
Abstract
Liver injury with concomitant loss of therapeutic transgene expression can be a clinical sequela of systemic administration of recombinant adeno-associated virus (rAAV) when used for gene therapy, and a significant barrier to treatment efficacy. Despite this, it has been difficult to replicate this phenotype in preclinical models, thereby limiting the field's ability to systematically investigate underlying biological mechanisms and develop interventions. Prior animal models have focused on capsid and transgene-related immunogenicity, but the impact of concurrently present nontransgene or vector antigens on therapeutic efficacy, such as those derived from contaminating nucleic acids within rAAV preps, has yet to be investigated. In this study, using Ad5-CMV_GFP-immunized immunocompetent BALB/cJ mice, and a coagulation factor VIII expressing rAAV preparation that contains green flourescent protein (GFP) cDNA packaged as P5-associated contaminants, we establish a model to induce transaminitis and observe concomitant therapeutic efficacy reduction after rAAV administration. We observed strong epitope-specific anti-GFP responses in splenic CD8+ T cells when GFP cDNA was delivered as a P5-associated contaminant of rAAV, which coincided and correlated with alanine and aspartate aminotransferase elevations. Furthermore, we report a significant reduction in detectable circulating FVIII protein, as compared with control mice. Lastly, we observed an elevation in the detection of AAV8 capsid-specific T cells when GFP was delivered either as a contaminant or transgene to Ad5-CMV_GFP-immunized mice. We present this model as a potential tool to study the underlying biology of post-AAV hepatotoxicity and demonstrate the potential for T cell responses against proteins produced from AAV encapsidated nontherapeutic nucleic acids, to interfere with efficacious gene transfer.
INTRODUCTION
Liver enzyme elevations after vector administration have been previously observed after treatment with adeno-associated virus (AAV) in a range of disease contexts. 1 –4 In disorders where expression from the liver is targeted for disease correction, such as hemophilia, this signature has been observed in parallel with a decline in therapeutic output. 1,5 –7 In some cases, this is controlled through the administration of steroids, either prophylactically or reactively. Transaminitis onset typically occurs within 2–3 months after vector administration.
Clinically, AAV capsid-reactive CD8+ T cells have accompanied this phenomenon. 5,6,8,9 However, this correlation is not absolute. Some patients have detectable AAV capsid reactive T cells without appreciable elevation in serum transaminases. 8,9 Others exhibit transaminitis and therapeutic expression loss without detectable AAV capsid reactive T cells. 3,10 It is possible that in the latter instances, a source of antigen other than the capsid, or indeed the transgene, is driving this reactivity. Despite transaminitis being a common clinical observation, it has been difficult to recapitulate in animal models.
Adoptive transfer of capsid-specific CD8+ T cells has been shown to induce transaminitis and loss of transgene expression in liver-targeted gene transfer, and to diminish transgene expression after intramuscular AAV delivery. 11,12 Conversely, studies have reported that pre-existing capsid immunity does not result in elevated liver transaminases after recombinant AAV (rAAV) delivery. 13,14
Prior study has also established that antitransgene-directed CD8+ T cell immunity can clear transduced hepatocytes in naive mice. In one study, this was achieved by incorporating the immunodominant ovalbumin (OVA) peptide into an expression cassette. 15 Hepatocyte clearance was dose dependent, with a low (1 × 108 vg) and medium dose (1 × 109 vg) resulting in a total loss of transgene expression after 12 weeks but with delivery of 1 × 1010 vg resulting in sustained OVA expression. A study into genome editing efficiencies also noted reduced efficacy and elevated transaminases when mice had pre-existing immunity to the CRISPR enzyme. 16
It is known that rAAV preparations exhibit considerable heterogeneity, in terms of both mutated and truncated genomes, and the aberrant packaging of nucleic acids from the production system. 17 Such nucleic acid species can be transferred, and persist, in vivo after rAAV administration. 18 –20 Furthermore, we have previously shown that highly represented contaminants, originating from plasmid DNA upstream of the AAV P5 promoter, have the potential to be transcribed and translated in the liver after AAV transduction. 21 Transcriptional activity from the inverted terminal repeat (ITR) sequences has also been shown to produce off-target transcripts in the transduced liver and brain. 22,23
T cells reactive to a cryptic peptide present within an expression cassette have also been detected in mice after AAV delivery. 24 Protein products, whether aberrant or intended, all could theoretically generate or contribute to a T cell-mediated clearance of AAV-transduced hepatocytes. Indeed, prior investigations of nucleic acid contaminants of rAAV have suggested and shown that the protein products of DNA impurities could be a source of antigen presentation. 19,21,25 However, whether T cell responses against any of these heterologous antigens could negatively impact therapeutic efficacy is currently unknown.
In this study, we examined whether memory T cell responses against nucleic acid contaminants delivered by rAAV could drive hepatic transaminitis endogenously and without the need for adoptive transfer of antigen-specific T cells. We demonstrate that, at clinically relevant doses, memory T cell responses against P5-linked contaminants packaged in AAV can induce both hepatic transaminitis and a reduction in the overall therapeutic protein output.
METHODS
Viral vectors
Adenovirus vectors were purchased from vector biolabs (Ad5-CMV_GFP: #1060; Ad5-CMV_Luciferase: #1000). rAAV vectors AAV8 serotype were produced in adherent 293T cells by triple plasmid transfection. Production and purification of these viruses are described in a previous study. 21 Vectors were assayed for titer by quantitative polymerase chain reaction (qPCR) using a linearized plasmid reference standard as previously described, 21 using primers directed against the FVIII transgene and contaminant amplicons (Supplementary Table S1). Quantitation of host–genome encapsidation was done through Alu element qPCR using a HEK293 cell standard. 26 Capsid titers were assayed by AAV8 titration ELISA kit (#PRAAV8; Progen). Measurements for ELISA were performed on a Biotek plate reader.
In vivo studies
All mice were housed in the animal facility at St Jude Children's Research Hospital. All procedures were conducted in compliance with the guidelines of the institutional animal care and use committee (IACUC). BALB/cJ mice and Albino C57BL/6J were used in the immunization studies. For BALB/cJ experiments, mice were intramuscularly injected with 2e7 pfu of Ad5-CMV_GFP, Ad5-CMV_Luciferase, or left uninjected. Twenty-eight days later, mice were injected by tail vein with 5e11 vg of an rAAV8 vector (AAV8GFP_P5 F8, AAV8Empty_P5 F8) or a lower (1.7e10 vg) dose of AAV8ssCMV_GFP.
Ten days after AAV8 delivery, whole blood and plasma were harvested for analysis. In Figure 6, Albino C57BL/6J mice were intramuscularly immunized with 2e7 pfu of Ad5-CMV_GFP or Ad5-CMV_Luciferase. Twenty-eight days later mice, were injected by tail vein with 5e11 vg AAV8GFP_P5 F8. Whole blood and plasma were harvested for analysis at days 10, 29, 56, and 141 post-AAV delivery.
Biochemical studies
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) values were assayed from mouse plasma by the St. Jude CPC diagnostic laboratory using commercial kits (Horiba, ABX Pentra ALT CP #A11A01627) (Horiba, ABX Pentra AST CP #A11A01629). Circulating FVIII was measured from mouse plasma by ELISA (Diagnostica Stago, Asserachrom VIII:Ag ELISA kit, #00280). ELISA was performed on Bio-Tek plate reader.
Anti-AAV8 capsid IgG ELISA: Plates were coated overnight with 5 × 1010 AAV8 vector genomes diluted in 5 mL 0.1 M NaHCO3 pH 9.2 and subsequently washed 5 × with 300 μL of Dulbecco's phosphate-buffered saline with Tween (DPBST). In total, 200 μL of DPBST with 6% bovine serum albumin (BSA) was added, the plate was then incubated at 37°C for 1 h. The plate was washed 5 × and incubated with 50 μL varying dilutions of mouse plasma (1:50, 1:200, 1:800, and 1:3,200) in DPBST +2% BSA, shaking gently for 2 h at 37°C.
The plate was washed 5 × and incubated with 100 μL of a 1:10,000 dilution of antimouse IgG (#A4416; Sigma) in DPBST +2% BSA. After 1 h shaking at 37°C, the plate was washed 5 × . Antibody binding is quantified by adding 200 μL per well of OPD peroxidase substrate (#P-9187; Sigma) resuspended in 20 mL of ddH2O. After 5 min, the reaction was halted with 50 μL of 3 M HCl. Signal was quantified at 492 nm on a Biotek plate reader.
T cell specificity assays
Spleens harvested from AAV-treated mice were placed in Hank's buffered salt solution (#14175095; Thermo-Fisher). Splenocytes were extracted, tetramer was stained, and analyzed as previously described. 21 In brief, isolated splenocytes were plated in 96-well plates at 5 million cells per well in phosphate-buffered saline. Plates were spun down, and cells resuspended in 150 μL of titrated tetramers (Fred Hutchinson Cancer center). Tetramer specificities: green flourescent protein (GFP) (H-2Kd restricted, EGFP200–208 HYLSTQSAL epitope); and AAV8 capsid (H2-Ld restricted, AAV8-CAP375–383 IPQYGYLTL epitope).
After 90 min, cells were washed 2 × , resuspended in 50 μL CD16/CD32 Fc-block (#553142; BD), and incubated for 10 min in the dark. In total, 50 μL of Ghost Dye™ (#13-0870-T100; Cytek) and antimouse CD8a antibody (#35-0081-U100; Cytek) were added to each well. Cells were incubated for 30 min in the dark at 4°C, washed 2 × in PBS, and resuspended in 150 μL of fluorescent-activated cell sorting (FACS) buffer. Cells were run on a BD LSR Fortessa FACS machine. In total, 2,000,000 events were collected per sample. FlowJo v.10 was used to analyze FCS files.
Graphs and statistical analysis
Figures were prepared in Adobe Illustrator. Graphs and statistics were generated in GraphPad Prism 10.0. and R studio. Error bars represent standard error of the mean (SEM).
RESULTS
Preimmunization to P5-associated contaminants can reduce therapeutic efficacy of an rAAV-delivered transgene
rAAV preparations are known to be heterogeneous in their nucleic acid content. 17 These contaminants can arise from elements of the production system that are erroneously packaged within rAAV particles, such as plasmid backbone sequences, host cell-line genomic DNA, or a combination thereof. 27 We have previously shown that sequences upstream of the AAV2 P5 promoter are preferentially incorporated nucleic acid contaminants in purified rAAV (Fig. 1a). 21 These P5-associated contaminants can be transcribed within the liver and the translation products of these contaminants can result in the generation of specific T cells in naïve mice after rAAV infection (Fig. 1b).
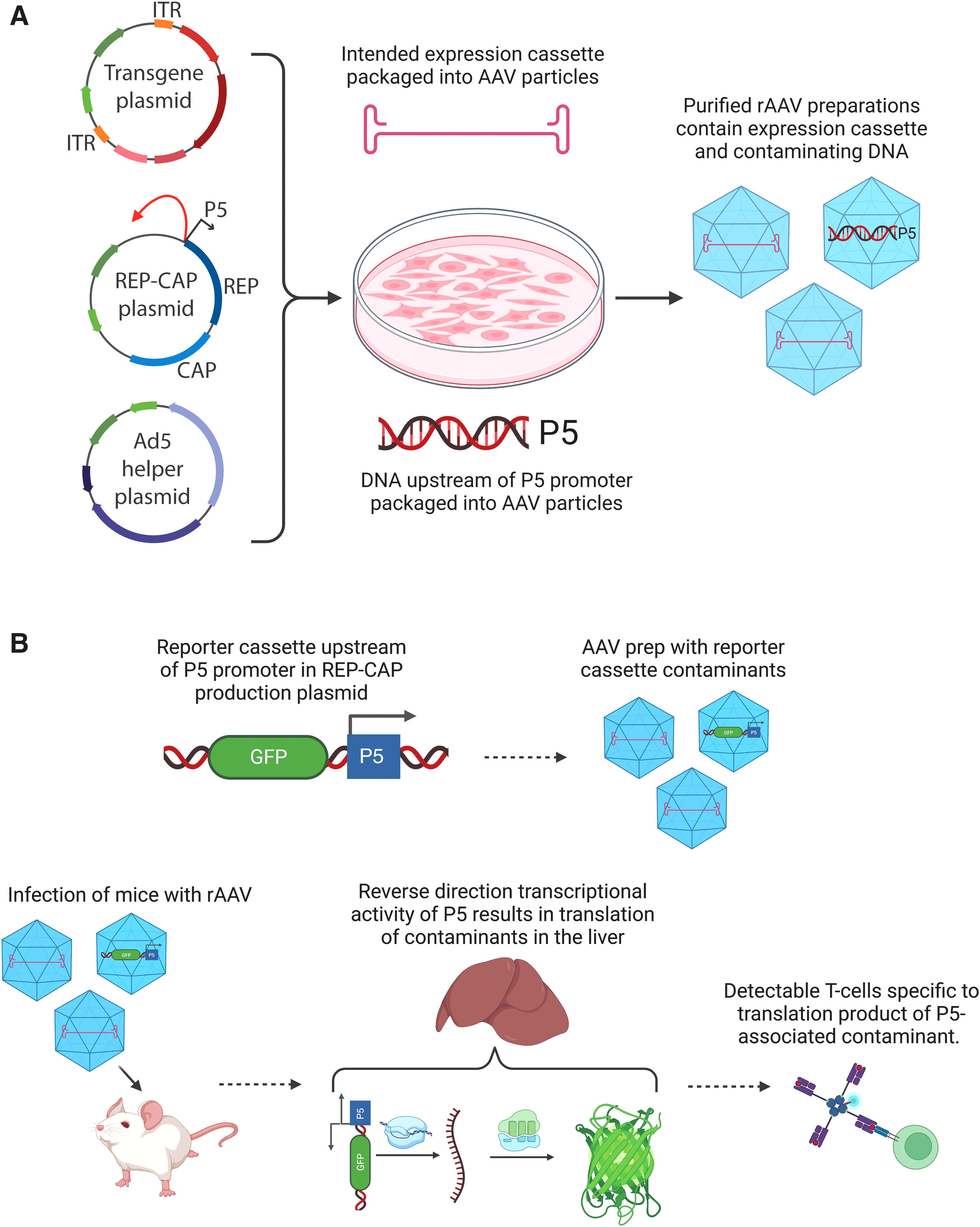
Incorporation and hepatic postinfection activity of P5-associated contaminants.
Here, we examined whether T cell responses against an rAAV contaminant-derived antigen could interfere with the therapeutic potential of the expression cassette. Using intramuscular injection of a recombinant adenovirus, packaging a ubiquitously expressed GFP reporter cassette (Ad5-CMV_GFP), immunity to the GFP protein was generated in BALB/cJ mice (Fig. 2a). Control mice did not receive an Ad5 injection. Twenty-eight days after immunization, an AAV8 vector encoding the coagulation factor VIII under a liver-specific promoter (AAV8-HLPhFVIII) was administered intravenously at a low (1e11 vg) or high (5e11 vg) dose.
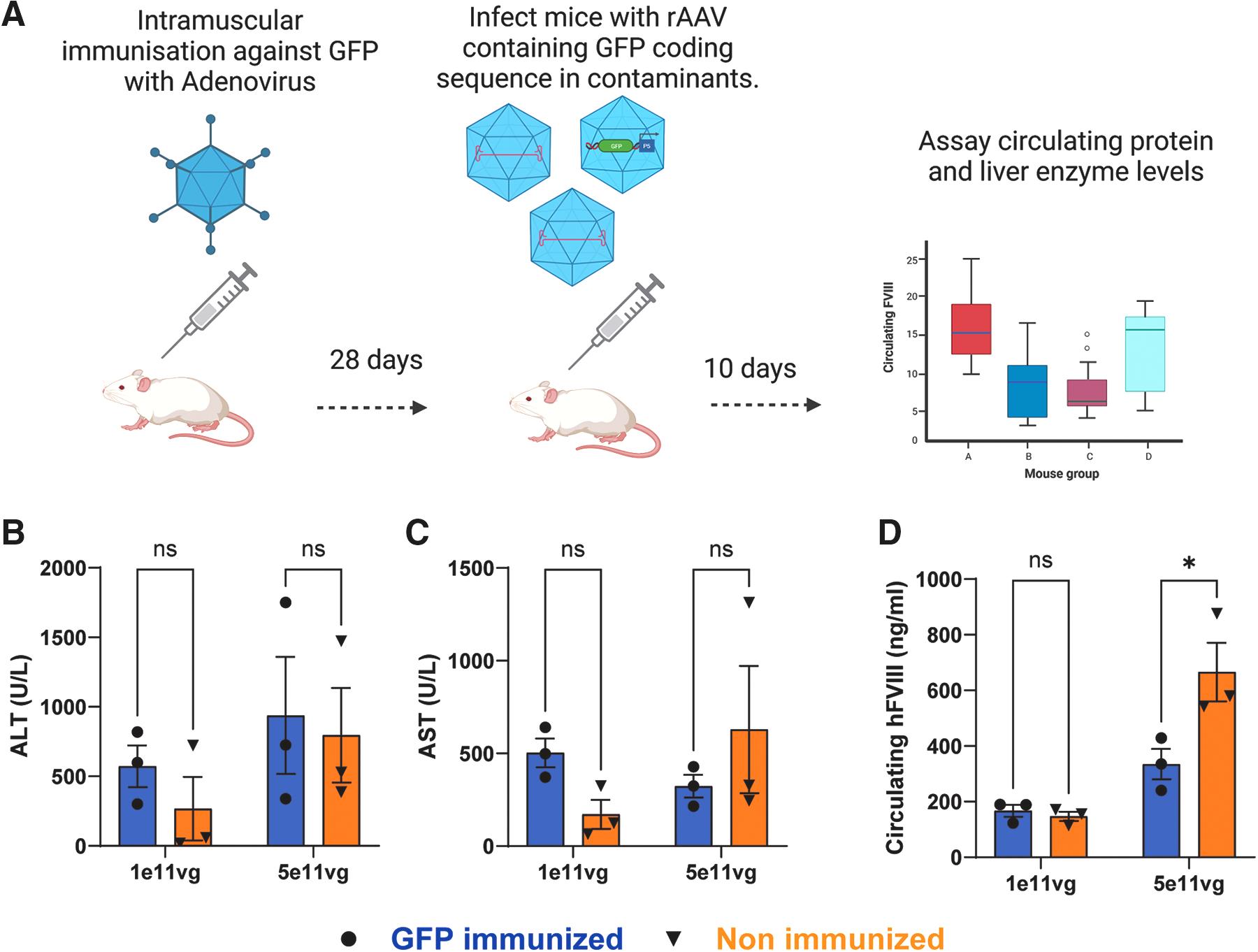
Preimmunization of Balb/cJ mice against P5-associated contaminants results in a dose-specific efficacy reduction of FVIII cassette.
The production plasmids used to make this construct were engineered to contain a GFP open reading frame upstream of the P5 promoter, thus this reporter cassette was packaged as contaminant DNA into rAAV particles within the FVIII preparation (GFP_P5). The abundance of full-length contaminant GFP aberrantly incorporated was determined to be ∼6% of the FVIII vector genome titer (Supplementary Table S2). Ten days after AAV delivery, mice were assayed for liver transaminase levels and circulating FVIII expression. Elevated transaminases were seen in both pre- and nonimmunized contexts (Fig. 2b, c).
We did not observe differences in the ALT and AST elevations between mice given the 5e11 vg dose of AAV (ALT: p = 0.94, AST: p = 0.47). Some GFP-immunized mice given the 1e11 vg dose had higher ALT levels and AST levels than the nonimmunized controls, but this was not statistically significant (ALT: p = 0.75, AST: p = 0.41). Interestingly, when circulating FVIII was assayed in the 5e11 vg dose group, the mice immunized to GFP had ∼50% lower circulating FVIII expression as was present in the nonimmunized group (p = 0.01) (Fig. 2d). This suggests that immunization against a heterologous AAV-delivered antigen (GFP) negatively impacted expression of the intended transgene (FVIII).
Elevated transaminases, FVIII expression reduction, and induction of CD8+ T cells reactive to P5-associated contaminants after rAAV delivery in preimmunized mice
Since the nonimmunized group in this experiment had not received an adenovirus vector, it is possible that circulating FVIII expression differences in the 5e11 vg group could be a nonspecific effect of Ad5 administration. To address this, we altered the strategy such that control mice would be administered with an Ad5 vector driving the expression of the firefly luciferase protein (Ad5-CMV_Luc) (Fig. 3a). Twenty-eight days postimmunization mice were administered 5e11 vg of AAV8HLPhFVIII that either contained a GFP cassette as contaminating DNA (AAV8GFP_P5 F8), or standard plasmid backbone sequences (AAV8Empty_P5 F8).
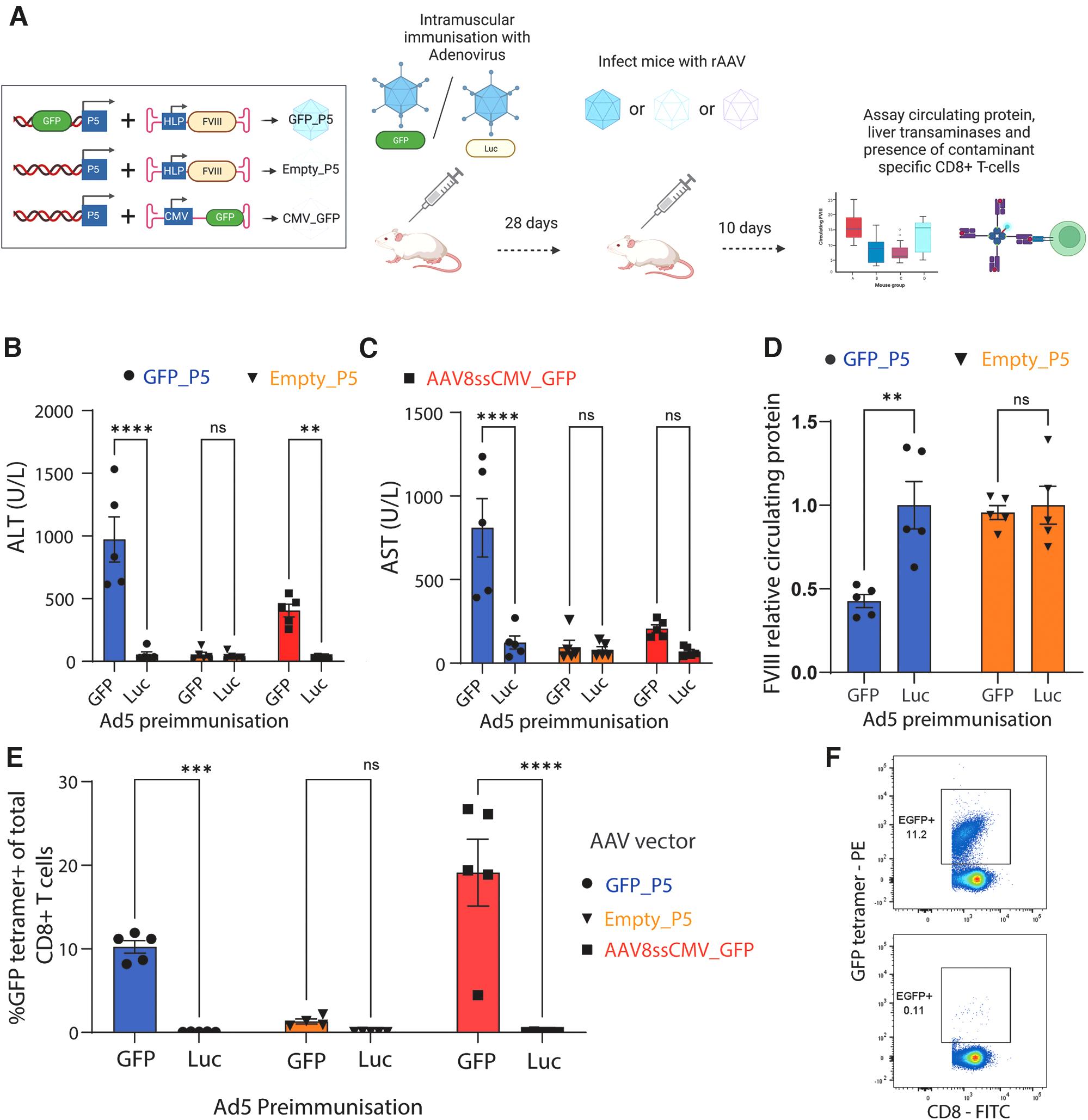
Transaminase elevations, transgene efficacy loss, and induction of contaminant-specific CD8+ T cells in preimmunized Balb/cJ mice.
A third group was given a lower dose of a control AAV8 vector in which an ITR-flanked CMV_GFP expression cassette was packaged (AAV8ssCMV_GFP). All AAV preparations were assayed for a range of known nucleic acid contaminants and for full capsid content (Supplementary Table S3). For mice dosed with AAV8GFP_P5 F8, significant elevations in both ALT and AST were observed in the GFP-immunized group (ALT: 972.9 ± 180.2; AST: 809.8 ± 174.6) as compared with Ad5-CMV_Luc-immunized mice (p < 0.0001) (Fig. 3b, c).
In contrast, at an equivalent dose of AAV8Empty_P5 F8, there were no significant elevations in either ALT or AST in either (Ad5-CMV_GFP)- or Ad5-CMV_Luc-immunized mice (ALT: 53.6 ± 19.6 vs. 44.6 ± 12.4; AST: 96.1 ± 41.0 vs. 80.4 ± 19.4) (p > 0.99). Elevations in ALT and AST were also seen when AAV8ssCMV_GFP was delivered to GFP-immunized mice, though to a lower magnitude than with the contaminant vector. When circulating FVIII was measured, the mice that received AAV8Empty_P5 F8 showed equivalent levels across the immunization conditions (p = 0.99) (Fig. 3d). However, when treated with AAV8GFP_P5 F8, the mice immunized to GFP exhibited less than half of the circulating FVIII observed in the Ad5-CMV_Luc-immunized control mice (42.7% ± 3.9%) (p < 0.0001).
Splenic T cells were harvested from the treated mice and analyzed for specificity against GFP and AAV8 capsid through tetramer staining (Supplementary Fig. S1). In Ad5-CMV_GFP-immunized mice treated with AAV8GFP_P5 F8, ∼10% of splenic T cells (10.2% ± 0.75%) were specific for the immunodominant GFP epitope, as compared with ∼1% when treated with AAV8Empty_P5 F8 (1.31% ± 0.31%) (Fig. 3e, f; Supplementary Fig. S2). In Ad5-CMV_GFP-immunized mice, the highest level of GFP-specific T cell generation was observed with the AAV8ssCMV_GFP delivery (19.11 ± 4.01), despite a lower elevation of transaminases than when GFP was only present as a contaminant.
As expected, levels of ALT correlated with AST in each mouse (Supplementary Fig. S3a). The overall frequency of GFP-specific T cells in AAV8GFP_P5 F8- and AAV8ssCMV_GFP-treated mice correlated with ALT elevations, whereas no significant correlation was seen for this in AAV8Empty_P5 F8 mice (Fig. 4a, b; Supplementary Fig. S3b), and circulating FVIII expression was negatively correlated with the frequency of GFP tetramer-positive T cells in AAV8GFP_P5 F8-treated mice but not AAV8Empty_P5 F8-treated mice (Fig. 4c, d).
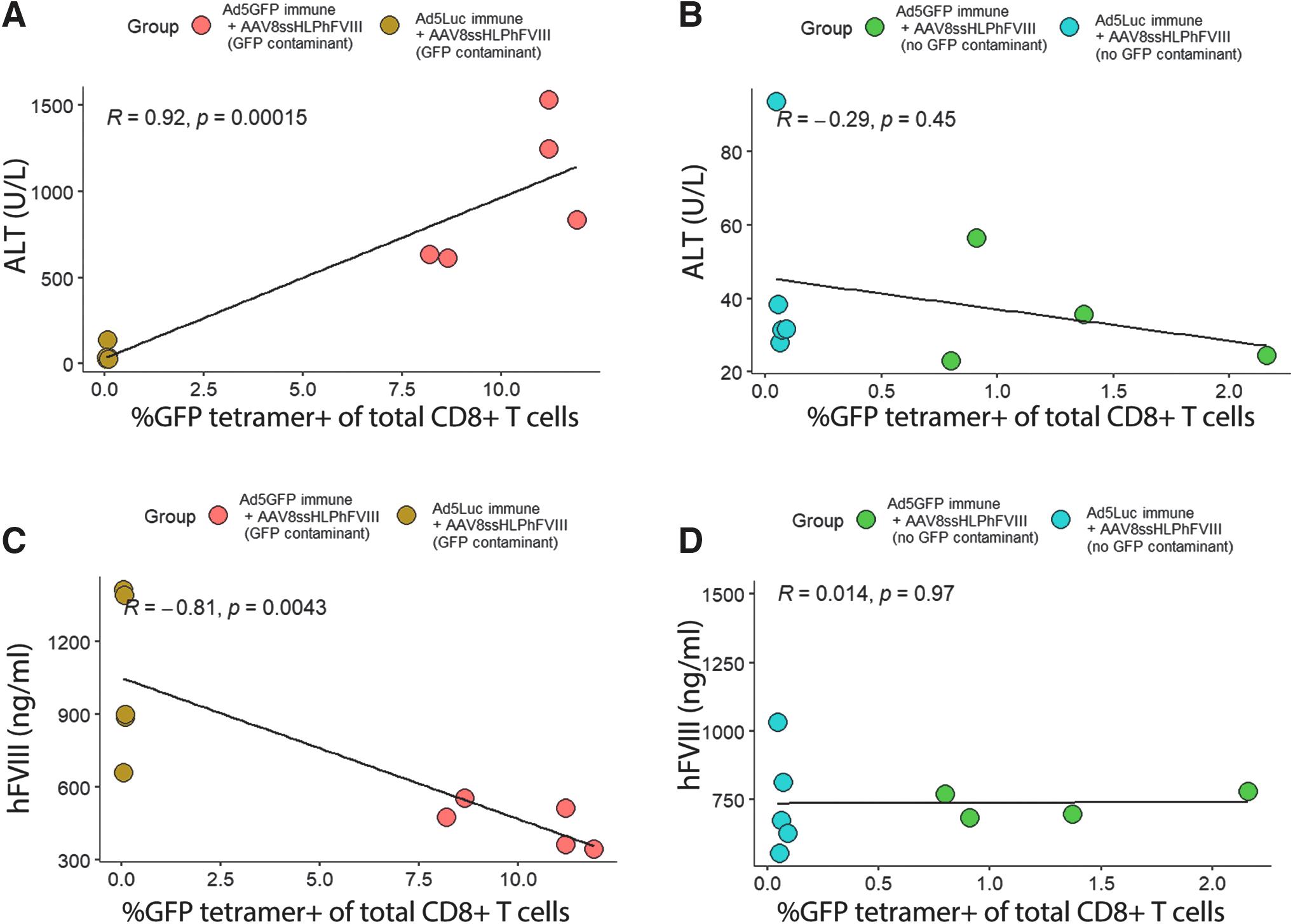
Correlation of transaminase elevations and FVIII transgene with detection of GFP-specific T cells.
Assessment of AAV capsid-specific CD8+ T cells in P5-associated contaminant-immunized mice
To determine whether the frequency of AAV8 capsid-specific T cells differed across treatment groups, we also probed for T cells specific for the immunodominant AAV8 capsid epitope (IPQYGYLTL375–383) in BALB/cJ mice. Although the frequencies of capsid-specific cells by tetramer staining were orders of magnitude lower than observed for GFP, we observed a significant increase in the frequency of capsid-specific T cells in GFP rechallenged mice (Fig. 5a, b; Supplementary Fig. S4). This occurred in both the AAV8GFP_P5 F8 and AAV8ssCMV_GFP administration groups, whereas no differences were observed between immunization groups after AAV8Empty_P5 F8 delivery.
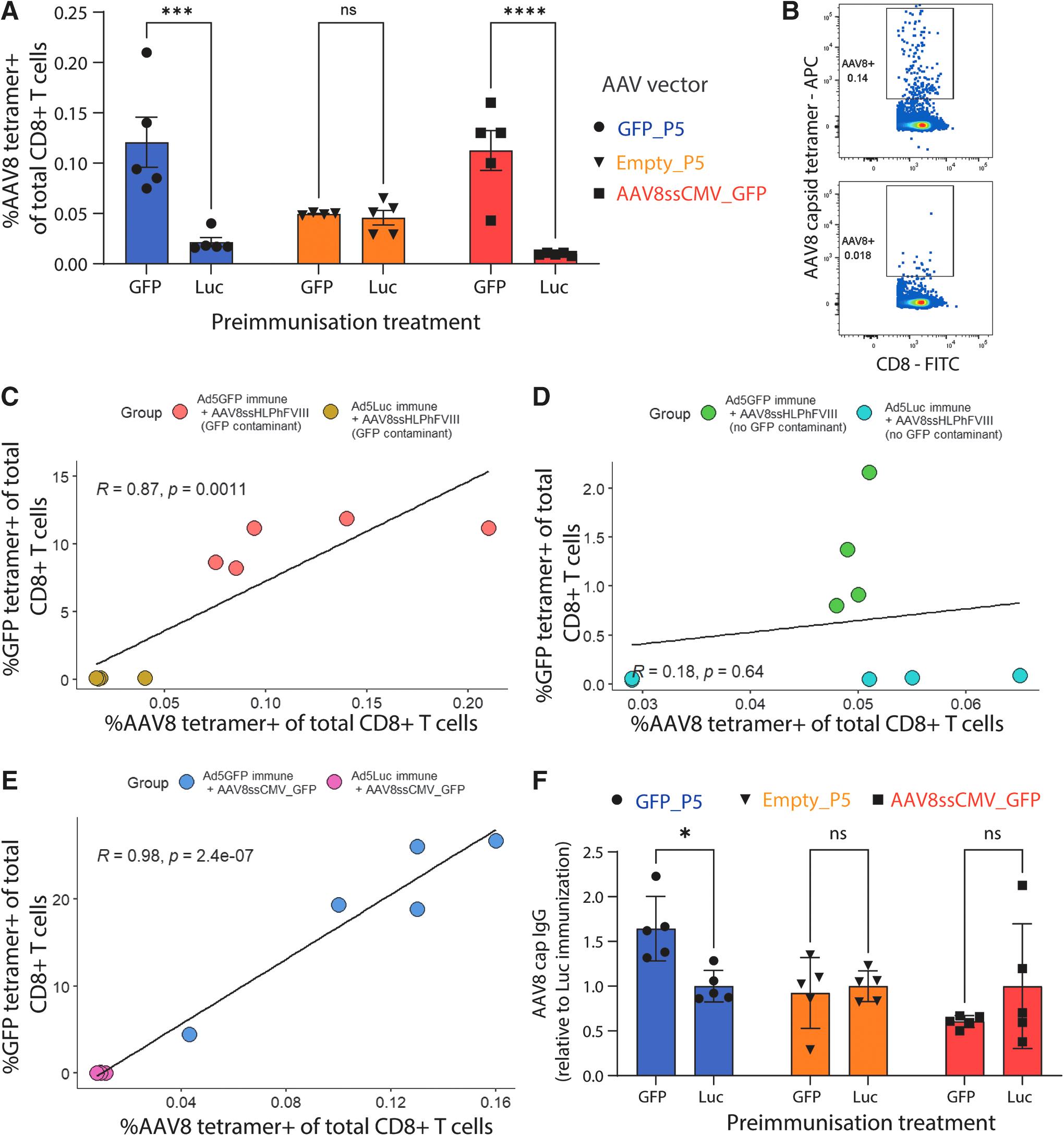
Capsid-specific T cells are elevated in GFP preimmunized mice dosed with a GFP containing rAAV.
When the frequency of GFP tetramer-positive cells was plotted against the capsid tetramer-positive frequency, again, a positive correlation was observed in AAV8GFP_P5 F8- and AAV8ssCMV_GFP-treated mice but not with AAV8Empty_P5 F8-treated mice (Fig. 5c–e). Then, to assess whether transaminitis induction led to alterations in antibody production, we assayed mouse plasma for the presence of anti-AAV8 IgG antibodies (Fig. 5f). IgG levels were 1.6-fold higher in GFP-immunized mice treated with AAV8GFP_ F8 as compared with luciferase, whereas equivalent levels were observed across the two AAV8Empty_P5 F8 groups. This effect was not observed in mice that received the AAV8ssCMV_GFP control vector.
No evidence of response to P5-associated contaminant immunization in Albino C57Bl6/J mice
BALB/cJ mice have both a well-characterized immunodominant CD8+ T cell epitope to the GFP protein (HYLSTQSAL200–208) and the AAV8 capsid (IPQYGYLTL375–383). 28,29 To determine how conserved this T cell response was, we next tested whether this model would be recapitulated in a different mouse strain, C57BL/6, which has different dominant epitopes for the GFP protein and AAV8 capsid (DTLVNRIEL118–126 and NSLANPGIA517–525, respectively). 28,30 Using adenoviral vector-mediated gene transfer, Albino C57BL/6J mice were immunized with either Ad5-CMV_GFP or Ad5-CMV_Luc.
Upon subsequent administration of AAV8GFP_P5 F8, mice were longitudinally measured for changes in liver enzymes and circulating FVIII protein (Fig. 6). In this instance, the model did not recapitulate the findings in BALB/cJ mice; no observable differences in circulating FVIII were seen across the groups. Elevated transaminases were detected but were observed in both immunization groups and a set of uninfected control mice from this strain (Supplementary Fig. S5).
In summary, these data show the potential for heterologous antigens delivered as AAV contaminants to negatively impact therapeutic efficacy in hepatically delivered rAAV. The model described here in BALB/cJ mice can be generated without the need for adoptive transfer of cells and could serve as a useful tool in studying the mechanisms driving CD8+ T cell-mediated clearance of hepatocytes after rAAV gene transfer.
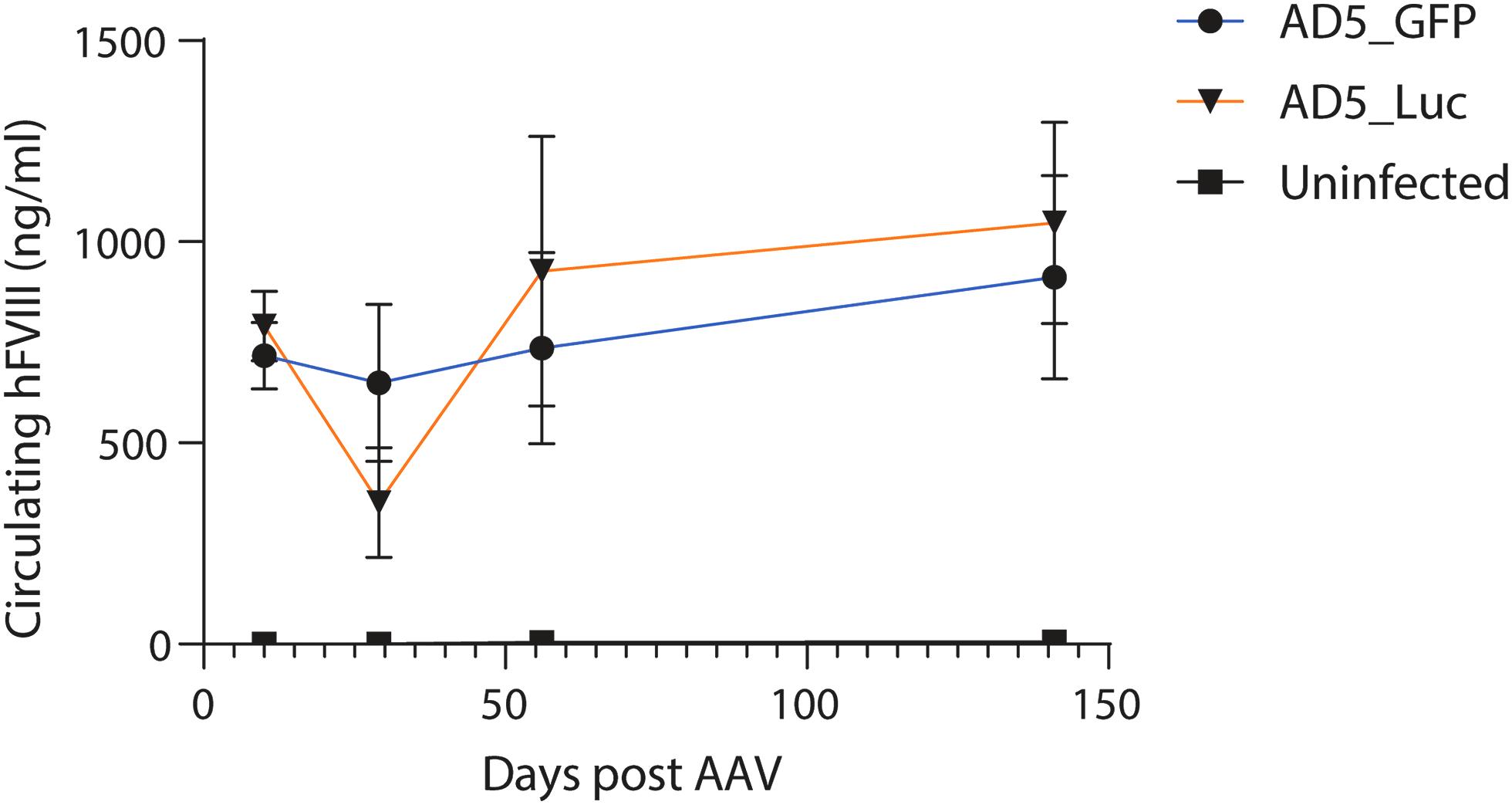
Preimmunization to P5-associated contaminants yields no difference of FVIII expression in Albino C57BL/6J mice. Longitudinal plot of circulating FVIII in Albino C57BL/6J mice preimmunized with Ad-CMV_GFP (blue) or Ad-CMV_Luciferase (orange). Nonimmunized non-AAV-treated mice were used as a control (black). Values were obtained at 10, 29, 56, and 141 days post-AAV8GFP_P5 F8 administration.
DISCUSSION
Contaminating nucleic acids are currently an inherent component of rAAV. Every reported rAAV preparation that has been analyzed for nucleic acid content has had readily detectable heterogeneity. 17 Here in BALB/cJ mice, we report that pre-existing immunity to a heterologous antigen (GFP) packaged as a DNA contaminant in rAAV elicits transaminase elevations and a reduction in the detectable levels of the therapeutic transgene (FVIII).
In doing so, we have generated a vector-inducible mouse model of transaminitis that could be used to interrogate mechanisms involved in CD8+ T cell-mediated clearance of AAV-transduced hepatocytes and develop strategies to circumvent this and avoid transgene expression loss. This model is relatively simple to generate, only requiring the administration of two heterologous viral vectors.
The tolerogenic environment of the liver lends itself to successful gene therapy. Indeed, higher hepatic expression of AAV-encoded transgenes induces a greater number of Tregs, 31,32 and it has been suggested for hemophilia that AAV gene therapy could serve as a tolerance-generating solution to patients who have antifactor inhibitors. 33 –35 The balance between tolerance and a functional CD8+ T cell clearance of hepatocytes is related to vector dose, with higher doses inducing transgene tolerance. 15 Yet, in published clinical studies for hemophilia, the primary determinant of whether patients on a trial will have detectable, delayed, transaminitis is being enrolled in the higher dose cohort. 5,8,10
The current evidence, at least with some patients, is that rAAV-induced transaminitis in humans is driven by an anticapsid T cell response, due to the detection of AAV capsid-specific T cells coinciding with this phenotype. 1,5,9 However, this may not be the full story. Delivering a high dose of the transgene may result in a lower dose of alternative antigens, such as contaminant protein products or cryptic antigens arising from within the expression cassette. 24 If such products encoded well-presented and recognized epitopes, then even with tolerance generated against the therapeutic protein, a deleterious response against the alternative antigens could result in the clearance of hepatocytes also containing a productive transgene, reducing overall treatment efficacy.
Although mouse models utilizing adoptive transfer of AAV capsid-specific T cells have shown successful induction of the transaminitis phenotype, 11,36 evidence from other animal models supports the notion that antigens expressed from within the cell are an equally valid mechanism. A previous study used intramuscular injection of an adenovirus expressing the AAV8 capsid and observed a B cell-dependent reduction in the resultant therapeutic output of an AAV2-FIX vector. 14 However, if capsid-specific cells were adoptively transferred after AAV delivery, no such reduction was observed.
More recently, a study in cynomologous monkeys showed that when AAV empty capsids or a promoterless expression cassette was administered, transaminitis did not occur. However, when the transgene was expressed, transaminitis was observed. 37 Several other studies have detected transgene-recognizing T cells after AAV delivery in a variety of contexts. 38 –41 Given that the human leukocyte antigen (HLA) genes, which determine which peptides are efficiently presented for T cell recognition, are the most diverse loci in the human genome, 42 in humans there may inevitably be different, patient-specific, answers to the hepatocyte clearance question. Rigorous monitoring for HLA associations with negative immune-mediated sequelae may be required to ascertain the precise cause(s) of T cell-mediated clearance of transduced cells after AAV delivery, on a case-by-case basis.
An interesting finding in our study was that although a greater number of CD8+ T cells were generated when mice were dosed with the AAV8ssCMV_GFP vector, transaminases were higher in the AAV8GFP_P5 F8 group. We hypothesize that because P5-associated contaminants in rAAV exist at a range of sizes, truncated forms of the GFP protein were being produced, resulting in easier presentation of the immunodominant epitope.
In addition, the greater detection of capsid-specific CD8+ T cells in mice that were rechallenged with GFP poses an interesting question about whether responses against gene therapy vectors should be viewed as being limited to a single epitope or antigen, but further study must be undertaken to address this question directly before any conclusion can be drawn.
The kinetics of the transaminitis within this model are typical of a memory recall phenotype, in that they occur quickly after the second exposure to the antigen. The clinical experience, however, is characterized by a delayed onset, often up to 3 months after AAV administration. 7,8 This would suggest that the mechanism behind the clinical phenotype is a delayed response to a de novo antigen, rather than a memory response. This notion is supported by the 2017 mouse model using an rAAV-delivered ovalbumin transgene, with observed delayed transgene expression loss. 15 GFP_P5 vectors from this study could potentially be used to assess if such an interference in transgene efficacy would occur in a delayed manner in the absence of preimmunization.
It should be noted that there are questions surrounding this model that should be answered before wider conclusions are drawn. First, if the mice were followed longer, would circulating FVIII expression drop to 0 or plateau around the level we observed at the 10-day timepoint, and would liver enzyme levels be restored to normal once such a plateau, or total loss, had been reached? Moreover, a limitation of the study is that livers were not assayed for infiltration of CD8+ T cells and their cognate reactivity. Such an examination would confirm the role of the observed GFP-specific T cells in clearing the transduced hepatocytes and help to answer whether the co-occurring increase in capsid-reactive T cells had any mechanistic contribution to the transaminitis phenotype.
It is also important to stress that the findings within this article were generated in an artificial capacity. A GFP cassette was purposely placed upstream of the P5 sequence to confer its encapsidation as a contaminant sequence. In this case, the abundance of full length GFP was ∼6% of the intended FVIII cassette, and so a high proportion of FVIII-transduced cells would likely contain a P5-linked GFP contaminant. A purer preparation, where GFP was present at 1%, or 0.1% of the transgene, may have a less pronounced impact on transgene efficacy after GFP preimmunization.
Furthermore, sequences upstream from P5 in clinical rAAV preparations may not have a coding sequence placed to be efficiently translated. However, plasmid backbones often contain coding sequences for antibiotic resistance genes, and even without a canonical coding sequence, there is the risk for production of cryptic peptide sequences from within the expression cassette. 24 The failure of the model to recapitulate the development of transaminitis in an additional mouse strain highlights that perhaps only under specific conditions can rapid, strong, deleterious responses against rAAV contaminant products drive a therapeutically consequential effect.
Although this investigation focused on P5-associated upstream contaminants within rAAV, there are several other sources of nucleic acid contamination that will be packaged within rAAV. This includes host–genome sequences, DNA from the replication or capsid gene of AAV, incomplete expression cassettes, and other plasmid or baculoviral sequences from the rAAV production system. 17 Clinically negative outcomes in rAAV-treated patients have not been directly linked to contaminants of any specific identity.
However, the artificial increase of CpG motifs within a construct used to treat hemophilia-B patients was attributed to complete efficacy loss in seven out of eight patients due to TLR9 activation. 6 Current AAV vectors are designed to minimize CpG motifs within transgenes, but the same does not necessarily apply to these contaminant species, as they are not intended to be packaged. In fact, sequences from plasmids contain bacterial elements that are often highly enriched in CpG motifs and if packaged could contribute to this activation.
A prior study was unable to detect in vivo transcription of either the capsid gene, adenoviral helper sequences, or the ampicillin resistance gene after AAV delivery. 19 However, since the capsid gene is downstream of P5 in a traditional plasmid-based production system, and other amplicons were not designed based upon proximity to P5, these findings are not in conflict with our data. Indeed, this may signify that, unlike P5-associated sequences, not all contaminants packaged into rAAV will be transcribed. Given the potential for widespread application of rAAV technology across disease indications, a more complete understanding of potential risks from preparation-related impurities is required. The model presented here is a potentially useful tool to facilitate this.
There are many open questions regarding how the immune system responds to the administration of rAAV for gene therapy. The model described here has shown that a T cell response after administration of an AAV vector does not need to be against the therapeutic transgene to exert a negative effect on therapeutic protein output. This further highlights the importance of in-depth product characterization and ensuring that unintended nucleic acids are kept to a minimum in rAAV preparations.
Moreover, by inducing rapid transaminitis through a memory recall response against rAAV-delivered antigens, we have developed a tool that can be used to investigate the mechanisms involved in hepatic transaminitis and present this model as an option to interrogate CD8+ T cell-mediated clearance of hepatocytes after the administration of rAAV.
Footnotes
ACKNOWLEDGMENTS
We thank Dr. Robert C. Mettelman for careful proofreading of the article. We thank the St Jude CPC Diagnostic Laboratory for analyzing the liver transaminase assays.
AUTHORs' CONTRIBUTIONS
M.A.B. contributed to conceptualization, methodology, formal analysis, investigation, data curation, writing—original draft, review, and editing, visualization, and funding acquisition. C.L.M. was involved in conceptualization, methodology, investigation, validation, resources, and project administration. S.W. and I.L.R. carried out investigation, resources, writing—original draft, review, and editing.
Y.S., P.-H.C., and J.Z. were involved in investigation and resources. A.C.N. and P.G.T. took charge of supervision and resources. A.S. was in charge of methodology, investigation, formal analysis, and writing—review and editing. A.M.D. was in charge of conceptualization, supervision, writing—review and editing, and project administration.
DATA SHARING AND AVAILABILITY STATEMENT
Plasmids used to make designed contaminant viral preparations are available upon completion of MTA with St Jude Children's Research Hospital. Data available upon request.
AUTHOR DISCLOSURE
A.M.D. and A.C.N. are listed inventors on patents relating to FVIII and FIX construct designs for AAV gene therapy and are entitled to royalty income from their licensing. M.A.B., P.-H.C., C.L.M., and A.M.D. are listed inventors on a pending patent that relates to reducing contaminants upstream of the P5 promoter. M.A.B., I.L.H., S.W., and A.M.D. are listed inventors on a provisional patent that relates to reducing contaminants upstream of the P5 promoter. A.C.N. acts as an advisor for Freeline Therapeutics, BioMarin Pharmaceutical, and Generation Bio; is a founder of, has a sponsored research agreement with, and owns equity in Freeline Therapeutics; and is a consultant to several biopharmaceutical companies.
FUNDING INFORMATION
Support for this research was provided by the American Society of Gene Therapy Career Development Award Fellowship in partnership with the Cystic Fibrosis Foundation (ASGCT CDA) (to M.A.B.).
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.