
Abstract
The ongoing advancements in CRISPR-Cas technologies can significantly accelerate the preclinical development of both
INTRODUCTION
Genome editing holds significant promise for establishing enduring therapeutic effects through precise manipulation of the genome. The introduction of CRISPR-Cas technology
1
has democratized genome editing, marking the dawn of a new era in gene and cell therapies. The evolution of CRISPR tools, including base editors,
2
prime editors, high-fidelity Cas enzymes,
3,4
and protospacer adjacent motif (PAM) variants,
5
has continued to expand the scope of genetic modification and enhanced accuracy, improving the safety and efficacy of therapeutic editing. These technological advancements have ushered a growing number of candidate therapies into clinical trials, both
One particularly exciting biomedical application is genetic engineering to create less immunogenic donor organs. The current standard post-transplant care necessitates lifelong strong systemic immunosuppression to prevent graft rejection in recipients. However, long-term whole-body immunosuppression carries significant morbidity by increasing the risk of complications, such as severe infections and malignancies, and adverse side effects such as renal dysfunction and diabetes. We hypothesized that genetically modified donor organs, designed to provide local long-lasting intragraft immunomodulation, could potentially reduce the need for maintenance high-dose systemic immunosuppression, thus addressing these critical clinical issues.
The translation of genome-targeting therapeutics faces a significant challenge in terms of human-relevant evaluation prior to initiating clinical trials. CRISPR-based therapies rely on sequence specific targeting of Cas enzymes.
6
As a result, interspecies differences of the genome can more substantially impact editing efficiency and the ensuing phenotypic changes compared with other interventions that don’t involve genome targeting.
7
In contrast to
In the field of solid organ transplantation, the advent of machine perfusion has opened new opportunities for evaluating potential interventions in human organs
In this study, we took the initial step of developing a genome-editing method to upregulate an immunomodulatory gene,
MATERIALS AND METHODS
Plasmid vector construction and adenoviral vector production
The guide RNAs (gRNAs) used in this study are listed in Supplementary Table S2. Oligos containing the spacer sequence of gRNAs were synthesized at ACGT Corporation (Toronto, ON, Canada) or Eurofins Genomics (Louisville, KY, USA). The annealed and phosphorylated oligos were cloned into
Cell culture
HEK293 cell line (CRL-1573, ATCC, Manassas, VA, USA) and PT-K75 cell line (CRL-2528, ATCC, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle medium (DMEM) (11995-065, ThermoFisher Scientific, Waltham, MA, USA), and L2 cell line (CCL-149, ATCC, Manassas, VA, USA) was cultured in Ham’s F-12K media (21127022, ThermoFisher Scientific), respectively, supplemented with 10% fetal bovine serum (FBS) (A3840302, ThermoFisher Scientific) and 100 U/mL of penicillin streptomycin (15140122, ThermoFisher Scientific) at 37°C with 5% CO2.
Plasmid transfection and adenoviral vector transduction to cultured cells
Cells were seeded in 6-well plates 1 day before transfection at a density of 2 × 105 viable cells/mL for HEK293 cells, 1.5 × 105 viable cells/mL for L2 cells, and 4 × 105 viable cells/mL for PT-K75 cells. The plasmid transfection was performed using 2.5 μg/well of plasmid and 3.75 μL/well Lipofectamine 3000 reagent (L3000150, ThermoFisher Scientific) according to the manufacturer’s instructions.
For adenoviral transduction, L2 cells were seeded in 12-well plate at a density of 1.5 × 105 viable cells/mL, transduced with adenovirus by changing the media to 500 μL/well virus-containing media. After 3 h, 500 μL/well of media was added. The media was left untouched for the initial 3 days and changed —two to three times a week afterward. The media was changed 72 h before sample collection.
Ex vivo lung perfusion
The study was approved by the Research Ethics Board of University Health Network (UHN) (06–0283) and Trillium Gift of Life Network, Ontario, Canada. Human donor lungs that were not used for clinical lung transplantation and from donors who consented to research were used in this study.
The donor lungs were retrieved using the same protocol as in clinical transplantation in the Toronto Lung Transplant Program and transported at 4°C. In Case 1, the lung was stored at 10°C 20 for approximately 5 h after retrieval before starting the cannula attachment for logistical reasons. The EVLP was performed based on the standard clinical Toronto EVLP protocol 21 with some modifications. 22 Briefly, the cannulas are attached to pulmonary artery and left atrium. An endotracheal tube was inserted into the right or left main bronchus and fixed with ties. Retrograde flushing was performed using low potassium dextran solution (Perfadex Plus, XVIVO Perfusion, Gothenburg, Sweden or LungProtect® from Traferox Technologies Inc., Mississauga, Canada) after the cannula attachment.
The circuit was primed with perfusate (LPD2A from United Therapeutics, Silver Spring, MD, USA), supplemented with methylprednisolone (Solumedrol, Pfizer, New York, NY, USA), heparin sodium (Heparin LEO; LEO Pharma Inc., Toronto, ON, Canada), and meropenem (Meropenem; SteriMax Inc., Oakville, ON, Canada) using the same dose as those in the standard Toronto EVLP protocol. During the initial 1 h of EVLP, the flow was increased stepwise. The temperature was increased incrementally, and ventilation was initiated when the temperature reached 32°C, which is around 30 min from the EVLP start. The perfusate from both the pulmonary artery and left atrium side were analyzed using GEM Premier 5000 system (Werfen, Bedford, MA, USA) every hour. Bronchial wash was performed by administering 20 mL of saline into the right or left main bronchus and collected using a bronchoscope. Perfusate and bronchial wash samples were snap-frozen and stored at −80°C until analysis. Tissues were formalin fixed or snap-frozen for downstream analysis.
Adenoviral administration during EVLP
AdVs were diluted with saline to a final volume of 5 mL. The virus-containing reagent was delivered through a catheter (NA-411D-1521, Olympus Corporation, Japan) using a bronchoscope (BF-Q190, Olympus Corporation). After delivery, the lung was recruited and then returned to the maintenance ventilation setting.
PCLS of the human lung after EVLP
A low-gelling temperature agarose was prepared at 1.5% in DMEM/F-12 media (10565018, ThermoFisher Scientific). Upon completion of the EVLP, pieces of lung tissues were inflated with the warm liquid agarose, solidified on ice, and sectioned at 500 μm thickness using vibrating blade microtome (VT1200 Vibratome, Leica, Wetzlar, Germany) in Hanks' balanced salt solution (14025092, ThermoFisher Scientific) with 10 mM N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (HEPES) (15630080, ThermoFisher Scientific). The slices were cultured in Glutamax-containing DMEM/F-12 media (10569010, ThermoFisher Scientific) supplemented with 10% FBS (A3840302, ThermoFisher Scientific), antibiotic-antimycotic (15240062, ThermoFisher Scientific), MEM Non-Essential Amino Acids Solution (11140050, ThermoFisher Scientific), 1 mM sodium pyruvate (600-100-EL, WISENT Inc., St-Bruno, QC, Canada), and 14.3 mM beta-mercaptoethanol (M6250, St. Louis, MO, USA). The culture media were changed daily. For the measurement of the secreted IL-10 protein, each slice was incubated with 1 mL/slice of culture media for 36 h. The slices were then fixed with formalin or snap-frozen for downstream analysis. Live cell staining was performed using the LIVE/DEAD™ Viability/Cytotoxicity Kit (L3224, ThermoFisher Scientific) according to the manufacturer’s instructions.
Rat study
The study was approved by the Committee of Animal Resources Center at UHN (Animal Usage Protocol # 6057). Male inbred Lewis rats (LEW/SsNHsd, 250–350 g) were purchased from Envigo (Huntingdon, United Kingdom).
The same method of viral delivery was used as in a previous work. 23 Briefly, rats were anesthetized by inhalation of isoflurane, orotracheally intubated with a 16G catheter (BD382258, BD, Franklin Lakes, NJ, USA), and ventilated (Harvard Apparatus, Holliston, MA, USA) at a tidal volume of 10 mL/kg, positive end-expiratory pressure 2 cm H2O, fraction of inspired oxygen (FiO2) 1.0, and a respiratory rate of 80 breaths per minute in the supine position. After placing the intubation tube in the left bronchus, the rat was changed to the left lateral position and 500 μL of vector containing buffer (HEPES 100 mM, MgCl2 20 mM) was delivered to the left lung through the intubation tube. After moving the intubation tube to the trachea, the rat was ventilated in the left lateral position until awakening.
Triple immunosuppression, which comprising cyclosporine (15 mg/kg/day), azathioprine (6 mg/kg/day), and methylprednisolone (30 mg/kg), was intraperitoneally injected 2 h before viral vector administration and on day 1 and day 2. After 2 h of viral delivery, methylprednisolone alone was additionally injected into the peritoneal space. From day 3 to day 28, triple suppression with a reduced dose of methylprednisolone (cyclosporine 15 mg/kg/day, azathioprine 6 mg/kg/day, and methylprednisolone 2.5 mg/kg/day) was subcutaneously injected daily.
Nucleic acid purification
In
Reverse transcription-quantitative polymerase chain reaction
cDNA was synthesized using 500 ng (cell culture and PCLS samples) or 1000 ng (rat tissue samples) of RNA and an iScript Advanced cDNA Synthesis Kit for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) (Bio-Rad, Hercules, CA, USA). The cDNA reaction was diluted in 1:6 (cell culture and PCLS samples) or 1:10 (rat tissue samples), and 3 μL was applied to 10 μL RT-qPCR reaction with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) and run on a CFX384 Real-Time System (Bio-Rad Laboratories). The Ct values obtained after 40 cycles were normalized to a housekeeping gene peptidylprolyl isomerase A (ppia) and used to calculate relative expressions. The primers used in this study are listed in Supplementary Table S3. For the samples that were not detectable after 40 cycles of RT-qPCR reaction, a Ct of 40 was applied.
Analysis of genome editing using Sanger sequencing
The target genomic region was amplified from the genomic DNA using Q5® Hot Start High-Fidelity 2X Master Mix (New England Biolabs, Ipswich, MA, United States) using the primers listed in Supplementary Table S4. The PCR products were purified using the QIAquick PCR Purification Kit (28104, QIAGEN). Sanger sequencing was performed at the Center for Applied Genomics (Toronto, ON, Canada), and the data were analyzed using DECODR software. 24
Enzyme-linked immunosorbent assay
Culture media and bronchial wash samples were applied to an enzyme-linked immunosorbent assay (ELISA) plate. Snap-frozen tissue samples were homogenized in lysis buffer (10 mM HEPES pH7.9, 10 mM KCl, 0.1 mM ethylenediamine tetraacetic acid, 0.1 mM ethylene glycol-bis(beta-aminoethyl ether)-N,N,N',N'-tetra acetic acid, 0.6% Nonidet P-40 with a protease inhibitor cocktail) using TissueLyser II (QIAGEN) and subjected to protein concentration measurement with a bicinchoninic acid Protein Assay Kit (23227, ThermoFisher Scientific), according to the manufacturer’s instructions. The ELISA kits used for cytokine detection were as follows: human IL-10 in
Wet-to-dry weight ratio
The weight of lung tissues was measured before and after drying at 65°C for 72 h. The ratio was calculated as the weight of the fresh tissue divided by the weight of dried tissue.
Histology
Formalin-fixed tissues were processed and embedded in paraffin. Thin sections were cut and stained for hematoxylin and eosin and terminal deoxynucleotidyl transferase dUTP nick-end labeling at the Pathology Core of Spatio-Temporal Targeting and Amplification of Radiation Response (STTARR, Toronto, ON, Canada) at the UHN. Bright-field images were obtained using a slide scanner (APERIO CS2, Leica Biosystems).
Next-generation sequencing
The next-generation sequencing was performed to analyze editing efficiencies. The target genomic locus was PCR-amplified from 100 ng of genomic DNA using Q5® Hot Start High-Fidelity 2X Master Mix (New England Biolabs, Ipswich, MA, USA). PCR amplicons were purified using magnetic beads (Sera-Mag SpeedBeads™ Carboxyl Magnetic Beads, Cytiva). The primers used for the PCR amplification were listed in Supplementary Table S4. Adaptors and indices (TruSeq DNA UD Indexes v2; Illumina, San Diego, CA, USA) were ligated in library preparation using TruSeq Nano DNA kit (Illumina). The library was validated and sequenced on the MiSeq Reagent Nano Kit v2 300-cycles (MS-102–2002; Illumina). A 15–20% volume of PhiX was spiked in to increase the diversity of the library. Paired-end readings (150 bp) with a target depth of 3,000–10,000 reads per sample were obtained at the Center for Applied Genomics. The data were analyzed using CRPSPResso2 software. 25
Fluorescence in situ hybridization
Fluorescence
Statistics
Data were displayed as mean ± standard deviation (SD) and analyzed using the Prism software (GraphPad Software, San Diego, CA, USA). An unpaired two-tailed Student’s
RESULTS
Genome editing of a regulatory element led to upregulation of IL-10
We first explored CRISPR genome-editing approaches to consistently upregulate an immunomodulatory gene in the whole organ. We selected the IL-10 gene, which encodes an anti-inflammatory and immunomodulatory cytokine, as one of the target genes for graft immunomodulation. Previous work demonstrated that the delivery of the IL-10 gene to the donor lung via AdV before implantation improved early graft function post-transplantation.
26
A limitation of this approach, however, is the short duration of transgene expression, lacking the ability to provide sustained immunomodulation. Therefore, we aimed to induce long-lasting
To achieve significant editing in the organ, we sought to optimize CRISPR nuclease methods that can be achieved by simple delivery of the editing enzymes. We hypothesized that targeted mutagenesis of the
Using SaCas9 nuclease and a series of gRNAs, we introduced targeted DNA double-strand breaks (DSBs) that led to insertion or deletion mutations (indels) in the
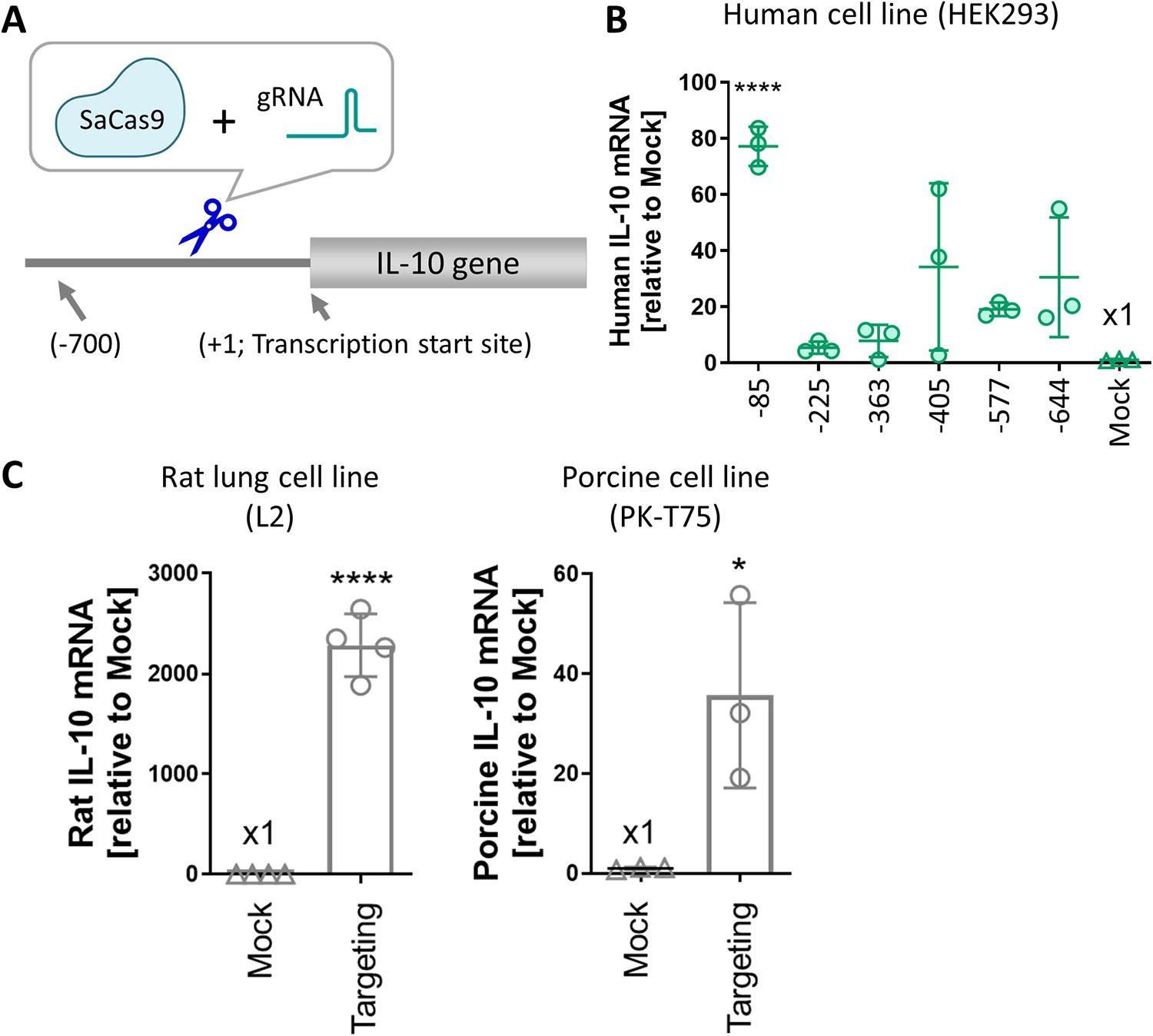
Genome editing of the regulatory region upregulated
A dual mode of IL-10 expression by combining AdV-IL-10 delivery with genome editing
We aimed to develop a method that would allow for both early and sustained IL-10 expression with a single intervention. Early ischemia-reperfusion-induced injury causes primary graft dysfunction, limiting short-term lung graft outcomes, and importantly is associated with chronic lung allograft dysfunction and rejection. Therefore, addressing both early and later graft injuries could enhance the therapeutic impact of long-term graft immunomodulation. Viral vectors have shown promise for
We developed a plasmid vector encoding the human IL-10 cDNA, SaCas9, and the gRNA targeting the −85 region of human IL-10 for AdV production. Both the exogenous human IL-10 and SaCas9 expressions were driven by a single promoter using a 2A peptide to express all three components from a single vector.
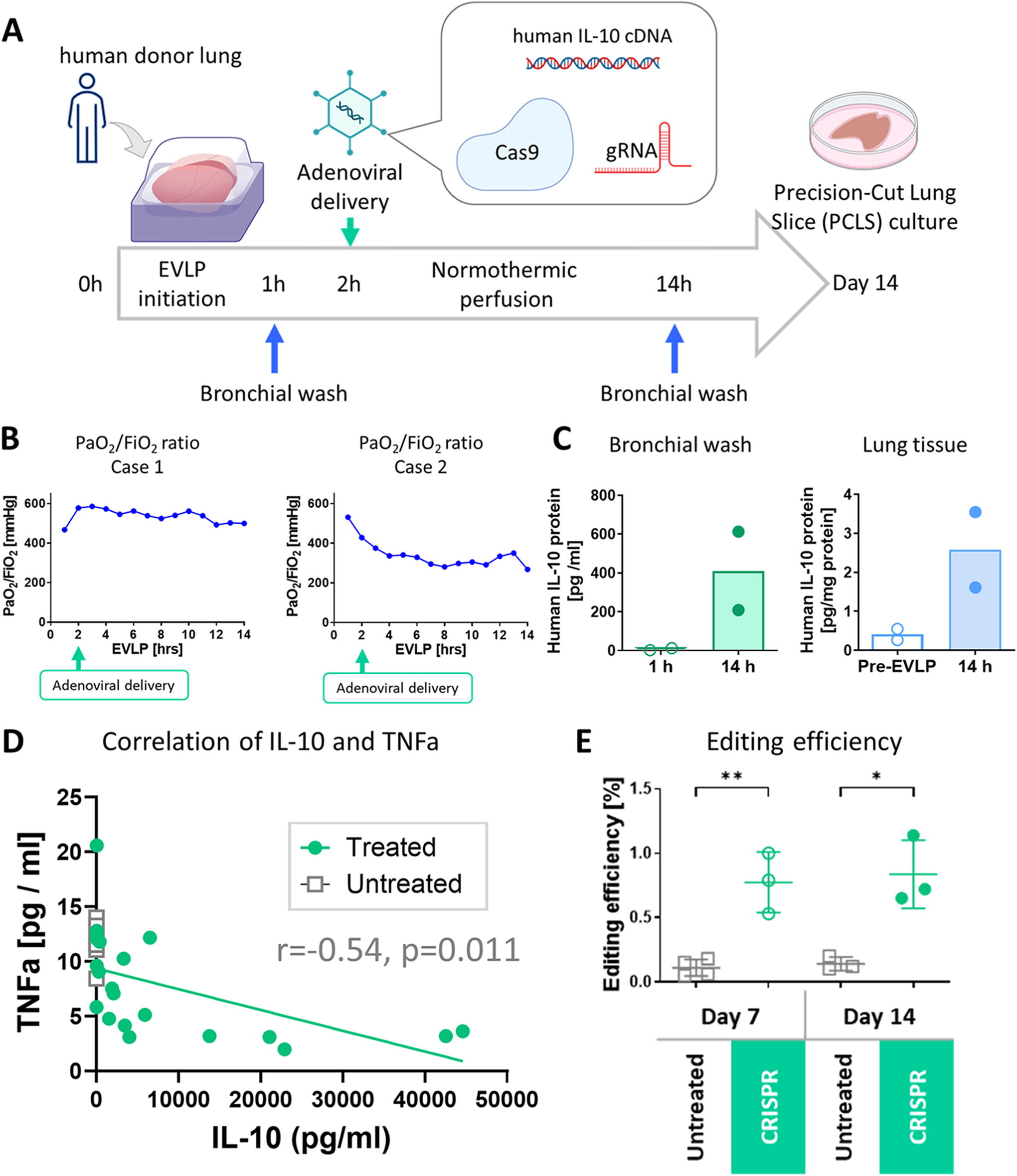
Evaluating CRISPR-based IL-10 modulation in ex vivo perfused human donor lungs
We next sought to assess CRISPR genome editing in
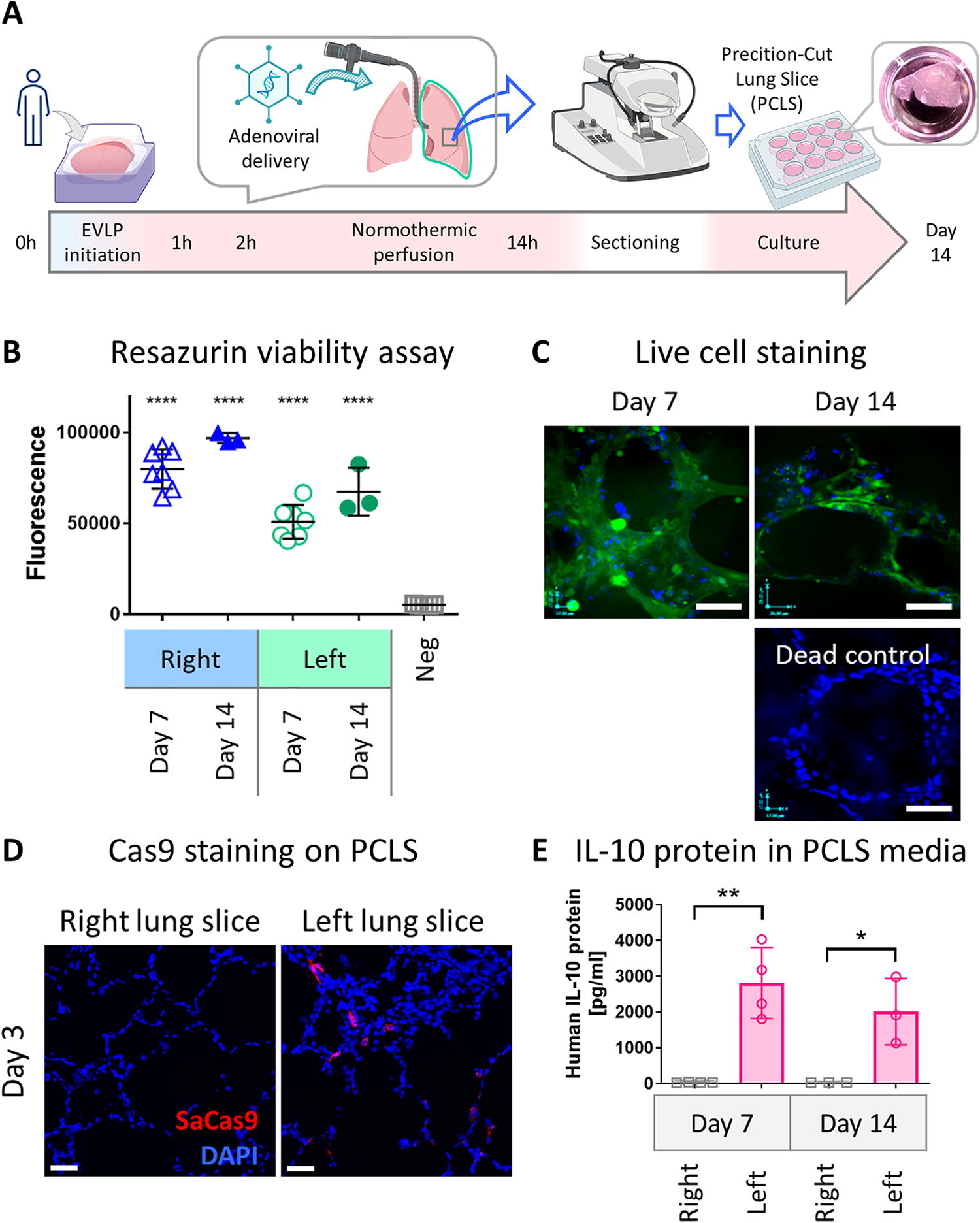
Viability and transgene expressions in PCLS generated after EVLP.
Evaluating genetic engineering in
Building on these findings, we divided human lungs from two donors and perfused individually using distinct circuits (Supplementary Table S1), a design that allows for assessment of the intervention minimizing the impact on the untreated lung. The lungs were then perfused for 12 h post-intervention, a standard duration for EVLP studies, followed by PCLS culture for 14 days (Fig. 3A).
In both cases, the lungs were stably perfused for 12 h following the delivery of editing enzymes and human IL-10 cDNA (Fig. 3B). The treated lungs showed an increase in IL-10 protein levels in the bronchial wash and lung tissue (Fig. 3C) during the 12 h post-intervention perfusion period, whereas the untreated lung from Case 1 showed no increase in IL-10 protein (18.4 pg/mL at 1 h vs. 17.6 pg/mL at 14 h for bronchial wash, 0.97 ± 0.44 pg/mg protein at 14 h for tissue, respectively). IL-10 protein levels in PCLS media varied among slices prepared from different lung regions, likely due to heterogeneous vector distribution after non-aerosolized delivery (Supplementary Figure S2). Most of the PCLS from the treated lungs exhibited higher production of IL-10 protein on day 7 that was negatively correlated with the levels of TNFα (Fig. 3D). Analysis of the genomic DNA of the PCLS with high IL-10 production using next-generation sequencing detected significant editing in the treated group on both days 7 and 14 compared with the untreated group (0.77% ± 0.24% and 0.84% ± 0.27% in the treatment group vs. 0.11% ± 0.06% and 0.14% ± 0.053% in the untreated group;
In vivo rat study for assessment in short- and long-term effects in the whole-body system
Although
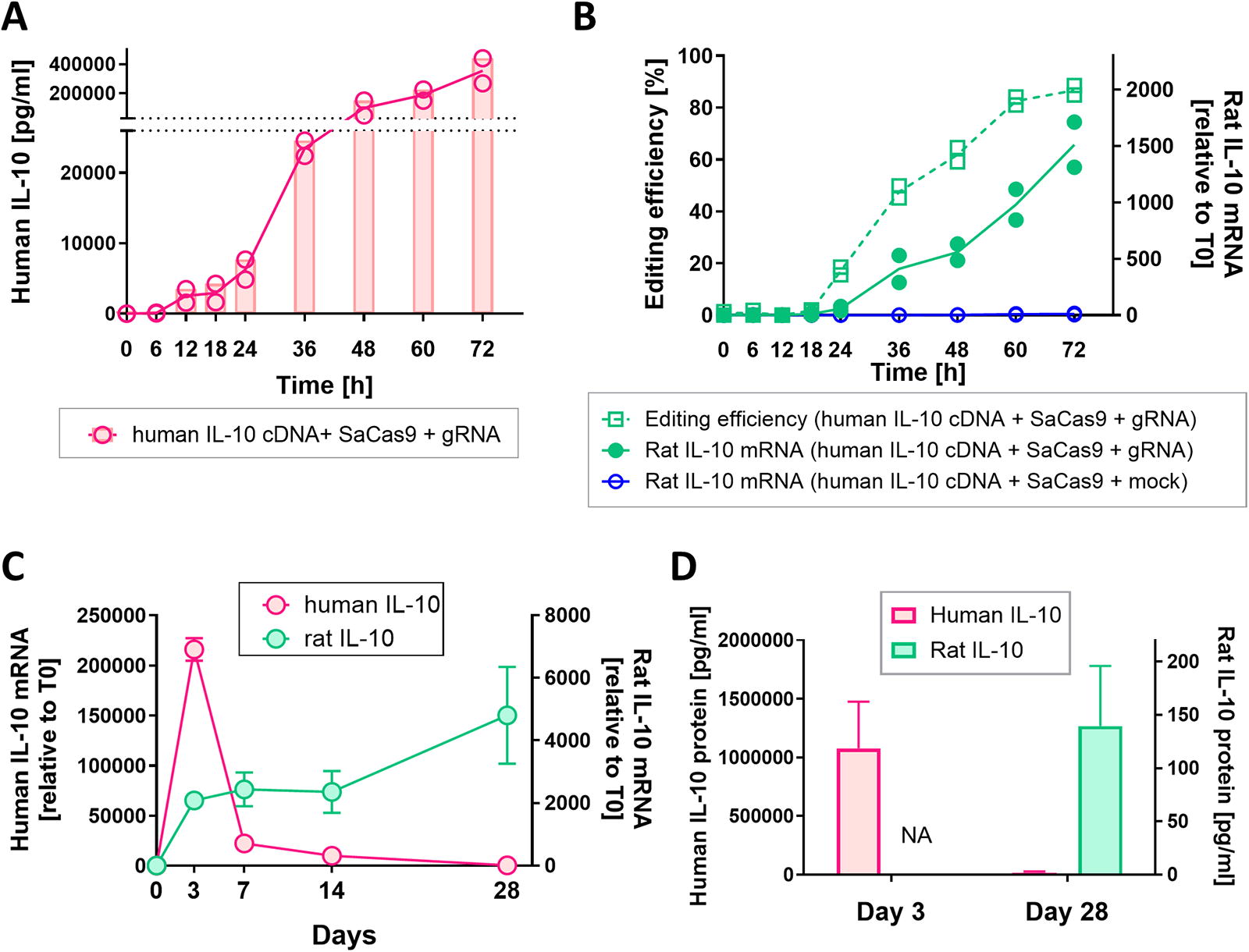
A dual mode of IL-10 expression after single dosing of adenoviral vectors in rat lung cells. L2 cells were transduced with adenoviral vectors, which express human IL-10, SaCas9, and gRNA, at a multiplicity of infection (MOI) of 1100. The culture media and cell lysate were collected at each time point. The IL-10 protein levels in the media and mRNA levels in cell lysate were measured by ELISA and RT-qPCR, respectively. In RT-qPCR analysis, relative expression to the start time (T0) is shown. Genome editing efficiency was analyzed by Sanger sequencing.
The AdV-hIL10-CRISPR was next delivered to the rat lung
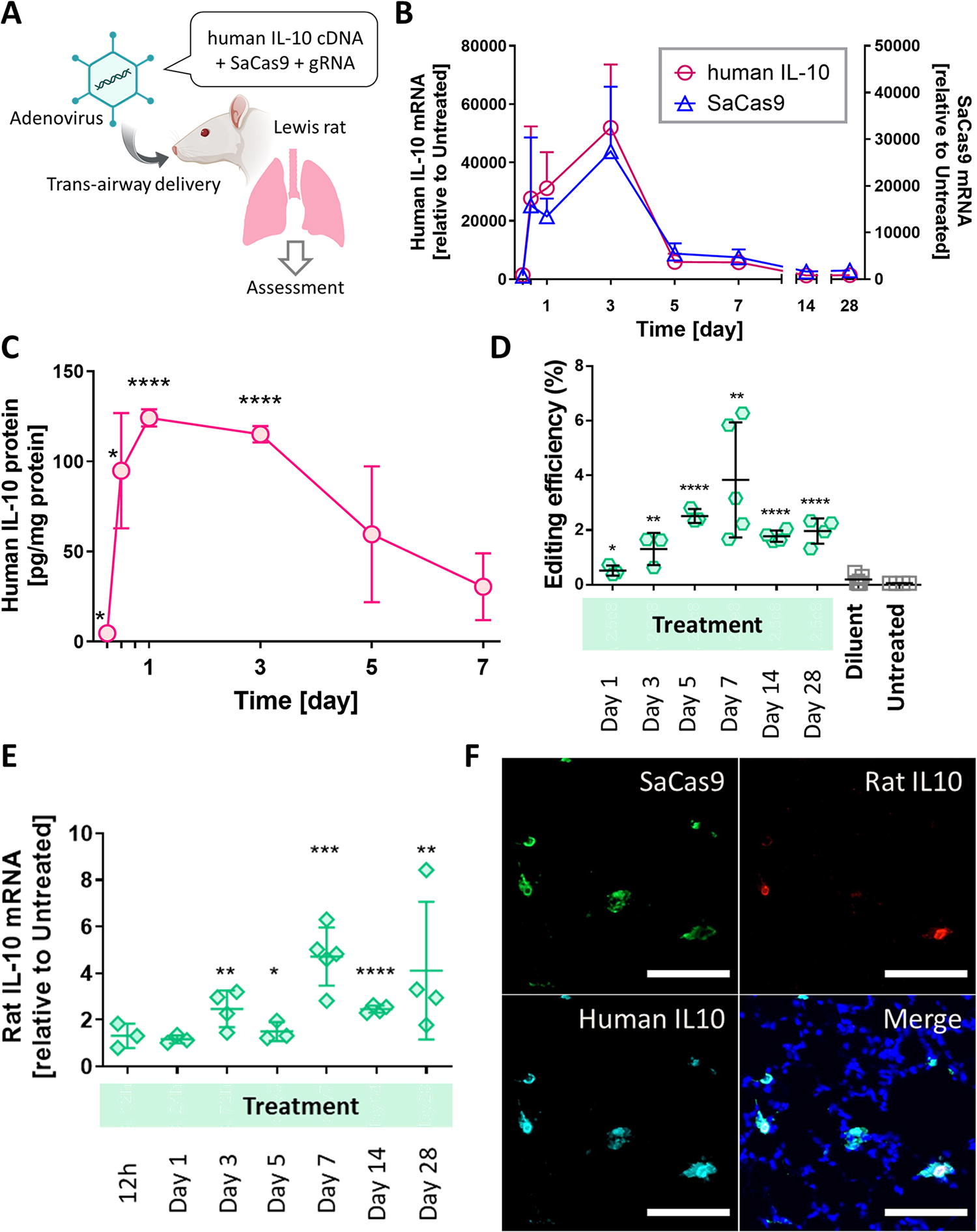
Assessment of genome editing combined with human IL-10 cDNA delivery
In summary, our
DISCUSSION
In this proof-of-concept study, we have successfully demonstrated the feasibility of genome editing in human lungs
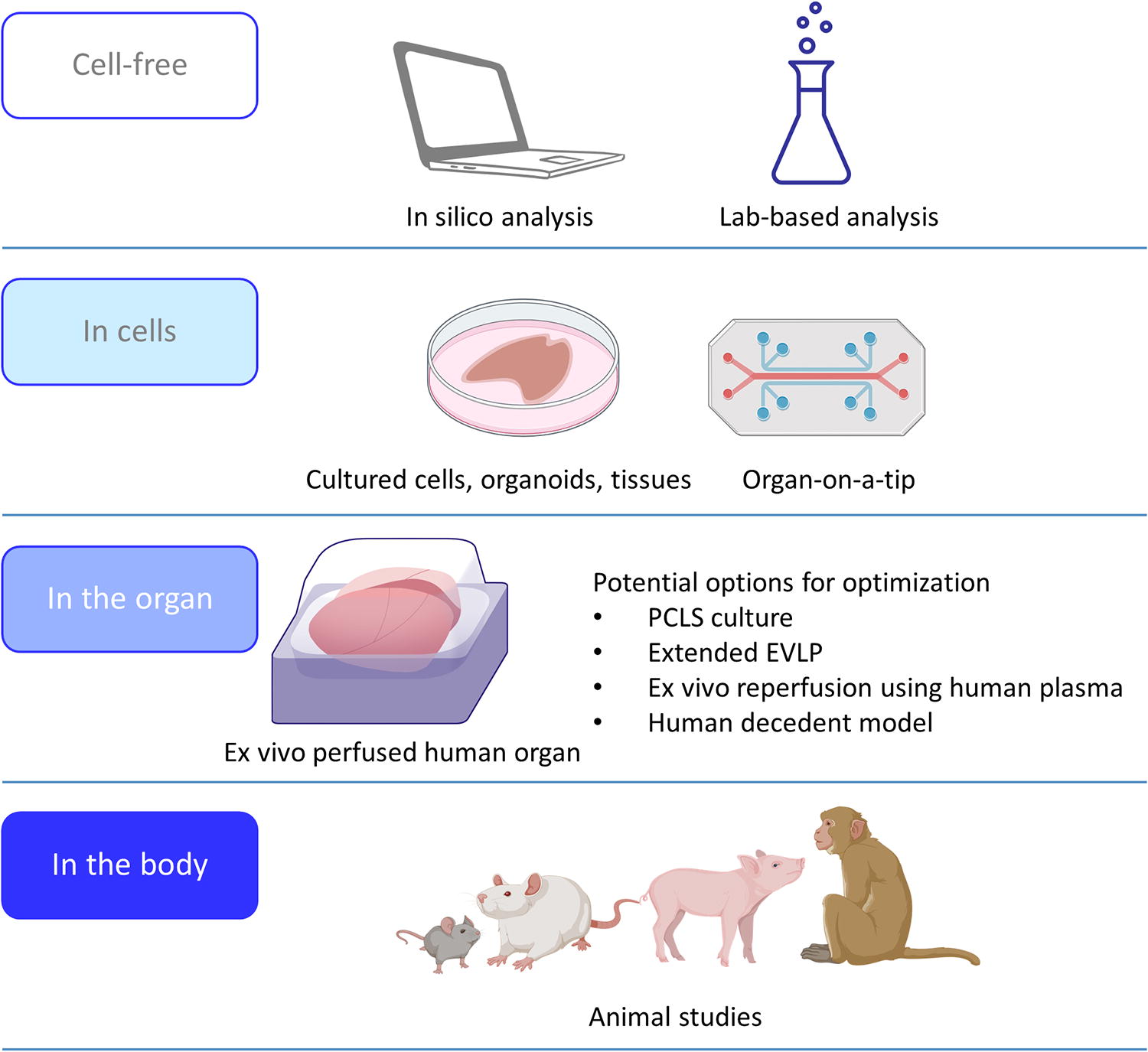
New modalities for future translational path of genome-targeting therapeutics. Studies using
Although genome editing demands long-term observations, the lack of suitable transplant models presents a challenge for extended follow-up of treated human organs. In this study, we incorporated PCLS culture technology, allowing us to monitor interventions in live lung tissue for 14 days. This integration of tissue culture techniques provided an excellent model for extended assessment of human genome editing, a previously unestablished endeavor. Furthermore, we conducted an
In this study, we discovered that genetic disruption of a regulatory element enhances IL-10 expression. Gain-of-function genome editing often faces the challenge of low-efficiency DNA integration, 38,39 which limits its application to entire organs. However, the derepression approach offers a solution, achievable through simple editing methods such as generating DSBs using Cas nucleases and introducing base substitutions with base editors. These highly efficient editing techniques significantly increase the success rate of therapeutic organ editing. Potential strategies to release natural repression include (1) reducing the binding affinity of the repressor by perturbing the regulatory element, (2) reducing repressor expression, and (3) recapitulating single nucleotide polymorphism (SNPs) associated with the desired phenotype. For the IL-10 gene, the mechanism of its native repression remains unknown, prompting us to identify target loci through phenotypic screening. The region we identified does not overlap with known repressor candidate-binding motifs 40 –43 or putative silencers. 44 In addition, the region targeted in this study differs from three SNPs in the regulatory region of the IL-10 gene, namely, −592 (rs1800872), −819 (rs1800871), and −1082 (rs1800896), which are associated with elevated IL-10. 45 Therefore, to the best of our knowledge, the regulatory element edited in this study represent previously unexplored regions. Further investigations, such as high-throughput screening, would greatly support deciphering the molecular basis of IL-10 regulation. The editing efficiency we observed in lung tissue is comparable with the transduction efficacy following AdV delivery to the lung via the airway. IL-10 enhances immunomodulatory function through both paracrine and autocrine mechanisms, indicating that even a small population of edited cells could yield therapeutic benefits for graft immunomodulation. Further optimization of transduction efficiency, potentially using AAV or non-viral delivery methods, as well as fine-tuning IL-10 upregulation at the cellular level by refining the editing approach, and enhancing editing efficiency, such as using base editors for more uniform mutations, 46 would allow more precise control of IL-10 levels in the graft.
CONCLUSION
In this study, we successfully developed a genetic engineering approach for lung graft immunomodulation and demonstrated genome editing in human lungs
Footnotes
ACKNOWLEDGMENTS
The authors thank STTARR at UHN for performing
AUTHORS’ CONTRIBUTIONS
K.M. and S.K. conceptualized the project. K.M. performed experiments, analyzed data, and wrote the article under the supervision of S.K. S.J., J.H., A.R.D., and S.K. guided the study through program advisory committee meetings of University of Toronto graduate program. K.M., H.Y., and A.M. conducted human lung study with the help of the Toronto EVLP team. J.Y., A.A., and G.W.W. supported next-generation sequencing. Z.G., Y.Y., H.M., and C.D. provided technical assistance in experiments. A.W., M.C., and M.L. guided the study through discussion at the meetings and supported article writing. B.P.K. advised on genome editing approaches and next-generation sequencing analyses. The article was reviewed by all authors.
AUTHOR DISCLOSURE
M.C. and S.K. are co-founders of Perfusix Canada and Scientific Advisors to Lung Bioengineering. S.K. is CMO of Traferox Technologies. UHN has a patent filed and pending for technologies related to donor organ modification, on which K.M., S.J., M.C., M.L., and S.K. are inventors. S.K. and M.C.’s interests were reviewed and are managed by UHN in accordance with their conflict-of-interest policies. A.R.D. is on the Scientific Advisory Board of Acrigen. B.P.K is an inventor on patents and patent applications filed by Mass General Brigham that describe genome engineering technologies. B.P.K. is a consultant for EcoR1 capital and is a scientific advisor to Acrigen Biosciences, Life Edit Therapeutics, and Prime Medicine. B.P.K. has a financial interest in Prime Medicine, Inc., a company developing therapeutic CRISPR-Cas technologies for gene editing. B.P.K.’s interests were reviewed and are managed by MGH and MGB in accordance with their conflict-of-interest policies.
FUNDING INFORMATION
This work was supported by the UHN Foundation, New Frontiers in Research Fund—Transformation Grant (NFRFT-2020-00787), and the Canadian Institutes of Health Research (CIHR PJT-173392).
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.