
Abstract
Adeno-associated virus (AAV) vectors represent a novel tool for the delivery of genetic therapeutics and enable the treatment of a wide range of diseases. Success of this new modality is challenged, however, by cases of immune-related toxicities that complicate the clinical management of patients and potentially limit the therapeutic efficacy of AAV gene therapy. While significant progress has been made to manage immune-related liver enzyme elevations following systemic AAV delivery in humans, recent clinical trials utilizing high vector doses have highlighted a new challenge to AAV gene transfer—activation of the complement system. While current in vitro models implicate AAV-specific antibodies in the initiation of the classical complement pathway, evidence from in vivo pre-clinical and clinical studies suggests that the alternative pathway also contributes to complement activation. A convergence of AAV-specific, environmental, and patient-specific factors shaping complement responses likely contributes to differential outcomes seen in clinical trials, from priming of the adaptive immune system to serious adverse events such as hepatotoxicity and thrombotic microangiopathy. Research focused on the interplay of patient-specific and AAV-related factors driving complement activation is needed to understand and identify critical components in the complement cascade to target and devise strategies to mitigate vector-related immune responses.
INTRODUCTION
As the leading platform for gene therapy, adeno-associated virus (AAV) vectors have safely and successfully been used to deliver long-term therapeutic gene expression to patients with a range of inherited and acquired diseases. 1 Clinical use of recombinant AAVs (rAAVs) for gene therapy takes advantage of the relatively weak immunogenic profile of this vector, which has a conveniently diversified tropism for many human tissues. Despite many advances, successful gene transfer in humans has been hampered by the activation of the immune system; pre-existing AAV neutralizing antibodies (NAbs) currently limit broader access of AAV therapies to a subset of seronegative patients. 2 With the use of progressively higher doses in AAV therapies (≥1 × 1013 vector genome [vg]/kg), adverse complement responses have also emerged in clinical trials (Table 1), prompting the need to better understand the interactions between the complement system and AAV both pre-clinically and clinically.
Complement-related thrombotic microangiopathy in adeno-associated virus gene therapy: clinical evidence
AAV, adeno-associated virus; CAG, cytomegalovirus (CMV) enhancer fused to the chicken beta-actin promoter; CK, creatine kinase; CMV, cytomegalovirus; DD, Danon disease; DMD, Duchenne muscular dystrophy; GLA, α-galactosidase A; IS, immunosuppression; IV, intravenous; LAMP2B, lysosome-associated membrane protein-2; MMA, methylmalonic acidemia; MMUT, methylmalonyl-CoA mutase; ND, not disclosed; SMA, spinal muscular atrophy; SMN1, survival of motor neuron 1; tMCK, truncated muscle creatine kinase; vg, vector genome.
The complement system is an ancient branch of the innate immune system that serves as the first line of defense against invading pathogens. The complement system is both highly reactive and tightly regulated so that it can rapidly respond to pathogens, while avoiding rampant activation resulting in bystander damage to host tissues. 3 Despite this, excessive or uncontrolled activation of the complement system has been linked to a diverse repertoire of human pathologies, including a rare complement-mediated form of thrombotic microangiopathy (TMA), atypical hemolytic uremic syndrome (aHUS). 4 So far, in the setting of gene therapy clinical trials, this form of TMA has only been observed following intravenous (IV) administration of high doses of AAV vectors (equal or above 1 × 1013 vg/kg).
The potential risks associated with aHUS, as with other forms of TMA, include hemolytic anemia, thrombocytopenia, acute kidney injury, progressive organ failure, and significant morbidity and mortality. 5 While complement-mediated serious adverse events (SAEs) in AAV gene therapy have been documented across trials and postlicensing, these events have yet to be comprehensively described in the literature. Due to the limited number of targeted studies investigating the interplay of complement and AAV biology, particularly in vivo, little is known mechanistically about complement activation and downstream signaling in response to AAV. Despite these AAV-specific knowledge gaps, extensive research has been conducted on the complement system that affords the gene therapy community a wealth of information that can be utilized toward developing studies to examine the interplay of complement and AAV, as well as mitigation strategies.
This review will cover the complement cascade with a focus on AAV-complement interactions observed in vitro, complement activation in preclinical and clinical studies, and potential mitigation of complement activation within the context of complex human biology.
THE COMPLEMENT SYSTEM
The complement system comprises over 50 soluble and membrane-bound proteins, most of which are produced in the liver, which circulate in the blood and through nearly every tissue of the body. In addition to extracellular activity, there is considerable evidence that intracellular complement activity (the “complosome”) and local secretion of complement proteins by immune cells are more pervasive than previously considered. 6 The complement system was first noted for its bactericidal activity in blood; however, it is now recognized to be involved in a broader range of functions, including surveillance and priming of the adaptive immune system, clearance of necrotic/apoptotic cells and debris, tissue regeneration, inflammation and homeostasis, and crosstalk with other pathways. 4
Activation of the complement system can be broadly grouped into three pathways—classical pathway (CP), lectin pathway (LP), and alternative pathway (AP)—that converge on a common terminal pathway (Fig. 1). Regardless of the initiating trigger, activation of zymogen precursors leads to a proteolytic enzymatic cascade that merges at the formation of C3 convertase and the cleavage of C3. On the most fundamental level, cleavage of C3 leads to the production of inflammatory anaphylatoxins C3a and C5a, opsonization of surfaces by C3b, and formation of the C5b-9 membrane attack complex (MAC). Central to the amplification of the complement cascade is C3 split product, C3b, which in addition to opsonization is essential for the formation of the AP C3 and C5 convertases and the initiation of the AP amplification loop. 7
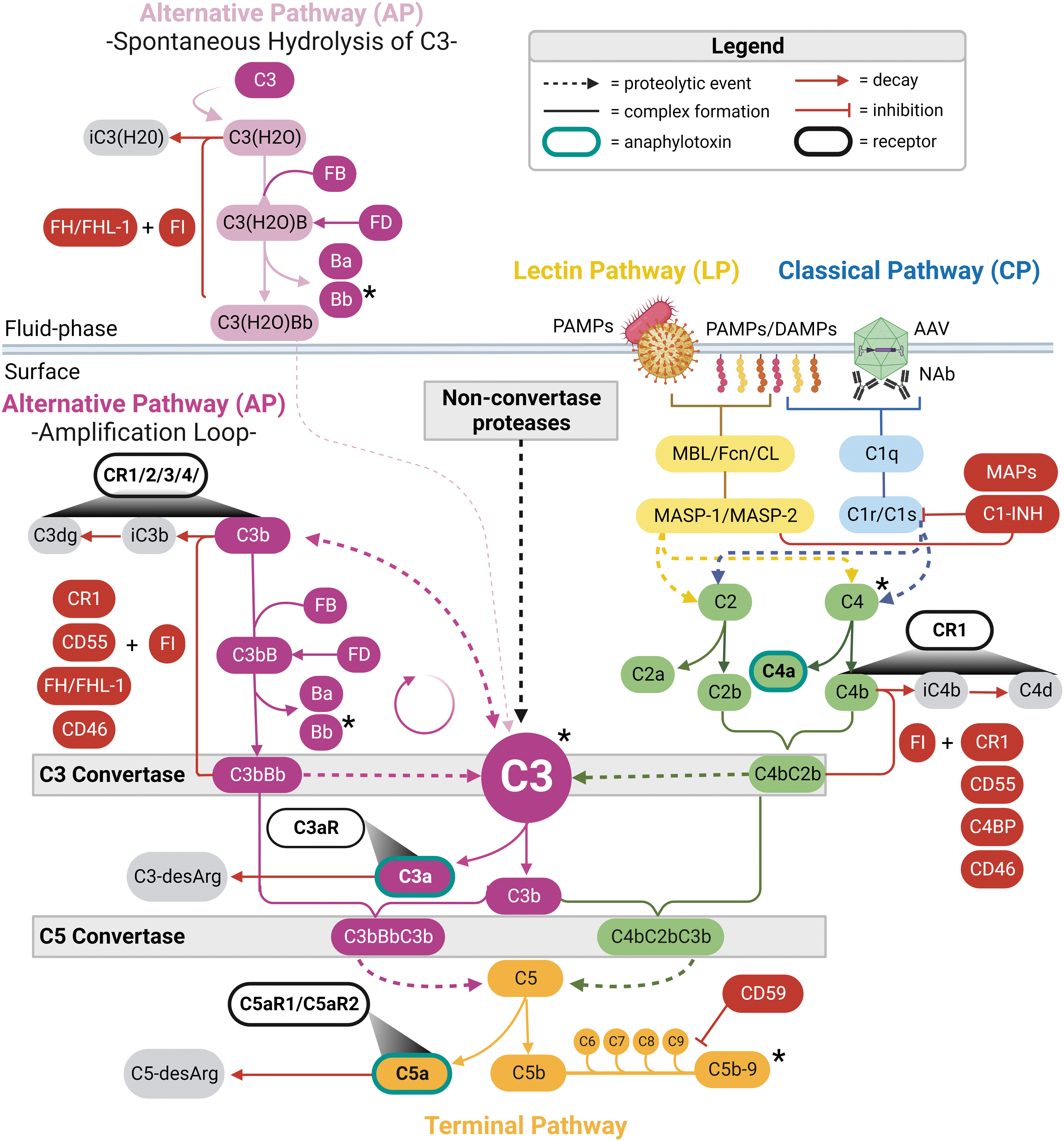
Schematic illustration of the complement cascade. The complement system cascade is initiated by PRMs of the classical pathway (C1 complex; C1q complexed with C1r and C1s) and the lectin pathway (MBL, Fcn, and CL complexed with MASP-1 and MASP-2). Complement system PRMs associate with non-self and altered-self molecular patterns on apoptotic cells, Fc regions of antigen-bound antibodies (e.g., anti-AAV antibodies bound to AAV), PAMPs, and DAMPs. In the fluid phase, the alternative pathway is constitutively active at low levels through the spontaneous hydrolysis of C3 to C3(H2O). Downstream activation of complement zymogen precursors results in a proteolytic enzymatic cascade that converges at the formation of C3 convertases [C4bC2b, C3bBb, and C3b(H2O)Bb] and the cleavage of C3 to anaphylatoxin C3a and opsonin C3b. Nonconvertase proteases can also cleave C3 and initiate the complement cascade independent of C3 convertases. Opsonization of target surfaces by C3b generates a positive feedback loop through the alternative pathway that generates additional C3 convertases and bioactive C3 split products, and drives the formation of C5 convertases (C3bBbC3b and C4bC2bC3b). C5 convertases cleave C5 to anaphylatoxin C5a and C5b, leading to the formation of the lytic MAC, C5b-9. CRs play an important role in regulating inflammation, leukocyte recruitment, and phagocytosis. Anaphylatoxins C3a and C5a engage with G protein-coupled receptors (C3aR or C5aR) to mediate inflammation and chemotaxis to sites of inflammation. Surfaces opsonized by C3 and C4 fragments (C3b, iC3b, C3dg, C3d, and C4b) interact with receptors CR1, CR2, CR3, and CR4 to mediate phagocytosis, antigen presentation, and enhance adaptive immune responses. The complement cascade is tightly regulated to prevent hyperactivation and bystander damage to host tissues. Soluble SP inhibitor FI along with RCA cofactors (FH, FHL-1, CD46, CR1, and C4BP) inactivate C3b and C4b to prevent C3 convertase formation, while decay-accelerating cofactors (CR1, CD55, FH, and C4BP) dissociate C3 convertases to limit their activity. Modified from Pouw and Ricklin. 4 *Denotes complement analytes observed to change in instances of complement-mediated SAEs following AAV gene transfer. AAV, adeno-associated virus; C4BP, C4 binding protein; CL, collectins; CR, complement receptor; Fcn, ficolins; FH, factor H; FHL-1, factor H-like 1; FI, factor I; MAC, membrane attack complex; MASP, MBL-associated serine protease; MBL, mannose-binding lectin; PAMPs, pathogen-associated molecular patterns; RCA, regulators of complement activation; SAE, serious adverse event; SP, serine protease.
AAV ANTIBODY-DEPENDENT COMPLEMENT ACTIVATION: THE CP
The seroprevalence of antibodies against AAV is estimated to be 40–80% of the world population, indicating that most people have been infected by wild-type AAV (wtAAV) in their lifetime. Broad cross-reactivity of anti-AAV NAbs due to structural homology between AAV serotypes is reflected in their ability to neutralize a range of diverse serotypes and recombinant vectors used for gene transfer. 8
Pre-existing NAbs present in individuals before AAV vector infusion can bind to the AAV capsid and limit the efficacy of AAV gene transfer. For this reason, the presence of pre-existing anti-AAV NAbs is an exclusion criterion for many systemic AAV gene therapy programs. However, screening assays to determine NAb titer cutoffs for inclusion in AAV gene therapy trials vary broadly in their sensitivity and methodology, complicating the analysis of seropositivity, even between closely related capsids, with clinical outcomes. An additional challenge presented by pre-existing anti-AAV antibodies is their potential role in complement system activation.
The conventional understanding of the CP is that it is initiated by antigen-antibody immune complexes. This, however, is a broad oversimplification that requires a more nuanced understanding of C1q as a pattern recognition molecule (PRM) that recognizes over 100 different targets, including damage-associated molecular patterns (DAMPs) on altered host cells, acute phase proteins such as C-reactive protein (CRP), and activated platelets. 3 To avoid erroneous complement activation by C1q, IgG oligomerization or clustering of DAMPs is needed for avid activation. 9 Multivalent IgM antibodies do not require oligomerization and are therefore strong activators of the CP.
Upon binding to its target, C1q domains in the C1 complex initiate a conformational change, resulting in the activation of associated serine proteases (SPs) C1r and C1s, which cleave C4 and then C2 to their respective a and b subunits. The CP C3 convertase (C4bC2b) is subsequently formed by noncovalent binding of surface-bound C4b to C2b (Fig. 1). Each molecule of C4bC2b can cleave up to 1,000 C3 molecules, which exist in circulation at high concentrations of ∼1.2 mg/mL. 7 Cleavage of C3 results in the release of short-lived bioactive anaphylatoxin C3a and opsonin C3b, the latter of which indiscriminately coats self and non-self surfaces in the immediate vicinity (Fig. 1).
Early in vitro studies by Zaiss et al. showed that AAV-induced complement activation by AAV2-GFP at >1 × 1012 particles/mL exclusively occurred in seropositive human serum. An antibody-dependent mechanism for in vitro complement activation by AAV was reinforced by demonstrating that IgG depletion or ethylene glycol tetraacetic acid (EGTA) treatment, to selectively block CP and LP activation, prevented AAV-induced complement activation. 10
Additional in vitro work by Smith et al. showed that pre-existing anti-AAV antibodies to AAV-Spark100 and AAV-LK03 preferentially activated complement and other components of the innate immune system only at high NAb titers (≥1:100). 11 Following human whole blood stimulation at 5 × 1011 vg/mL, high titer donors, but not low NAb titer (≤1:10) donors, had significantly elevated C3a in the serum and inactive C3b split product (C3d) opsonization on neutrophils, indicative of CP activation (Fig. 2).
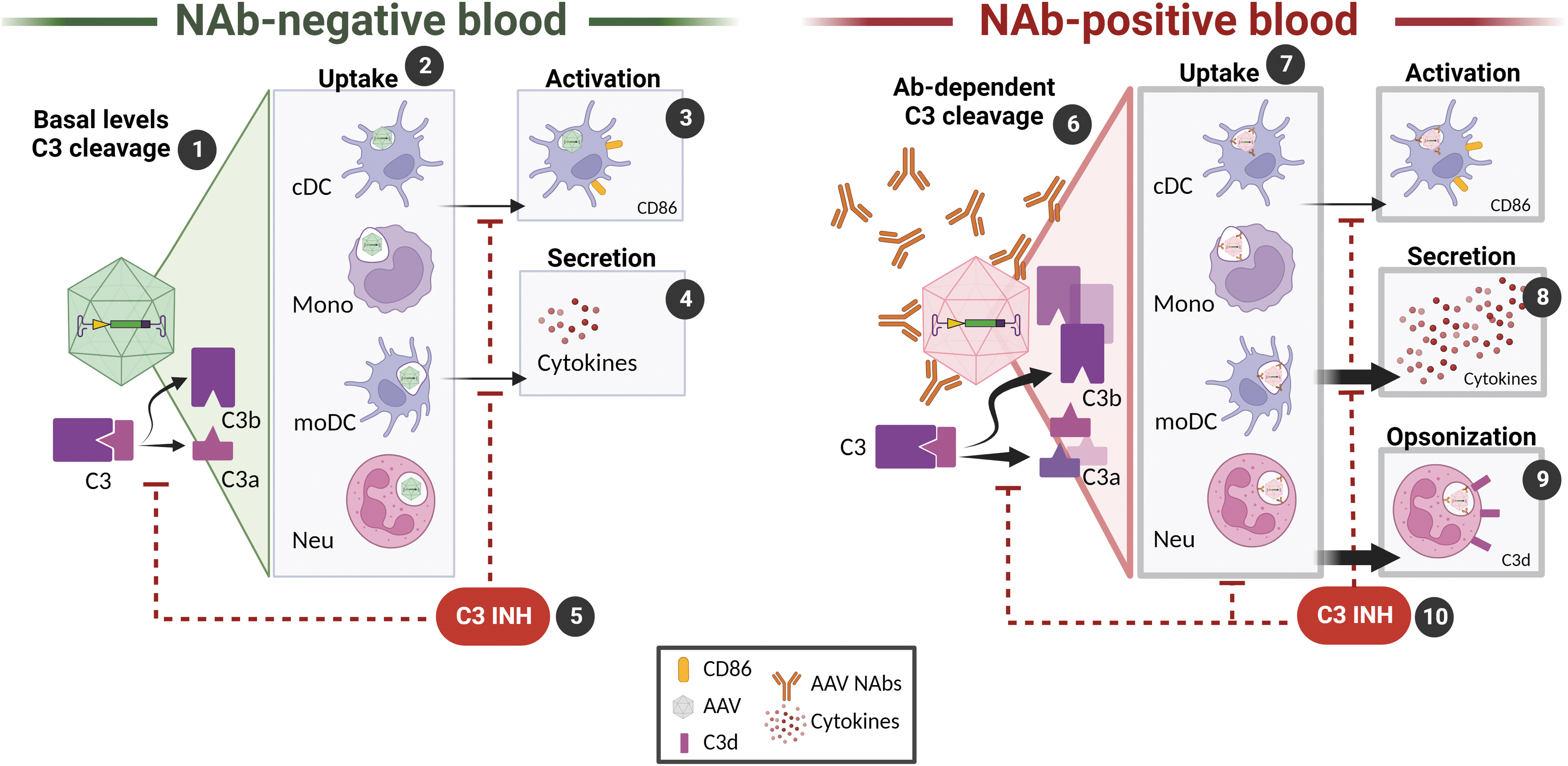
Interplay of AAV with complement in NAb-positive and NAb-negative human blood in vitro.
In addition to complement activation, high titer anti-AAV NAbs enhanced the uptake of AAV by blood phagocytes and dendritic cells (DCs). The uptake of AAV resulted in the upregulation of activation marker CD86 on monocyte-related DCs (moDCs) and elevated cytokine/chemokine secretion (Fig. 2). Addition of C3 complement inhibitor, APL-9 (Apellis), at the time of AAV stimulation, resulted in a statistically significant decrease in AAV uptake, moDC activation, and cytokine/chemokine release in seropositive as well as seronegative blood (Fig. 2). Together, these findings support antibody-dependent complement activation and the dependence on complement for the propagation of other AAV-induced innate immune responses in vitro.
The in vitro findings that AAV uptake and activation by myeloid cells are enhanced in the presence of complement and anti-AAV antibodies are further supported by a recent in vivo AAV re-administration mouse study by Emami et al. 12 In this study, dystrophin-deficient mice were retro-orbitally dual-dosed 4 weeks apart with ∼1.16 × 1014 vg/kg of an AAV9 vector expressing either a Cas9 or microdystrophin (μDYS) transgene.
Plasma analysis revealed significant consumption of C3 and C4 and release of proinflammatory cytokines and chemokines only after the second vector infusion, when high levels of both AAV and anti-AAV antibodies were in circulation. Because the upregulation of Fc-gamma receptors (FCγR) and complement receptors (CRs) was observed exclusively on activated monocytes, the authors hypothesized that both complement activation and AAV uptake were antibody dependent. However, the finding that two successive doses of an AAV9 vector at >1 × 1014 vg/kg are required to detect moderate complement consumption in mice strongly suggests this model has limited translational value to study complement responses to AAV.
Recent studies performed by West et al. demonstrated a NAb titer threshold of ≥1:5 for in vitro complement activation by 2 × 1012 empty AAV9 capsids in human serum, which could be reduced or eliminated through immunoglobulin removal or IgG degrading enzyme, immunoglobulin G-degrading enzyme of Streptococcus pyogenes (IdeS). 13
Interestingly, IdeS treatment, which cleaves IgG at the hinge region, but leaves potent complement activator IgM intact, reduced complement activation by >50% in most NAb-positive donors, despite the presence of anti-AAV IgM antibodies. It is important, however, to distinguish between low levels of pre-existing anti-AAV IgM antibodies analyzed in this context and high-affinity capsid-specific IgM antibodies that arise following AAV gene transfer, which may be more effective at activating the complement cascade. Mass spectrometry analysis of serum incubated with AAV9 revealed that key components of the CP such as C1q associate with the AAV capsid only in the presence of anti-AAV antibodies.
Together, these findings support the original hypothesis by Zaiss et al. that the antigen-antibody complexes are the primary driver of CP activation by AAV. While no evidence of LP or AP activation was observed in these studies, dependence on antibodies for complement activation in humans and nonhuman primates (NHPs) remains to be determined. Importantly, in vitro measurement of complement is complicated by anticoagulants used for whole blood and plasma collection, which chelate Ca2+ and Mg2+ and inhibit complement activation.
Conversely, blood clotting steps required for serum isolation can prematurely activate complement and affect subsequent measurement of complement levels. 14 While the use of the direct thrombin inhibitor, hirudin, as an anticoagulant for complement activation analysis is recommended, it is not widely available. 15 Recombinant hirudin derivatives such as desirudin, bivalirudin, and now discontinued lepirudin are alternatives to natural hirudin; however, procurement, short half-life, and the preparation of custom blood collection tubes can present challenges for routine use. 16
AAV ANTIBODY-INDEPENDENT COMPLEMENT ACTIVATION: THE AP
The AP was characterized in the 1970s following experiments using cobra venom factor (CVF) that revealed an antibody-antigen-independent mechanism by which C3 was cleaved by a complex consisting of C3b, Factor B (FB) and Factor D (FD) (Fig. 1). In the following years, this unique C3-cleaving enzyme was identified as C3bBb, or the AP C3 convertase. Importantly, it was shown that C3bBb is stably formed on surfaces where it recruits properdin (Factor P), which increases the half-life of the AP C3 convertase >10-fold and protects it from degradation by complement inhibitors. 7
Fluid-phase C3bBb is rare because it cannot bind properdin and is rapidly inactivated by soluble complement inhibitors. The source of the C3b fueling the AP, however, was not clear and became a source of speculation and dispute in the field. In 1975, the term “C3 tick-over” was proposed by Lachmann et al. 17 as the mechanism by which low levels of C3 were cleaved to generate small amounts of C3b capable of initiating the AP. This theory is supported by the fact that low, but constant levels of C3a (∼100 ng/mL), which has a half-life of 30 min, are present in the blood of normal healthy individuals at baseline. 18
In the 1980s, the “C3 tick-over” theory was further modified by Pangburn et al. 19 to incorporate the recent discovery of “C3b-like” C3(H2O), generated through the continuous spontaneous hydrolysis of C3. Like C3b, C3(H2O) can bind FB and subsequently FD to form fluid-phase C3 convertase, C3(H2O)Bb (Fig. 1). Mounting evidence, however, suggests that C3(H2O) might not be as “C3b like” as previously proposed and that the C3b that drives the AP is not produced by C3(H2O)Bb convertase activity. 20
These findings have ushered in a movement to rebrand the AP as an amplification loop instead of a pathway. Using this model, even small amounts of C3b generated by the CP and LP, in addition to C3 cleavage by noncomplement proteases, are enough to initiate a powerful amplification feedback loop through the AP. Significantly, it is believed that up to 90% of complement activation occurs through the AP even when initiated through the CP and LP. 21
Despite clear findings from in vitro studies regarding the role of AAV-specific antibodies in CP initiation, data from NHP studies indicate antibody-independent mechanisms may be driving complement activation in response to high-dose AAV in vivo. Following toxicities involving thrombocytopenia, elevations in liver transaminases, coagulopathy, and hepatocellular loss in NHPs IV administered >1014 vg/kg of an AAV9 variant (AAVhu68), 22 several studies have shown transient complement activation peaking 3–4 days postinfusion, alongside decreased platelet counts and elevations in AST and ALT in NHPs and mice. 23 –25
Interestingly, in all these studies, complement activation was observed before the development of detectable capsid-specific IgG and IgM antibodies and showed a marked increase in AP complement marker Bb (FB split product) and soluble MAC, sC5b-9, compared with CP-associated markers. 24 –27 These findings and toxicities observed in the clinic may highlight a potential role of tissue damage, DAMPs, and nonconvertase proteases in the initiation of the complement system in vivo, in the absence of anti-AAV antibodies. Of note, similar discrepancies in antibody dependence between in vitro and in vivo activation of complement were noted in studies utilizing adenovirus. 28 One of the major drawbacks to in vitro modeling of complement activation, however, is the lack of cytotoxic damage to cells and tissues observed in vivo, which not only exposes complement-activating DAMPs but also releases noncomplement proteases such as trypsin, chymotrypsin, and elastase, which can proteolytically cleave C3 independent of C3 convertase. 29
Due to the kinetics of complement activation and TMA observed in presumed NAb- and total antibody (TAb)-negative NHPs and humans administered AAV gene therapy, measures have been taken to analyze AP-specific complement activation markers. This analysis, as mentioned earlier, has revealed peaks in AP-specific analyte Bb and terminal complex sC5b-9, which coincide with biomarkers of liver toxicity and TMA. 22 –24,26,27 The finding that the AP may be driving complement activation in response to high-dose AAV, while contrary to what has been seen in vitro, reveals that antibody-independent mechanisms might be contributing to complement activation in vivo.
While it has been hypothesized that C3 tickover, specifically C3(H2O), could be initiating the AP response to AAV, there is no evidence that this convertase is formed efficiently in vivo or that it has enough stability to drive an AP response of the magnitude observed in high-dose AAV gene therapy. Collectively, these findings indicate that, while complement activation in response to AAV may be amplified through the AP in an antibody-independent manner, the driver of this response is unlikely to be C3(H2O).
THE LECTIN PATHWAY
Activation of the LP also results in the formation of C3 convertase C4bC2b; however, the initiating triggers are different from the CP. The LP is activated by the binding of soluble PRMs, including mannose-binding lectins (MBLs), collectins (CL), and ficolins (Fcn), to carbohydrate ligands on some microorganisms, as well as DAMPs on damaged or apoptotic cells (Fig. 1). 3 Complexes of PRMs associate with inactive MBL-associated SPs (MASP-1, MASP-2, and MASP-3), much as C1q associates with SPs C1s and C1r. Upon activation, MASP-1 and MASP-2 cleave C4 and C2 to generate C3 convertase, C4bC2b. Due to the intersection of the CP and LP pathways following cleavage of C4 and C2, the resulting C3 convertase is often referred to as the CP/LP C3 convertase (Fig. 1). To avoid aberrant formation of the CP/LP C3 convertase, C1 inhibitor (C1-INH) and sMAP/MAP-1 (Map19 and Map44) inhibit the initiation of the CP and LP, respectively. 3
While glycosylation of nonenveloped viruses is not as common as enveloped viruses, post-translational modifications of AAV capsids can introduce glycosylated peptides. Recent characterization of AAV serotypes 1–9 has revealed a high abundance of mannosylated N-glycans, which could activate the LP following AAV gene transfer. 30 While there is no evidence of LP activation clinically or preclinically, it is still possible that some of the activation that has been attributed to the CP could be coming from the LP. Independent of LP PRMs, DAMPs produced by endothelial injury to host cells have been shown to activate LP SP MASP-2, which can cleave prothrombin to thrombin, resulting in the activation of both the complement and coagulation cascades. 31 As with the CP, activation of the LP by DAMPs and crosstalk with the coagulation cascade may potentially present an alternative mechanism by which high-dose AAV could be activating complement.
THE TERMINAL PATHWAY
The addition of C3b to preformed C3 convertases from any of the complement pathways results in the formation of C5 convertases C4bC2bC3b and C3bBbC3b, initiating the terminal pathway of the complement cascade (Fig. 1). Importantly, as with C3, C5 can be cleaved by noncomplement proteases, including thrombin and human factors XIa, Xa, and IXa from the coagulation cascade. 3 Particularly, on surfaces densely coated in C3b and C3bBb, there is a preferential affinity for the formation of C5 convertases (vs. C3 convertases), resulting in an escalation of proinflammatory signaling by potent C5a and the formation of the lytic pore complex, MAC (C5b-9). The primary function of the MAC is to osmotically lyse Gram-negative bacteria, enveloped viruses, parasites, and metabolically inert erythrocytes. Metabolically active nucleated cells largely resist lysis due to stringent regulation by fluid-phase and membrane-bound (CD59) inhibitors, as well as active shedding or internalization of the MAC. 3
While identification of C5b-9 deposits on nucleated cells has been observed in patients with aHUS as well as preclinical AAV studies in NHPs, there is considerable evidence that C5b-9 deposits can also have non-lytic functions that are poorly understood. 25,32 Non-pore-forming MAC deposits on nucleated cells, particularly endothelial cells, can activate cells and induce downstream intracellular signaling. These activities have been shown to activate the coagulation process as well as adaptive immune responses and enhance disease pathogenesis through the activation of immune cells and the production of proinflammatory cytokines. 33
Bystander damage to host cells due to hyperactivation of the complement system can occur when excessive complement activation overpowers complement inhibition, resulting in the assembly of MAC on host cells. 34 This phenomenon of C5b-9 bystander lysis of host cells, which is observed in a range of human diseases, including aHUS, offers one possible explanation for C5b-9 assembly on host tissues following AAV gene transfer. 35 Additional research is ongoing to understand the promiscuous roles of C5b-9; however, the lytic activity of C5b-9 is only one of its terminal pathway functions that could be at play following AAV gene transfer.
ANAPHYLATOXINS AND EFFECTOR MECHANISMS OF COMPLEMENT ACTIVATION
The effector mechanisms of the complement cascade form an essential link with the adaptive immune system to amplify and accelerate the humoral and cellular response to pathogens. The bridging of the innate and adaptive responses by complement is carried out primarily by the engagement of anaphylatoxins (C3a and C5a), opsonins (C4b, C3b, and iC3b), and the terminal MAC with CRs on immune and nonimmune target cells. 36 In addition to their interactions with lymphoid cells of the adaptive immune system, fragments from C4, C3, and C5 cleavage can engage in crosstalk with other innate immune mechanisms such as TLRs and coagulation. While these activities may not result in observable toxicity or disease, their contribution to the immune response to AAV gene therapy is potentially significant.
Anaphylatoxins C3a and C5a are small (∼10 kDa) bioactive fragments that mediate proinflammatory functions through binding of their respective G-coupled receptors, C3aR and C5aR (Fig. 1). While anaphylatoxins are produced through the cleavage of C3 and C5 by convertase and nonconvertase proteases, they are also known to be generated locally by T cells and other cell types where they participate in intracellular signaling and cell homeostasis.
Inflammation induced by anaphylatoxins results in vasodilation and migration of macrophages, neutrophils, activated B and T cells, basophils, and mast cells toward the gradient established by C3a and C5a. C3aR and C5aR engagement together with TLR co-stimulation (TLR-2, TLR-4, and TLR-9) have been shown to induce production of inflammatory cytokines interleukin-6 (IL-6), IL-1β, and tumor necrosis factor-α (TNF-α). 36 Conversely, C5a/TLR co-stimulation can have anti-inflammatory activities such as reduced IL-12, IL-23, and TNF-α production, which stunts the development of Th17 and Th1 T cells. Importantly, C3aR and C5aR engagement on T cells and antigen-presenting cells (APCs) can enhance T cell survival and proliferation, influence the development of interferon-γ (IFN- γ) producing Th1 cells, and suppress the development of regulatory T cells (Tregs). 36
Surfaces opsonized by C3 and C4 fragments (C3b, iC3b, C3dg, C3d, and C4b) interact with CRs CR1 (CD35), CR2 (CD21), CR3 (CD11b/CD18), and CR4 (CD11c/CD18), and the complement receptor of immunoglobulin (CRIg) on leukocytes. Complement opsonization leads primarily to phagocytosis, antigen presentation, and cytokine release that activates cells of the adaptive immune system. The interaction of C3b- and C4b-opsonized surfaces with CR1 on red blood cells results in shuttling of these complexes to the liver and spleen where they are phagocytosed by Kupffer cells or marginal zone macrophages and DCs. CR3 on macrophages, monocytes, neutrophils, and other leukocytes mediates the phagocytosis of iC3b-opsonized pathogens.
In addition, iC3b- and C3d-opsonized surfaces interact with CR2 on B cells to lower the threshold for B cell activation up to 10,000 times and together with CR2 on follicular DCs contribute to memory B cell formation. 36 Importantly, monomeric deposition of C3 fragments does not result in the engagement of CRs, which requires clustering at supraphysiological concentrations. 18
While many of the effector functions involved in AAV-complement interactions remain to be elucidated, studies by Zaiss et al. and Smith et al. have characterized important interactions of AAV and complement impacting humoral and cellular adaptive immune responses. Work by Zaiss et al. found that incubation of AAV2-GFP, AAV1, and AAV8 vectors with human macrophage-like phorbol myristate acetate (PMA)-differentiated THP-1 cells, in the presence of complement-intact human serum, resulted in macrophage activation and increased the expression of proinflammatory cytokines and chemokines. 10 Opsonization by iC3b, which is known to enhance uptake through CR3 on phagocytes, was found to associate with the AAV capsid in an antibody-independent manner, indicating that AAV uptake was likely enhanced by C3 split product deposition.
Similar findings by Smith et al. mentioned earlier, demonstrated that uptake of AAV by phagocytes and APCs in human whole blood resulted in the activation and secretion of proinflammatory cytokines and chemokines that were enhanced in the presence of high anti-AAV NAbs. 11 In vivo studies in C3 and CR1/2 knockout mice IV injected with 2.5 × 1011 particles of AAV2-GFP found that the development of anti-AAV NAbs was delayed by ∼1 week and titers were significantly reduced in the absence of CR1/2 and C3. These findings are supported by research showing that the interaction of CR2 on B cells with surfaces, such as AAV, opsonized by C3b split products lowers the threshold for B cell activation. 36
CONVERTASE-DIRECTED COMPLEMENT REGULATION
Soluble and membrane-bound complement inhibitors provide checkpoints that confine the potentially destructive activity of the complement cascade to the surfaces of foreign pathogens and protect healthy host tissue from bystander damage. To avoid erroneous C3 convertase assembly on the surface of host cells, C3b and C4b that bind to self surfaces are degraded to inactive split products (iC3b, C3dg, iC4b, and C4d) by factor I (FI) and cofactors FH, FHL-1 (Factor H-like 1), C4BP (C4 binding protein), MCP (membrane cofactor protein, CD46), and CR1 (complement receptor 1, CD35) belonging to the regulators of complement activation (RCA) family (Fig. 1).
The requirement of RCA cofactors for the proteolytic activity of FI determines FI's specificity for C3b, whereby C3b bound to host cells is inactivated, while C3b bound to non-self surfaces is spared. While the inactive forms of C3b can still engage with receptors on immune cells to facilitate phagocytosis, the split products of C3b and C4b lose their ability to form stable C3 and C5 convertases. To inhibit the assembly and shorten the half-life of C3 and C5 convertases, decay acceleration factors (DAF, CD55), CR1, FH, and C4BP destabilize or displace the enzymatic components of the C3 convertases. 37
Some pathogens have evolved mechanisms to avoid complement activation by developing cofactor functions or directly associating with FH. While AAV does not have cofactor activity, AAV has been demonstrated to recruit FH to the capsid surface to inactivate C3b. 10 Importantly, FH and its family of FH-related proteins (FHR1–5), which are thought to act as FH antagonists, are the most frequently mutated complement proteins in humans. 38
While some mutations in the complement Factor H (CFH) gene can result in truncated or absent levels of FH in the plasma, other FH point mutations can alter the binding of FH to surfaces and C3b itself, resulting in suboptimal complement regulation to varying degrees. 39 The term “complotype” has been used to describe patterns of inherited complement polymorphisms found in individuals. Central to these designations is the finding that plasma concentration levels, binding strength, and functionality of complement proteins such as FH, which determine the ability of host cells to regulate complement, can vary widely between individuals. In the case of complement activation following AAV gene therapy, patient complotype is likely to impact the degree of complement amplification.
COMPLEMENT ACTIVATION AND aHUS IN AAV CLINICAL TRIALS
Hepatotoxicity and SAEs, including TMA, have been observed recently following high dose (>1 × 1013 vg/kg) AAV gene therapies employing a range of capsid serotypes, transgenes, promoters, and routes of delivery (Table 1). In all instances, a rare and life-threatening complement-mediated form of TMA associated with overactivation of the AP, known as aHUS, appears to be involved. Significantly, while dose appears to play a significant role in the development of these SAEs, factors such as AAV serotype, disease indication, and manufacturing methods do not appear to correlate with the development of SAEs.
As with other forms of TMA, aHUS is characterized by microangiopathic hemolytic anemia, thrombocytopenia, and acute renal failure. Pathological features of TMA include red blood cell fragments (schistocytes), decreased haptoglobin, decreased hemoglobin, low platelet count (thrombocytopenia), and elevated lactate dehydrogenase. aHUS can be triggered by genetic defects in complement genes in the case of primary TMA or environmental triggers such as pregnancy, bacterial or viral infections, transplants, cancer, and drugs in complement-mediated secondary TMA. 40 Recent findings also support a “multiple hits” hypothesis, whereby aHUS is initiated due to concomitant environmental triggers and predisposing genetic risk factors such as loss-of-function mutations in genes encoding complement regulators and other variants of unknown significance.
In acute aHUS, complement dysregulation resulting in terminal pathway activation, generation of C5a, and deposition of C5b-9 on damaged endothelial surfaces promotes the procoagulation and proinflammatory microenvironment that leads to apoptosis and TMA. 41 Definitive diagnosis of aHUS is complex and involves ruling out other forms of TMA such as thrombotic thrombocytopenic purpura (TTP), which presents with the same symptoms as aHUS, but is caused by a deficiency in enzyme ADAMTS13. 5 This distinction is important, given both syndromes are life-threatening, but require markedly different treatments. Treatment with anti-C5 monoclonal antibody, eculizumab, has been shown to significantly alleviate symptoms of TMA in 80% of aHUS patients. 42
Adding to the complexity of diagnosing aHUS is the poor correlation of complement protein measurements in the blood of aHUS patients with disease pathology. In a recent study of 44 individuals in acute phase aHUS, it was found that only half had abnormal serum levels of complement proteins. 32 Interestingly, even though 50–60% of aHUS patients have mutations or autoantibodies to AP regulator FH, low serum levels of complement regulators are nonpredictive for aHUS, indicating that loss-of-function variants may be affecting the regulatory activity of key complement regulators. 39
Functional characterization of variants to determine their pathogenic significance is a relatively new area of research to identify mutations of previously unreported significance. 43 Additional ex vivo functional assays using patient serum have also been developed as a diagnostic tool to measure complement deposition on cultured endothelial cells. 44 These assays, while labor intensive, have been used to identify abnormal deposition of C3 and C5b-9 from acute aHUS patient serum with unidentified genetic variants. Identifying patients with complotypes that put them at risk for developing complement-mediated SAEs presents a currently underexplored area of investigation in AAV gene therapy, but could be used to screen patients before receiving AAV gene transfer.
AAV gene therapy for Duchenne muscular dystrophy
The first cases of aHUS-like TMA following AAV gene transfer were observed in two separate high-dose clinical trials for Duchenne muscular dystrophy (DMD), which systemically delivered AAV9-encapsidated vectors delivering a μDYS gene through muscle-specific promoters. 45 In the Phase 1/2 IGNITE DMD trial (NCT03368742), three pediatric patients dosed with SGT-001 at 5 × 1013 vg/kg and 2 × 1014 vg/kg experienced complement-related SAEs after dosing (Table 1).
Improvements to the SGT-001 manufacturing process to reduce empty capsid content and clinical protocol updates to include prophylactic use of eculizumab, a C1-esterase inhibitor, and increased corticosteroid dose enabled continued program development following a clinical hold. 46 As of January 2022, nine patients have been dosed with SGT-001, and most patients demonstrated improvements from baseline in ambulatory function. In a Phase 1b trial (NCT03362502), 2 of 16 patients dosed with 2 × 1014 vg/kg PF-06939926 exhibited complement-mediated TMA. The two pediatric DMD participants developed thrombocytopenia, hemolytic anemia, and renal dysfunction related to complement activation after dosing. 47 Both patients recovered following treatment with eculizumab; however, one patient required additional hemodialysis and platelet transfusion for acute kidney injury. 48
While dose appears to play a role in complement activation by AAV in the four reported cases of aHUS-like TMA in DMD, AP complement activation is a known pathological component of DMD and other proinflammatory myopathies. Necrotic muscle fibers from DMD patients have been found to have significant deposits of C5b-9, which are not found on healthy muscle fiber, indicating that complement activation and a heightened inflammatory state are present in DMD patients, which could be exacerbated by AAV gene transfer. 49 Eculizumab has been proposed as a treatment for inflammatory myopathies, indicating that prophylactic C5 inhibition in AAV gene therapy trials aligns both with current treatment regimens for DMD and a multi-hit induction of aHUS in situations where patients are already predisposed to complement activation. 49
AAV gene therapy for spinal muscular atrophy
Complement activation and TMA have also been observed in infant and toddler-aged patients with spinal muscular atrophy (SMA) dosed with Food and Drug Administration (FDA)-approved onasemnogene abeparvovec (OA [Zolgensma; Novartis, Basel, Switzerland]) at 1.1 × 1014 vg/kg. 50 –54 OA utilizes a self-complementary AAV9 vector under the control of ubiquitous chicken β-actin (CBA) promoter and cytomegalovirus (CMV) enhancer to deliver survival of motor neuron 1 (SMN1) gene (Table 1). While no instance of aHUS was observed in 101 patients across 5 clinical trials for OA (NCT02122952, NCT03306277, NCT03505099, NCT03461289, and NCT03837184), 90% had some degree of ALT and/or AST elevation peaking at week 1 following infusion and again at month 1. Importantly, impaired hepatic function is a comorbidity associated with SMA, and >60% of OA trial participants had elevated liver transaminase levels at baseline before dosing, despite criteria to exclude patients with >3 × or >2 × upper limit of normal (ULN) ALT and AST. 55
Postlicensure, the FDA has reported 9 cases of complement-mediated TMA in 1,400 patients, and the European Medicines Agency (EMA) has reported 5 cases in 800 patients dosed with OA. 56 In all cases, TMA was observed ∼1 week after infusion and proceeded by hypertension, vomiting, dehydration, decreased urine output, transient elevation in liver transaminases, and decrease in platelets. All patients had normal ADAMTS13 activity, and AP-specific (sC5b-9) complement activation was observed in most instances. 50 –54 Symptoms of TMA were resolved through treatment with plasmapheresis, glucocorticoids, and eculizumab. In 2022, the first fatality associated with TMA was reported in a 6-month-old patient with SMA following treatment with OA. 51 Sequencing of complement genes in this patient revealed a CFI variant of unknown significance, prompting treatment with eculizumab and temporary recovery.
However, prolonged hospitalization and a hospital-acquired Staphylococcus epidermidis infection resulted in death. Following this fatality, it was suggested that OA could be a “second hit” in situations where patient complotype predisposes to more severe forms of complement dysregulation and aHUS. 50 Concurrent infection, recent vaccination, body weight >8 kg, and prior use of antisense oligonucleotide nusinersen (Biogen) have been suggested as potential cofactors in the induction of TMA in OA patients. However, pre-existing liver injury and transaminitis elevations combined with patient complotype are also potential drivers of hepatotoxicity, AP complement activation, and TMA.
AAV gene therapy for Fabry disease
In the INGLAXA Phase 1/2 clinical trials for patients with Fabry disease (NCT04519749, NCT05629559), three of six patients developed aHUS 3–7 days following IV administration of 4D-310 at a dose of 1 × 1013 vg/kg. 4D-310 is composed of a novel muscle-tropic C102 capsid that delivers an α-galactosidase A (GLA) transgene under a ubiquitous promoter (Table 1).
All cases of aHUS resolved within a week of treatment with eculizumab, although a more serious case required dialysis and prompted an enrollment hold by the FDA. An investigation by the Sponsor determined that the 69-year-old male patient with the more severe (Grade 4) case of aHUS requiring dialysis had predosing AP complement activation and a heightened IgM response following dosing. 57 While enrollment in the clinical trial was set to resume following updates to the immunosuppressive regimen to include rituximab/sirolimus and prescreening of participants for AP complement activation, the program remains on clinical hold by the FDA. 58
Inflammation and DAMPs produced in patients with inflammatory lysosomal storage disorders (LSDs) such as Fabry disease could predispose patients to adverse complement-mediated responses to AAV gene transfer. The accumulation of glycolipids within lysosomal deposits contributes to chronic inflammation thought to be driven, in part, by PRM recognition of glycolipid DAMPs as well as autoantibodies. Proinflammatory signaling through cytokines, chemokine, complement, autoantibodies, and growth factors can result in tissue damage and further amplification of the inflammatory pathways. Although the role of complement has not been well studied in LSDs, there is evidence of complement activation in a mouse model of Fabry disease, and elevated levels of serum C3, iC3b, C4, C4b, and FB, as well as renal C3 deposition in human patients with Fabry disease. 59
AAV gene therapy for Danon disease
In a Phase 1 gene replacement study of patients with Danon disease (DD; NCT03882437), TMA with complement activation and acute renal failure was observed following IV dosing of RP-A501 at 1.1 × 1014 vg/kg. RP-A501 is an AAV9 vector expressing lysosome-associated membrane protein-2 (LAMP2B) transgene (Table 1). Of the two patients who received 1.1 × 1014 vg/kg RP-A501, one participant developed complement-mediated TMA and renal failure, which resolved following eculizumab and temporary hemodialysis.
Following this case of TMA, enrollment in the high-dose cohort was ceased and an enhanced prophylactic immunosuppressive regimen was put in place, including rituximab to deplete peripheral B cells, low-dose steroids, and sirolimus to inhibit complement activation. Frequent monitoring for TMA was implemented to facilitate early use of eculizumab in the event of an SAE. 60 Following these changes, a new pediatric low-dose cohort consisting of two participants who received a dose of 6.7 × 1013 vg/kg showed minor sC5b-9 complement activation, but no complement-related SAEs. 61
Although DD has been long thought to be a glycogen storage disorder, newer evidence suggests that pathogenesis results from disruption of autophagy and accumulation of glycogen in autophagic vacuoles. 62 Myopathy in DD presents as muscle weakness and atrophy, and muscle biopsies reveal accumulation of autophagic vacuoles and complement C5b-9 deposits; however, little is known about the role of complement activation and deposition in DD. 62
AAV gene therapy for methylmalonic acidemia
In the SUNRISE Phase 1/2 clinical trial (NCT04581785) for patients with methylmalonic acidemia (MMA), two cases of TMA were observed following IV infusion of an LK03-encapsidated vector expressing methylmalonyl-CoA mutase (MMUT), LB-001, at 5 × 1013 vg/kg (Table 1). 63 Both patients who experienced TMA were part of the younger patient cohort, 6 months to 2 years of age.
Both cases resolved with hospitalization, fluids, and in one case—eculizumab. While complement activation was not noted, the classic symptoms of TMA as well as the early onset of TMA 1–2 weeks following dosing implicate complement involvement. Following the second case in February 2022, the FDA put a clinical hold on the trial, which was then lifted in May 2022, requiring frequent testing for complement activation and the use of a complement inhibitor in the event of TMA onset. The SUNRISE trial has since been terminated due to “low likelihood of clinical benefit” for patients.
Since most of the metabolic conversion of MMA takes place in the liver, one of the treatment options for MMA is liver and/or kidney transplantation. Proteomic analysis of liver specimens from MMA patients found that differentially expressed enzymes resembled those upregulated in fatty liver disease, indicating that hepatic injury may be present in patients with MMA. 64 It is possible that a secondary assault on the liver following AAV gene transfer could result in tissue damage and inflammation, which could contribute to complement activation and TMA in susceptible patients.
COMPLEMENT ANALYSIS FOR AAV GENE THERAPY
Complement proteins, regulators, and split products have not traditionally been measured or reported in subjects receiving AAV gene therapy. With incidences of complement-mediated TMA in some subjects administered high-dose AAV, and the limited reporting of complement activation markers in these subjects, there is a new interest in analyzing complement in subjects receiving AAV gene therapy. Accurate assessment of complement, however, is highly complex and requires special blood collection, sample handling, and storage considerations to avoid spontaneous activation of complement. 65 Serum and plasma collected from AAV gene therapy subjects for other immune profiling purposes may not be suitable for complement measurements based on the protocol used for collection. Thus, scrutiny of the complement analysis methodology is essential for correct interpretation of clinical data. The challenges associated with complement measurement and analysis are highlighted in a recent publication from Salabarria et al. 66
In a retrospective analysis of 38 subjects across 4 AAV9-based gene therapy platforms, Salabarria et al. concluded that TMA following AAV gene transfer was CP driven (antibody dependent) with a secondary contribution from AP amplification. 66 These conclusions were drawn from dividing the patients into two groups based on their prophylactic immunomodulation (IM) regimen and resulting anti-AAV antibody levels in the first weeks following gene transfer. It is important to note that the conclusions reached by Salabarria et al. are based on a heavily skewed distribution of subjects. Of primary concern is the uneven grouping of subjects by AAV platform, disease indication, and vector dose. While the authors provide biomarker measurements, including anti-AAV antibodies, complement proteins, and hematology markers across a 30-day period following AAV gene transfer, the utility of these findings is diminished by the reporting of group means instead of individual responses.
Additional concerns stem from the unspecified use of both plasma and serum for complement measurements. For complement, nonspecific activation of complement following blood collection can vary widely between serum and plasma. Plasma collected with different anticoagulants, freeze-thaw cycles, and sample handling has been shown to contribute to significant differences in complement measurements. 14,65 For these reasons, it is difficult to make conclusions on whether antibody complexes, or a myriad of other patient-specific or sample-specific factors, are driving the observed changes. Despite these limitations, the comprehensive measurement of complement analytes in human subjects and efforts to contextualize complement activation within the larger immune response to AAV represents an important milestone.
MITIGATION OF COMPLEMENT ACTIVATION
Therapeutic complement modulation by C5 inhibitor, eculizumab, has been used reactively following AAV gene transfer for aHUS and in some cases recommended for future prophylactic use (e.g., SGT-001). Complement inhibition directed at the central hub of the complement cascade, C3, is an attractive new therapeutic target to prevent the release of C3 split products, AP amplification, and initiation of the terminal pathway.
Pegcetacoplan, a PEGylated compstatin-derived C3 inhibitor, which is FDA approved for the treatment of paroxysmal nocturnal hemoglobinuria (PNH), a disorder associated with dysregulated complement on red blood cells, has been demonstrated to be superior to eculizumab for the treatment of PNH. 67 Relevant to AAV gene therapy, C3 inhibition by APL-9, a derivative of pegcetacoplan, has been demonstrated by Smith et al. to effectively reduce complement activation, AAV uptake, and cytokine/chemokine release following in vitro AAV stimulation in human whole blood. These findings suggest that future testing of C3 inhibition to reduce host immune responses to AAV vectors may be worth exploring.
While complement inhibitors are safely used for the treatment of acute and chronic complement dysregulations, they require a meningococcal vaccination and prophylaxis with antibiotics to mitigate the risk of infection. Pathway-specific complement modulators that selectively inhibit complement activation are being explored as more targeted alternatives to inhibitors that broadly prevent complement activity. 4 An additional area of research in complement modulation is tissue-targeted complement therapeutics. Targeted complement drugs carry a lower risk of infection and bind directly to tissue-specific markers or DAMPs to provide local complement inhibition. 68
Development of an IM regimen for AAV gene therapy that includes complement modulation requires an understanding of complement-AAV interactions that goes beyond CP activation and AP amplification and examines the role of tissue damage, inflammation, intracellular complement, and crosstalk with other pathways in the initiation of complement in AAV gene therapy. Further studies are needed to elucidate these interactions to determine the best-suited inhibitor, to control complement activation in response to AAV gene transfer.
CONCLUSIONS
Use of high AAV vector doses resulting in hepatotoxicity and SAEs such as complement-mediated TMA has brought to light understudied mechanisms of the innate immune system, which could impact the efficacy and safety of AAV gene therapy. The role of complement in host defense and homeostasis is multifactorial and intersects not only with other pathways of the innate immune system but also other enzymatic cascades, including coagulation and the adaptive immune system.
The oversimplified model of complement as a cascade initiated by antigen-bound antibody complexes and glycoproteins displayed by pathogens ignores the more nuanced role of complement as a highly responsive pattern recognition pathway that can be initiated at multiple junctions. While dysregulation of the complement cascade can result in tissue damage and death, under normal circumstances, the complement system is tightly regulated to protect host cells from the damaging effects of complement activation. In the event of tissue damage, the complement cascade also plays an essential role in tissue regeneration and return to homeostasis, indicating that the complement system not only has dual roles in clearing pathogens and apoptotic host cells but is also active in cellular proliferation and repair.
So far, complement-mediated TMA has only been observed following high-dose AAV administration. Below the threshold of SAEs, asymptomatic, but measurable complement activation in AAV gene therapy could be potentiating AAV clearance and anti-AAV adaptive immune responses, as suggested by in vitro studies. The challenge for the AAV gene therapy community will be to identify not only signs of adverse complement activation like aHUS but also more subtle implications of complement opsonization and boosting adaptive immune responses. While liver enzyme elevations have been noted to manifest alongside symptoms of aHUS following AAV gene therapy, the impact on long-term transgene product expression is unknown.
Incorporating short-term prophylactic complement modulation into the immunosuppressive regimen of patients receiving AAV gene therapy is an area of active research. The benefits of reactive eculizumab treatment following complement-mediated SAEs in AAV gene therapy, while effective, are limited to the terminal pathway of the complement cascade. Based on evidence that the AAV capsid associates with C3b and iC3b and concerns surrounding adaptive immune priming by C3 and C5 split products, complement modulation for AAV gene therapy should at a minimum include inhibition at the level of C3 cleavage.
The central hub of the complement cascade, C3, is an attractive target for prophylactic intervention in AAV gene therapy due to the role of C3 split products in inflammation, phagocytosis, priming of the adaptive immune system, and the lytic terminal pathway. As more pre-clinical data emerge, which tie the kinetics of complement activation to other measurable markers of inflammation and tissue damage, clinical use of C3 complement inhibitors may find a place in prophylactic immune regimens for AAV gene therapy. Compatibility of infection risk mitigation strategies with AAV gene therapy will need to be weighed against the benefits of complement inhibition to ensure the best outcomes for patients.
Footnotes
ACKNOWLEDGMENTS
We thank Frances Xin for assistance with scientific writing. Figures were created with BioRender.com
AUTHORs' CONTRIBUTIONS
E.K.: Conceptualization, writing—original draft, writing—review and editing, data curation, and visualization. D.M.: Conceptualization and writing—review and editing. A.M.: Conceptualization and writing—review and editing. F.M.: Writing—review and editing. K.K.: Conceptualization, writing—review and editing, and visualization.
AUTHOR DISCLOSURE
All authors are employees of Spark Therapeutics, Inc.
FUNDING INFORMATION
This work was funded by Spark Therapeutics, Inc.