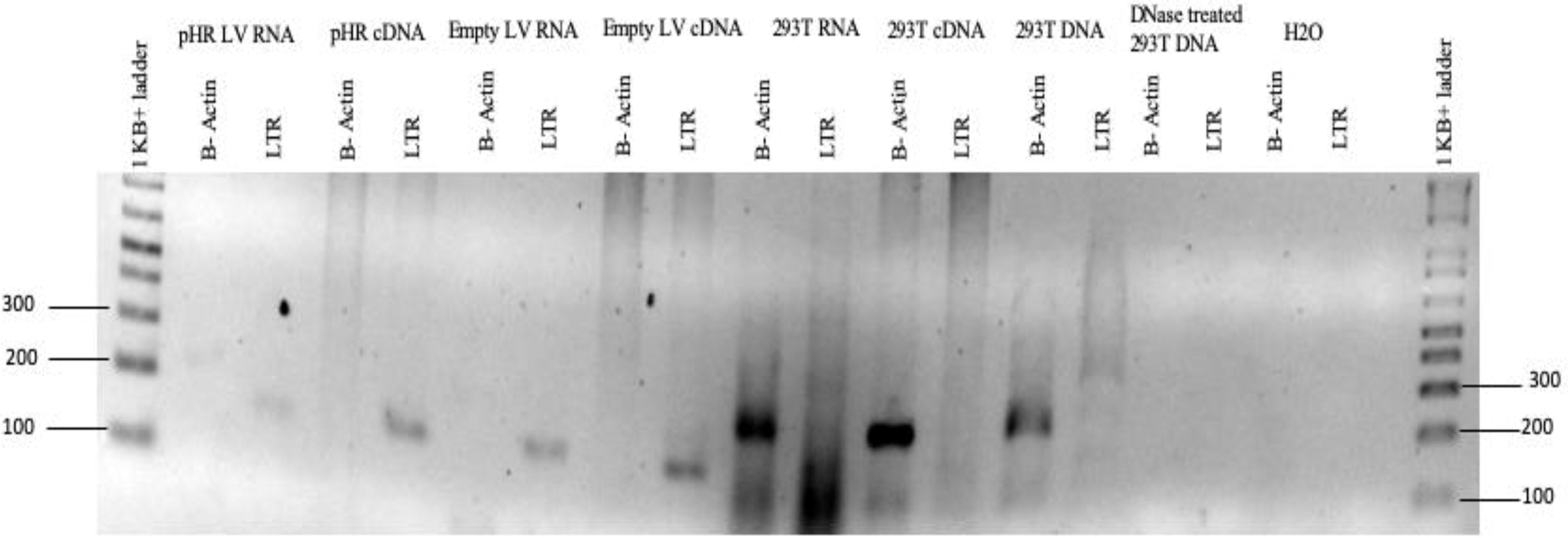
Agarose gel electrophoresis of the viral vector RNA following PCR amplification to identify 293T cell components in virus preps. Virus particles were treated with DNAse I and then lysed. Purified RNA was reverse transcribed before PCR amplification. PCR primers identifying b-actin were used to identify HEK293T genomic DNA or cDNA. The absence of b-actin in viral samples and the presence of cellular nucleic acid indicate the vector to be free of gDNA contamination. Positive bands were identified for reverse-transcribed LTRs. Weak LTR bands for nonreverse transcribed samples suggest reverse transcription within particles by HIV-1 RT during maturation. Positive LTR bands in the empty virus particles suggest the presence of contaminating trans-plasmid used to generate virus particles. Samples were run on a 2% agarose-TBE gel at 70V for 35 min. HEK, human embryonic kidney; PCR, polymerase chain reaction.
Analysis of LV particle contents following long-distance PacBio (IsoSeq) sequencing
Extracted DNAse-1 treated viral RNA samples were examined for RNA integrity using an Agilent 6000 Pico Bioanalyser and RNA pico chips. RNA was depleted for rRNA, then cleaned and reanalyzed for diminished rRNA profiles. cDNA libraries were synthesized and purified before examination with a DNA HS Bioanalyser chip. PacBio sequencing of the extracted samples identified various reads and the sequence metrics of each prep.
As expected, the pHR LV vector had 3,956,793 greater number of subreads than the empty, nongenome carrying, LV preparation (6,617,371 vs. 2,660,578). The sequence data were processed using the IsoSeq pipeline within the SMRT Link software suite (v5.1.0.26412). As the primary aim of this experiment was to sensitively detect transcripts, the final IsoSeq output files were not used, as these involved iterative clustering of sequences resulting in reduced sensitivity. Instead, the intermediate Circular Consensus Reads (CCS) generated as part of the IsoSeq pipeline were used. These reads were generated by creating a consensus sequence from multiple sequencing passes of the same template molecule that improved sequence quality and reduced errors. As the sensitivity was critical, no lower limit to the minimum number of sequencing passes was specified.
When processed, a similar number of CCS reads were identified between the vector particles; 657,675 CCS were identified in full viral prep (pHR LV) and 691,011 CCS reads in empty particle prep (Table 1). The mean CCS length was similar between full (1,272 bp) and empty (1,823 bp) LV vectors. The distribution of CCS read length in both pHR and empty LV particles followed a double exponential curve with the slope flatter for empty LV preps, as expected (Fig. 3).
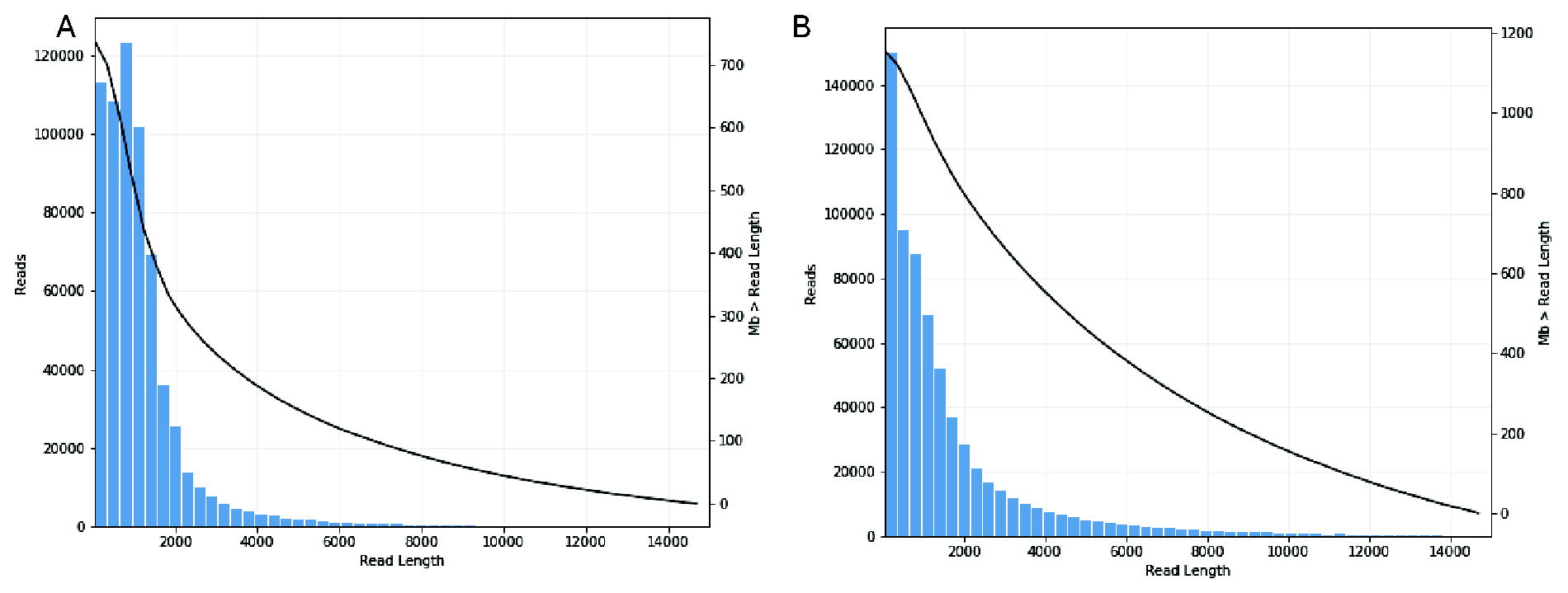
Read length (bp) distribution for pHR and empty vector LV RNA. Read length (bp) distribution for pHR
Read Metrics for PacBio Sequenced Viral RNA Samples
Metrics for pHR and empty LV RNA samples are shown.
CCS, Circular Consensus; LV, lentivirus.
Taxonomic assignment based on filtered BLAST analysis shows sequences present of multiple viral origins
To determine whether library DNA sequences were from RNA contained in virus and not packaged from the HEK293T cell transcriptome, BLAST was used to determine the highest probable matches. To confirm this, BLAST searches were made against the pHR viral backbone, human genome, and virus RefSeq genome library. These sequences were found to align with HIV-1 abundantly as well as other viruses.
To investigate the coverage of sequences for full genomic sequences, viral sequences were separated by species and mapped and CCS reads were determined for each. Samples were taxonomically assigned by using them as a query for nucleotide BLAST 21 against the viral RefSeq database (https://www.ncbi.nlm.nih.gov/genome/viruses/). Alignments were filtered to retain the single best hit for each CCS read, hits of 80 bp or longer, hits derived from reads that are 100 bp or longer, and hits where the alignment matched over at least 10% of the CCS read length. Postfiltering, 23.6% of reads from pHR were taxonomically assigned. However, a much smaller proportion of empty LV reads were taxonomically assigned (4.3%) due to the alignment lengths generally being extremely short and because there is much lower content in this sample. The top 10 species identified based on filtered reads alone are shown (Table 2). A full representation of the alignments is also provided (Supplementary Table S1). Note that the Woodchuck hepatitis virus and SFFV reads represent the presence of these elements in the pHR LV vector and may also represent the presence of the transfected cloning vector (HR’SIN-cPPT-SEW-eGFP-W). As this was not used in the production of empty particles, this sequence was not identified in the empty particle preparation. Furthermore, two aligned taxonomical classes identified in empty viruses (Bovine viral diarrhea virus 3 Th/04 KhonKaen and Cafeteria roenbergensis virus BV-PW1) were not identified in the pHR LV prep. Conversely, 23 taxonomical classes were identified in full viral particles but not in empty viruses, suggesting increased aberrant packaging of sequences may be triggered by vector genome production and packaging.
PacBio Reads of pHR LV and Empty LV
A—Taxonomic assignment based on filtered BLAST hits. Hits show the number of reads aligned to pHR and empty virus samples.
Screening for packaging sequences and cloning vectors
No sequences carrying the HIV-1 ψ packaging sequence were identified in empty LV particles, as expected. In full HIV-1 virions, however, 3,370 CCS reads contained significant hits to this sequence. Of these, 3,347/3,370 (99.32%) were identified with pHR, HIV, and human reference databases; 3,289 (98.27%) sequences align with pHR; however, only 26 (0.78%) were associated with HIV and 32 (0.96%) with the human reference databases suggesting that the ψ packaging sequences were derived from pHR and the RNA used in this analysis did not derive from a human source. As only 0.51% of all CCS reads (657,675) were identified carrying the ψ packaging sequence, this suggests the majority of sequences packaged both in full and empty particles were aberrantly selected. When mapping the sequences identified in full and empty viral preps to the pHR plasmid or the trans-acting gag/pol carrying plasmid pCMVR8.74, this identified numerous sequence alignments suggesting the plasmid vectors used in vector production are packaged at low level (Fig. 4).
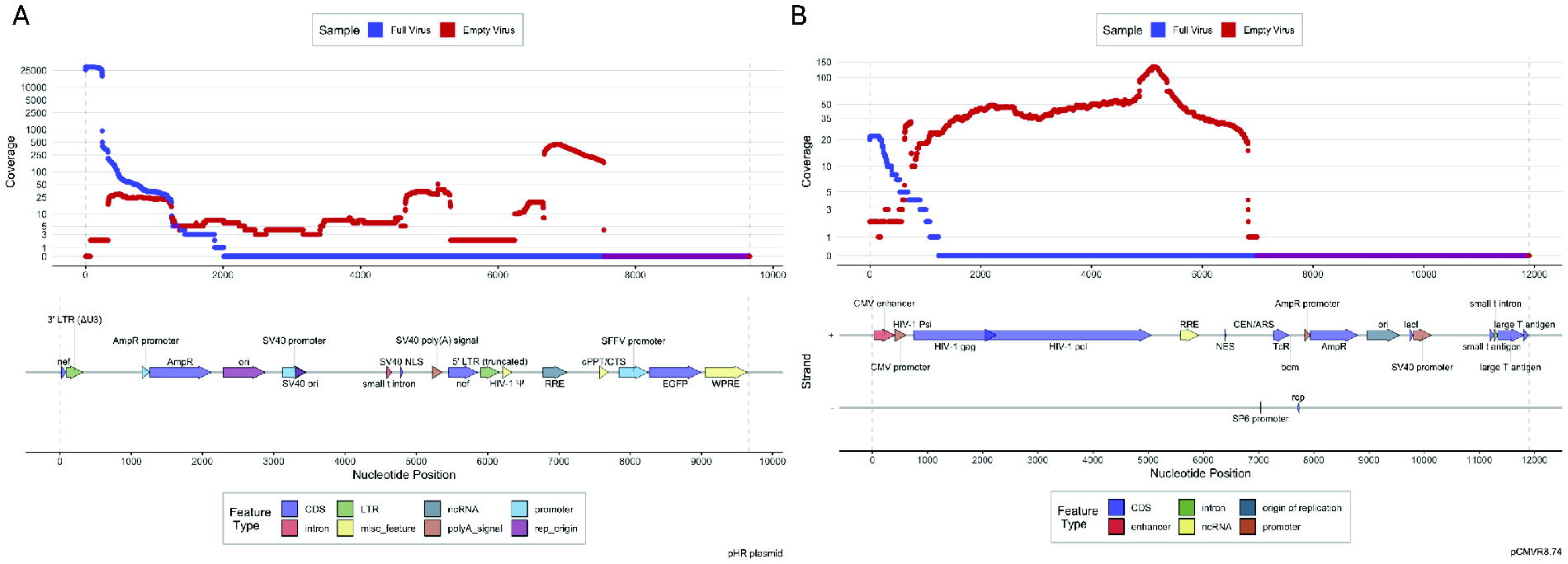
PacBio sequence reads aligned construct plasmids. Reads identified in viral preps were aligned to the pHR plasmid construct
Detection of HERV contamination of virus preparations
We noticed that HERV-K113 were detected at low levels in both pHR (seven hits with a relative abundance of 4.15 × 10−5) and empty particles (four hits with a relative abundance of 1.33 × 10−3), suggesting these are also packaged upon virus production. The identity of these reads is provided (Table 3). By identifying the relative abundance of human and HERV sequences, we sought to determine whether there is a disproportionately high amount of HERV, or equivalent amounts, as the latter would suggest that the HERV identified could be from 293T human genomic DNA contamination.
PacBio Reads of pHR LV and Empty LV
B—HERV sequence identity in pHR and empty LV samples. HERVK113 reads identified in viral samples were mapped to the HERVK113 and human genomes, respectively, to identify percentage identity. Higher percentage identity is found in the human genome, determining this as the source of the transcripts. A full list of hits and abundances are provided in Supplementary Table S1.
To address this, all CCS reads were aligned to the human reference genome, filtered in the same way as described earlier, and counts were calculated. Following this, the counts to human and HERV-K113 sequences were normalized based on the size of the respective genomes, and as a frequency per million reads, that is, reads per kilobase of genome per million reads (RPKM). In the pHR LV prep, the relative abundance of human sequences was calculated as 37.0 RPKM compared to HERV-K113 at 739.2 RPKM. In empty LV preps, human and HERV sequence relative abundance was calculated as 11.6 and 422.4 RPKM, respectively. This identifies significantly greater reads for HERV-K113 in proportion to genome size and suggests HERV-K113 sequences are not derived from human genomic DNA contamination. However, it is important to note that the number of HERV-K113 reads is very low for each sample.
Additionally, in all cases, the HERV-associated reads aligned better to the human reference sequence than to the viral database sequences, specifically, the length and BLAST alignment score of matches were better in all cases, and the percentage identity was better in most cases. This suggests the reads actually would be derived from the human genome, which would be a good indication that there are no nonintegrated HERV sequences. A summary of the key alignment metric differences between HERV and human alignments is shown (Supplementary Table S2).
DISCUSSION
RV and LV offer efficient and permanent delivery of therapeutic genes for gene therapy. Several improvements to vector manufacture that include high titer particle production and purity in addition to improved safety design have resulted in their successful use in the clinic for rare genetic diseases and cancer immunotherapy. 22 Most efforts to generate LV for gene therapy have used cell lines or transient transfection protocols of producer cells to yield pseudotyped particles. To remove contaminants, such as producer cell debris that may be toxic or elicit a host immune response, particles can be concentrated by ultracentrifugation. 5,23 More recently, more refined downstream purification protocols have generated LV particles of high quality. 6,7 Although these technologies provide vector batches that have external impurities removed, it is unclear to what extent impurities exist within particles other than the therapeutic genome.
Following infection of the host, two copies of the vector RNA genome are used for successful reverse transcription before genome integration as therapeutic cDNA. RV and to a lesser extent LV genotoxicity are known to be associated with the vector genome following integration as highlighted by several in vitro and in vivo nonclinical applications and clinical trials to correct genetic disorders. 24 Understanding the cause of vector-associated side effects on the host is, therefore, of utmost importance, and models have been developed to identify and measure vector genome/host interactions that alter cancer gene expression and may lead to oncogenesis. Risk assessment has been restricted to the vector genome as it has been assumed that packaging is specific and determined by recognition of the packaging ψ site present on the vector backbone. 25 –28 It has, however, been shown that RVs can also package spliced virus RNA, several tRNAs, and varying amounts of cellular mRNAs in a concentration-dependent manner. 14,15
It has also been shown that HERVs devoid of a ψ site can be packaged into virions by human producer cells at low levels. 15 HERVs being of RV origin exist as vestigial elements and represent ∼8% of the human genome. They have had a profound influence on human evolution and have been modified over time to prevent their activity that including their propagation and influence on gene expression. 29 Unlike RVs, most endogenous RV elements cannot transfer between cells, and most HERVs exist in deleted forms. However, an example of a preserved element with full-genome sequence is HERV-K113 that belongs to the HERV-K family, 30 which retains its RV structure with gag, pol, and env genes bordered on either side by LTRs. Importantly, HERV LTRs can inhibit or upregulate nearby promoter/enhancer activity and can promote novel splice activity, and under specific conditions such as cancer and autoimmune disease, can become active. 31 During HIV-1 infection, HERV-K113 expression has been shown to increase to the point where its RNA can be detected in patient plasma. 32,33 Although little complementation activity exists between HIV and HERVs, 29,34 RV-specific microarray has identified HIV-1 derived packaging cells able to package HERV transcripts. 35,36
After we demonstrated the removal of HEK 293 genomic DNA and unpackaged RNA from culture supernatants using DNaseI and RNase treatment and PCR and q-RT PCR analysis of the β-actin gene and vector LTRs, we chose the highly sensitive Illumina PacBio long-range RNA sequence platform to determine the nucleic acid composition of LV, with and without vector genomes. Interestingly, PacBio sequencing identified incomplete HERV-K113 sequences packaged in empty and to a lesser extent full LV particles at relative frequencies of 1.33 × 10−3 and 4.51 × 10−5. However, longer HERV-K113 of more than 1 kb were identified in pHR LV. We assume competition between HERV-K113 and pHR genomes for integration would be biased to pHR favored by the HIV-1 integrase, which should exclude HERV sequences that have no HIV-1 homology. In a clinical application, with an increased viral load of greater than 1 × 1010 TU, HERV-K113 sequences may become more common, especially when delivered by empty LV particles. 37 Importantly, though, we confirmed that the HERV-K113 sequences found were at values greater than background human reads with values at 36.4 RPKM times more prevalence than human genome reads in empty virus and 19.9 RPKM times for full virus samples. This difference in the relative quantity and RPKM in empty viruses is worth investigating to identify whether this difference is the result of sequencing bias or due to less competition for HERV-K113 sequence packaging in LV particles. An investigation as to whether HERV K113 gene transfer to nonhuman cells by LV could be beneficial using sensitive PCR, whole-genome next generation sequencing (NGS) analysis, TES, inverse PCR, or LAM PCR. As yet, to what extent HERV-K113 gene transfer presents a potential genotoxic hazard is unknown.
Aberrantly packaged nucleic acid sequences represented by the alignment of PacBio reads to the virus database appeared with more frequent reads in the full virus compared to empty particles and suggested potential promotion of contaminating sequence packaging alongside the vector genome. Sequencing of pHR and “empty” particles identified the Woodchuck hepatitis virus sequences only in the pHR vector used for optimal RNA export during vector production by HEK 293 cells; however, the presence of HIV-1 virus sequences in both pHR and empty LV suggests identification of sequences remaining in the plasmid vector used for virus packaging. Interestingly, sequences belonging to the Friend murine leukemia virus and Spleen focus-forming virus were only found in pHR and sequences of Macaca mulatta polyomavirus 1, Salmonella virus SP6, BeAn 58058 virus, Vesicular stomatitis Indiana virus, human adenovirus 5, Chrysochromulina ericina virus, and HERV K113 were present in both vectors. Unwanted packaged sequences would be expected to reduce effective viable vector titer, even though read length was mainly short with reads of up to 9 kb rarely identified. To improve LV batch quality, it would be interesting to remove empty particles during vector production. In addition, production without ectosomes and exosomes could improve effective vector and safety if, indeed, the contaminants identified in this study were provided by these vesicles. 38 Methodologies to remove these contaminants from LV preparations exist and should be considered. 39
In conclusion, by using sensitive PacBio NGS, we have identified multiple aberrantly packaged nucleic acid species in full genome carrying and empty LV particles. Work to determine whether these contaminants are integrated would be useful, as would the exosome contribution to gene transfer and side effects. We propose to reach a more characterized clinical grade LV, some of the potential issues raised in this report be considered with the aim to improve the safety profile and titer of LV vectors.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank Centre for Genomic Research, University of Liverpool, for the PacBio sequencing service platform used in this study and a GlaxoSmithKline (GSK) studentship funding award.
AUTHOR DISCLOSURE
The authors have no conflicts of interest to declare.
FUNDING INFORMATION
GSK funded the studentship.
SUPPLEMENTARY MATERIAL
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.