
Abstract
Immune cell-based therapies can induce potent antitumor effects but are also often associated with severe toxicities. We previously developed a PD-1-based small molecule-regulated reversible T cell activation switch to control the activity of cellular immunotherapy products. This chemically regulated and SH2-delivered-inhibitory tail (CRASH-IT) switch relies on the noncovalent interaction of switch SH2 domains with phosphorylated ITAM motifs in either chimeric antigen receptors or T cell receptors. After this interaction, the immunoreceptor tyrosine-based inhibition motif/switch motif (ITIM/ITSM) containing PD-1 domain present in the CRASH-IT switch induces robust inhibition of T cell signaling, and CRASH-IT-mediated suppression of T cell activity can be reversed by small molecule-induced switch proteolysis. With the aim to develop improved second-generation switch systems, we here analyze the possibility space of both the immune cell receptor docking and inhibitory signaling domains that allow control over T cell activity. Importantly, these analyses demonstrate that the inhibitory domains that most potently suppress antigen receptor signaling in primary human T cells are not derived from inhibitory receptors, such as PD-1 and BTLA, that are endogenously expressed in T cells, but include ITIM/ITSM containing inhibitory domains derived from receptors present in myeloid cells. In addition, we demonstrate that physical proximity of the inhibitory domain to the antigen receptor is crucial to efficiently suppress T cell activation, as only switch designs that employ SH2 domains directly interacting with ITAM motifs in antigen receptors efficiently and reversibly inhibit T cell functionality. These data demonstrate the flexible and interchangeable nature of immune cell signaling domains, and inform the design of a synthetic proximity-based switch system with a superior dynamic range.
Introduction
Adoptive T
To abrogate T cell activity, or to transiently halt T cell activity, a number of irreversible and reversible switch designs have been developed. 9,12 –14 Those strategies that allow reversible control of T cell activity all rely on modification of the antigen receptor, for instance by splitting the receptor into multiple parts, or by its fusion with regulatory domains. 15 However, this design is not compatible with control of endogenous TCRs, and such regulatory systems cannot be “retrofitted” into existing CAR T cell therapies. To address these issues, we designed a chemically regulated and SH2-delivered-inhibitory tail (CRASH-IT) switch that does not require hard modification of the antigen receptor. 16 This design allows CRASH-IT switches to be combined with T cells employing their endogenous TCRs, or T cells modified with transduced TCRs or CARs.
Inhibition of T cell activity by the first-generation CRASH-IT switch was achieved by the immunoreceptor tyrosine-based inhibition motif/switch motif (ITIM/ITSM) containing intracellular domain of the PD-1 receptor. Recruitment of this inhibitory domain to activated antigen receptors by the Zap70 SH2 domains present in the switch induces proximity of the SHP2 phosphatase, thereby suppressing T cell activation through dephosphorylation of the CD28 costimulatory receptor 17 or direct inhibition of TCR signaling. 18 Finally, the ability to control the level of T cell activity is provided by a small molecule-regulated protein stability control domain, such as the small molecule-assisted shutoff (SMASh) domain 19 that can be regulated by asunaprevir, or the FKBP12F36V [FKBP(V)] domain 20 that can be regulated by dTAG-13. Thus, CRASH-IT switches can be considered tripartite regulators of T cell activity, in which one domain controls vicinity to activated antigen receptors, one domain controls recruitment of phosphatase activity, and the final domain is used to influence the levels of this designed regulatory protein. This modular nature of the system makes it an attractive platform to dissect the rules for optimal control of T cell activity, and to test whether second-generation switch designs with improved properties can be developed.
Immune cell signaling pathways are frequently based on a common blueprint, in which the information of a ligand binding event is propagated by a cascade of phosphorylation and dephosphorylation events. Short recognition motifs (ITAM/ITIM/ITSM) are phosphorylated, followed by the highly specific recognition of these phospho-motifs by SH2 domains. 21 ITAM containing activating receptors associate with SH2 domain containing tyrosine kinases such as Zap70, whereas ITIM/ITSM containing inhibitory receptors generally recruit SH2 domain containing phosphatases such as SHP1/2 to counteract ITAM signaling.
To design synthetic T cell activity regulators, the use of intracellular domains that are derived from proteins that are physiological inhibitors of TCR signaling forms a logical first choice. However, the fact that different immune receptors and immune receptor signaling domains can influence cell activity when introduced into other cell types 22 –24 argues for a level of interchangeability. Based on this potential interchangeability, and the abundance of inhibitory signaling and SH2 domain containing proteins in diverse immune cell types, we set out to investigate the possibility space of CRASH-IT-based T cell switch designs, by replacing both the ITAM docking domain and inhibitory signaling domain with structurally or functionally homologous domains.
Comparison of switch activity of designs employing different docking domains underscores the importance of physical proximity of the inhibitory fusion protein to the antigen receptor complex. Furthermore, by exploring the activity of 21 inhibitory domains, we reveal that ITIM/ITSM containing inhibitory domains that are derived from myeloid cell inhibitory receptors, such as SIGLEC11 and SIRPA, yield synthetic regulators of T cell activity with a significantly improved dynamic range, as compared with regulators based on T cell inhibitory receptors. Specifically, both the improved T cell inhibition in the absence of asunaprevir or dTAG-13 and the near full restoration of T cell function in the presence of these drugs identify SIGLEC11- and SIRPA-based switches as optimal systems to control T cell activity. Finally, the observation that some of the most efficient regulators of TCR signaling capacity are found in proteins expressed in different cell types underscores the interchangeable nature of protein modules in phospho-ITIM/ITSM/ITAM-based signaling networks.
Methods
Retroviral DNA constructs
Codon optimized DNA sequences were synthesized by IDT as gene fragments and cloned into the MP71 vector by Gibson assembly. MP71-Zap70 2xSH2-PD1tail-SMASh-IRES-EGFP, MP71-Zap70 2xSH2-SMASh-IRES-EGFP (no signaling domain), MP71-PD1tail-SMASh-IRES-EGFP (no docking domain), MP71-Zap70-PD1-FKBP12F36V-IRES-EGFP, MP71 CD19 ScFv-CD28-CD3 ζ CAR-IRES-huEGFRt vectors, 16 and the HLA class I-restricted CDK4 TCR (TCR 17) 25 have been described previously. To generate CRASH-IT variants encoding alternative docking domains, the Zap70 (2xSH2 domains) in MP71-Zap70 2xSH2-PD1tail-SMASh-IRES-EGFP were replaced with the codon optimized sequences listed in Supplementary Table S1. CRASH-IT variants encoding alternative inhibitory domains were created by replacing the PD-1 signaling domain in MP71-Zap70 2xSH2-PD1tail-SMASh-IRES-EGFP and MP71-Zap70-PD1-FKBP12F36V-IRES-EGFP vectors with the codon optimized sequences listed in Supplementary Tables S2 and S3, respectively, or the variant sequences described in Figs. 1D and 4A. A CAR panel encoding various ITAM containing signaling domains was created using the signaling and structural domains described in Supplementary Table S4, using the same vector backbone and CD19 ScFv antibody sequence as used in the MP71 CD19 ScFv-CD28-CD3 ζ CAR-IRES-huEGFRt vector.
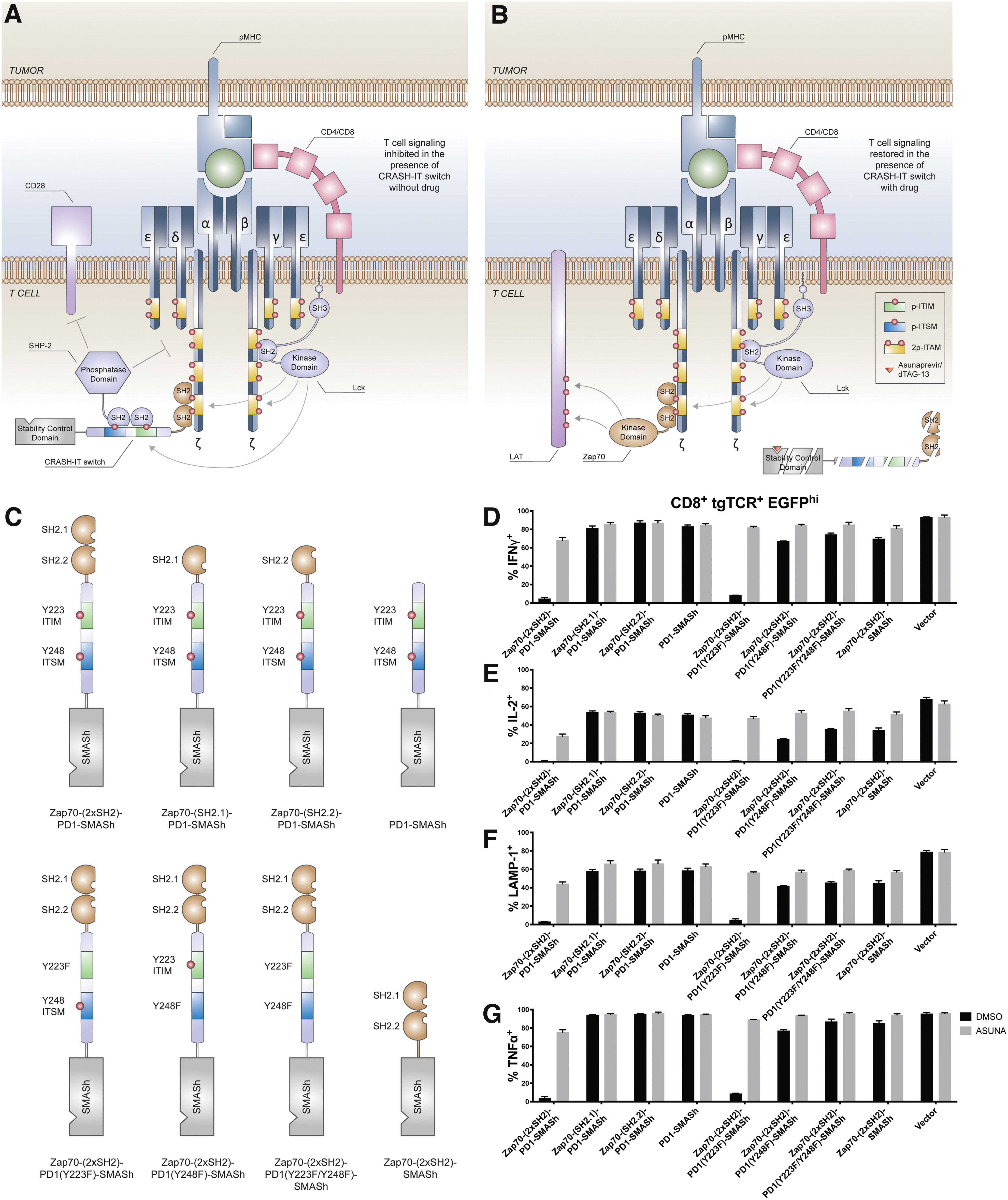
CRASH-IT components required for efficient and reversible inhibition of T cell functions.
Cell lines and cell culture
FLYRD18, NKIRTIL006 26 and Daudi cells were cultured in IMDM (Thermo Fisher Scientific), supplemented with 8% FCS (Thermo Fisher Scientific) and penicillin–streptomycin (100 IU/mL penicillin, 100 μg/mL streptomycin; Sigma-Aldrich). FLYRD18 and NKIRTIL006 cells were passaged every 2–3 days with trypsin-EDTA (Thermo Fisher Scientific). The human NK cell line KHYG-1 (DSMZ) was cultured in RPMI supplemented with 8% FBS and penicillin–streptomycin (100 IU/mL penicillin, 100 μg/mL streptomycin) containing 500 IU/mL IL-2 (Novartis). All cell lines were tested for mycoplasma using PCR-based screening and found negative.
Retrovirus production
Retroviral particles were produced in FLYRD18 packaging cells. In brief, 700,000 FLYRD18 packaging cells were plated per 10 cm dish 1 day before transfection. The next day, cell culture medium was refreshed with IMDM supplemented with 8% FCS without antibiotics. Twenty-five microliters of X-tremeGENE 9 (Roche) was mixed with 800 μL Opti-MEM (Thermo Fisher Scientific) and incubated for 5 min. Subsequently, the Optimem-X-tremeGENE 9 mixture was added on top of 10 μg retroviral plasmid DNA dissolved in water and incubated for 15 min, and the resulting transfection mixture was added dropwise onto the packaging cells. Retrovirus containing supernatant was harvested 48 h after transfection and immediately used or snap-frozen in liquid nitrogen.
T cell isolation and activation
Peripheral blood mononuclear cells (PBMC) were isolated from buffy coats from healthy donors in accordance with local regulations (Sanquin, Amsterdam, NL) by Ficoll-Isopaque density centrifugation 27 and were stored frozen until further use. To generate activated T cell populations, PBMCs were thawed in PBS containing 5% FCS, counted, and mixed with CD3/CD28 Dynabeads (CTS) at a 1:1 cell to bead ratio, at a density of 107 cells/mL. After a 30-min incubation at room temperature on a tumbler, the mixture was put on a magnet and unbound cells were removed. Bead bound T cells were subsequently resuspended in RPMI supplemented with 10% human serum (Sigma-Aldrich) and penicillin–streptomycin containing 100 IU/mL IL-2 and 5 ng/mL IL-15 (Peprotech), and plated at a density of 0.75 × 106 cells/mL.
Spin-transduction of T cells
Twenty-four-well nontreated cell culture plates were covered with 10 μg/mL RetroNectin (Takara) overnight at 4°C. The next day, the RetroNectin solution was removed and wells were blocked with 2% BSA (Sigma-Aldrich) in PBS for 30 min. Activated T cells (0.25 × 106 cells/mL in RPMI/10% human serum/penicillin–streptomycin/200 IU/mL IL-2 and 10 ng/mL IL-15) were then mixed with retroviral supernatant at a 1:1 ratio (volume/volume) in RetroNectin-coated 24-well plates and centrifuged at 2,000 RPM for 90 min at room temperature. Transduction efficiencies and estimated multiplicity of infection (MOI) values are listed in Supplementary Tables S5–16. MOI values were calculated using Poisson distribution equation P(0) = e −λ , where P(0) is the fraction of untransduced cells, and λ is the MOI.
Small molecule compounds
Asunaprevir, an orally available drug for the treatment of hepatitis C virus (HCV) infection, 28 was obtained from MedChemExpress (No. HY-14434, supplied as 10 mM stock solution in DMSO). dTAG-13 was obtained from Tocris (No. 6605) as powder, and a 10 mM stock solution in DMSO was prepared (No. D4540–500 ML; Sigma).
Cytokine release assay and antibody staining
T cells were pretreated with 10 μM asunaprevir, 0.5 μM dTAG-13 or DMSO control in T cell medium (RPMI/10% human serum/penicillin–streptomycin, 100 IU/mL IL2 and 5 ng/mL IL15) for 24 h before coculture experiments. 100,000 T cells and 100,000 of the indicated tumor cells were mixed in T cell medium supplemented with golgi-plug (1:1,000 dilution; BD) and anti-LAMP1-APC (No. 328620; 1:100 dilution; Biolegend), in the presence of indicated drugs or DMSO control in 96-well plates and incubated for 5 h at 37°C. In all coculture experiments, transduced cell populations from the same donor were for each experimental condition (i.e., DMSO control and either asunaprevir or dTAG-13 treatment) split into three wells before coculture with target cells. After incubation, cells were washed once with PBS and stained with IR dye (No. L34976; Invitrogen,) at 1:400 dilution for 5 min at 4°C. Subsequently, cells were washed once with FACS buffer (PBS plus 0.5% BSA) and stained with anti-CD8-PerCP Cy5.5 (No. 341050, 1:20 dilution; BD), anti-CD4 BV711 (No. 317440, 1:50 dilution; Biolegend), anti-murine constant TCR-PE (in experiments using TCR transduced T cells, 1:200 dilution, No. 553172; BD) or cetuximab-PE (in experiments using CAR transduced T cells that express the truncated human EGFR reporter, 1:200 dilution, No. FAB9577P; R&D Systems) for 20 min at 4°C. Cells were then washed once with FACS buffer and fixed with BD fixation and permeabilization solution for 20 min at 4°C. After permeabilization, cells were washed twice with BD perm/wash buffer and stained with anti-IFNγ-BV421 (1:100 dilution, No. 564791; BD), anti-IL-2-PE-Cy7 (1:100 dilution, #560707; BD), and anti-TNFα-BV650 (1:100 dilution, #502938; Biolegend) diluted in perm/wash buffer for 20 min at 4°C. Cells were then washed twice, resuspended in 100 μL FACS buffer and analyzed directly on a Fortessa Special Order analyzer. Data were analyzed using FlowJo and Prism 7 software. Flow gating strategy and representative flow data are provided in Supplementary Fig. S7. Absolute cytokine expression and degranulation profiles are listed in Supplementary Tables S17–34.
Results
Characterization of CRASH-IT components required for efficient and reversible inhibition of T cell function
Upon pMHC ligation to the TCR, Lck binds to CD4/CD8 costimulatory receptors and phosphorylates ITAM motifs present in the CD3zeta chain. The ITAM-phosphorylated CD3zeta chain subsequently forms a docking platform for the tandem SH2 domains of the Zap70 tyrosine kinase. To characterize components of the CRASH-IT switch (Fig. 1A, B) that are required for efficient and reversible inhibition of T cell signaling, we first investigated whether CRASH-IT switches require bivalent binding to the CD3zeta chain, or whether either of the two individual Zap70 SH2 domains suffices to bring the PD-1 inhibitory tail domain to the TCR complex. Primary human T cells were modified with a high affinity CDK4 neoantigen-specific class I-restricted TCR together with CRASH-IT variants that either contain one of the individual or both SH2 domains of Zap70.
Analysis of cytokine production and degranulation of modified T cells cocultured with NKIRTIL006 melanoma cells endogenously expressing the CDK4 neoantigen revealed efficient control over T cell activity in T cells equipped with the full-length first-generation CRASH-IT switch (a 15-, 33-, 20-, and 14-fold difference in intracellular IFNγ, IL2, TNFα, and surface LAMP-1 levels in absence or presence of 10 μM of the HCV NS3/4A protease inhibitor asunaprevir that is used as to induce degradation of SMASh containing CRASH-IT switches). In contrast, suppression of cytokine production and degranulation in T cells equipped with CRASH-IT variants containing a single SH2 domain was <1.2-fold (Fig. 1C–G).
To subsequently understand which domains within the PD-1 tail are critical for T cell inhibition by CRASH-IT switches, we focused on the PD-1 ITIM and ITSM motifs that are phosphorylated after PD-L1 ligation, and thereby form a docking platform for the SH2 domains of the inhibitory SHP-2 phosphatase. 29 Mutation of Y248 to phenylalanine identified a critical role for the PD-1 ITSM, with very limited control over all T cell activities in the absence of asunaprevir (Fig. 1C–G). In contrast, mutation of Y223 to phenylalanine resulted in only a mild impairment in switch activity, indicating a modest but significant contribution of the PD-1 ITIM, with a 1.8-, 1.5-, 2.3-, and 1.7-fold increase in intracellular IFNγ, IL2, TNFα, and surface LAMP-1 levels in T cells equipped with the ITIM deficient switch, relative to cells expressing the parental switch in the absence of asunaprevir (p = 0.0097, 0.0206, 0.0077, and 0.0336, respectively). In line with expectations, asunaprevir addition did not influence the activity of non-switch modified (i.e. EGFP-negative) CDK4 TCR+ T cells present in the same cultures, indicating that control over T cell function is fully cell-intrinsic (Supplementary Fig. S1).
Docking domain requirements to achieve efficient inhibition of T cell signaling
Having established the importance of both individual Zap70 SH2 domains, the PD-1 ITSM and, to a lesser extent, the PD-1 ITIM to allow efficient inhibition of T cell function, we explored the possibility space for domains facilitating efficient docking of inhibitory domains to the TCR complex by replacing the Zap70 SH2 domains of the CRASH-IT switch with interaction partners of the CD3zeta chain, with direct fusion to the CD3zeta chain, or with signaling/adaptor proteins involved in proximal or more distal TCR signaling. Using protein interaction information from the Uniprot database and literature, 30 a total of 27 putative switch constructs was designed employing the docking domains or full-length sequences from 19 signaling or adaptor proteins (Supplementary Table S1).
Analysis of switch function in primary human T cells modified to express the CDK4 TCR plus individual CRASH-IT variants indicated that robust inhibition of antigen-induced T cell activation was highly dependent on the docking domain used (Fig. 2 and Supplementary Fig. S2). Specifically, although Nck2-, TRAT1-, Grb2-, and CD3z-based switch designs showed a low-level inhibitory effect on T cell function, particularly IL2 production, the magnitude of these effects was minimal as compared with Zap70 SH2-based CRASH-IT switches.
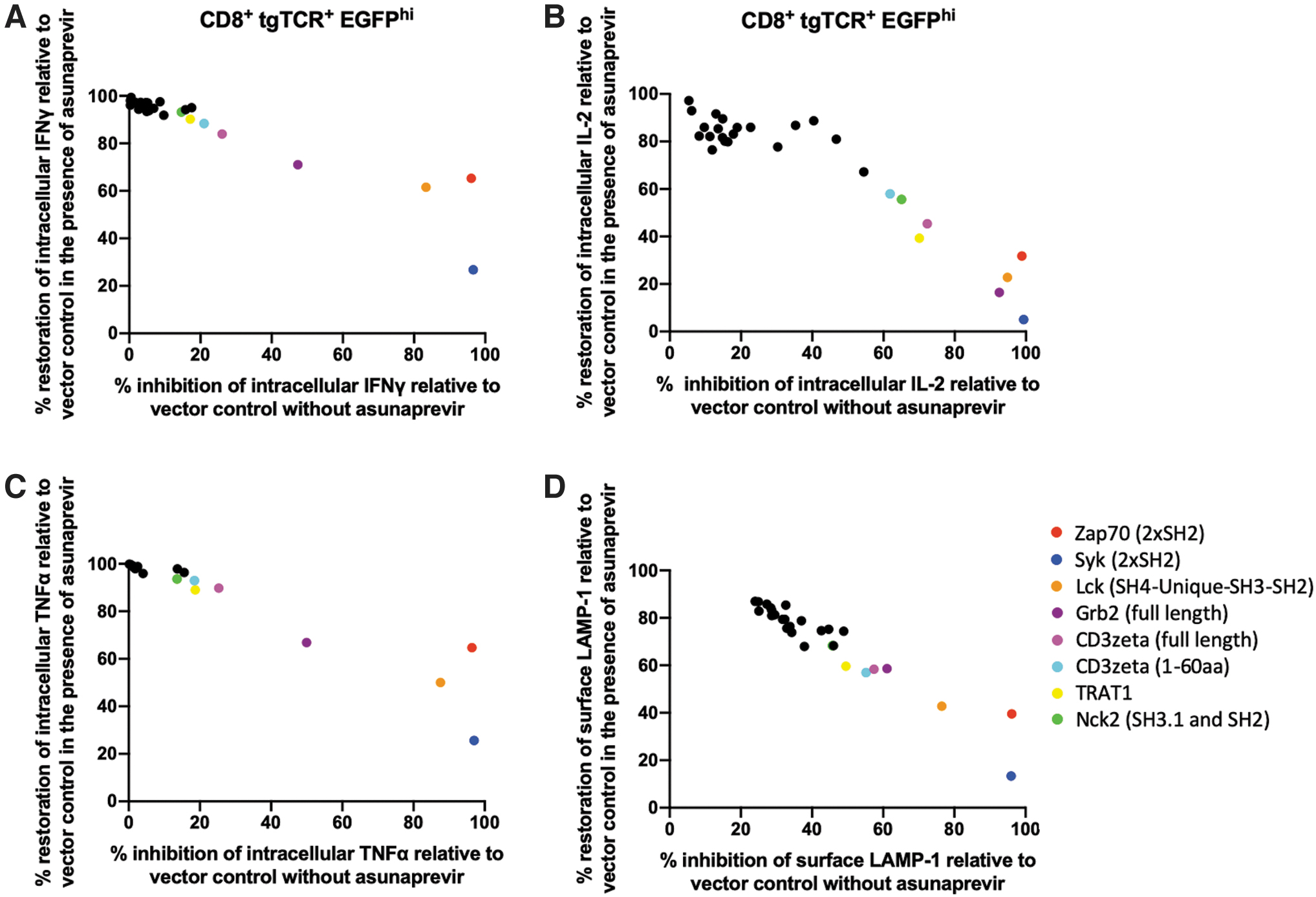
Docking domain requirements to achieve efficient inhibition of T cell signaling. Primary human T cells modified with the CDK4 TCR plus vector control, plus a switch without docking domain, or plus a docking domain-PD1-SMASh switch in which the docking domain was chosen from Zap70 (2xSH2), Syk (2xSH2), Lck (SH4-Unique-SH3-SH2), Nck1 (SH3.1 and SH2), Nck1 (SH2), Nck2 (SH3.1 and SH2), Nck2 (SH2), SLA (SH3-SH2), SLA2 (SH3-SH2), SHB (SH2), SHC1 (SH2), TRAT1, DOCK2 (SH3), SH2D1A (SH2), SH2D2A (SH2), SH2D1B (SH2), CRK (SH2), Grb2 (full-length), Grb2 (SH2), Numb (PTB), SLP76 (SH2), truncated CD3zeta (1-51aa, 1-56aa, 1-60aa), or full-length CD3zeta, were pretreated with 10 μM asunaprevir or DMSO control. Data depict percentage inhibition and percentage restoration of intracellular IFNγ
Notably, a CRASH-IT switch employing the SH2 domains from the Syk protein, a close homolog of Zap70, was shown to allow reversible inhibition T cell activation. Similar to Zap70, the Syk protein contains 2x SH2 domains for interaction with phosphorylated ITAM motifs. 31 Despite the conserved functions of the SH2 domains in both proteins, T cell reactivation levels in cells expressing the Syk SH2-based switch was reduced, relative to cells expressing the Zap70-based switch. A switch design that contained the SH4-Unique-SH3-SH2 domains of Lck likewise allowed reversible inhibition of T cell functions. However, transduction efficiencies with the Lck-based CRASH-IT switch were substantially lowered relative to other switch designs, suggesting a detrimental effect in producer cells, and thereby limiting its clinical utility. The observation that a large majority of switch designs employing binding domains from different levels in the TCR signaling pathway provide little control over T cell activity, whereas Zap70, Syk, and Lck-based CRASH-IT switches that contain SH2 domains that directly interact with phosphorylated ITAM motifs present in CD3zeta chain do allow such control, suggests a strict spatial or temporal requirement to efficiently delivery inhibitory signals to the T cell signaling pathway.
Improved CRASH-IT switch designs with ITIM/ITSM containing signaling domains derived from non-T cell inhibitory receptors
We subsequently explored the possibility space of the inhibitory module in proximity-based T cell switches by replacing the PD-1 tail with signaling domains derived from a diverse set of inhibitory receptors. In this analysis, we did not restrict the inhibitory receptor set to proteins that are endogenously expressed in T cells or proteins containing ITIM/ITSM, but also sampled signaling domains from inhibitory receptors with dominant expression in either T cells, B cells, or myeloid cells. Side-by-side comparison with the PD-1-based CRASH-IT switch revealed that a CRASH-IT switch based on the ITIM/ITSM containing BTLA signaling domain inhibited cytokine production and degranulation with similar efficiency in the absence of asunaprevir, and this suppression was reversed in the presence of 10 μM asunaprevir (Supplementary Fig. S3). In contrast, CD22-, FCGR2B-, CTLA4-, TIM3-, LAG3-, and TIGIT-based switches all failed to efficiently suppress T cell function. Notably, use of a switch system based on the 3xITIM 1xITSM containing SIRPA signaling domain resulted in a suppression of T cell activity that exceeded that observed with the PD-1-based switch. Considering that substantial inhibitory activity, in one case exceeding that achieved with the PD-1 inhibitory domain, was observed with three different ITSM containing inhibitory receptor domains, we created an additional panel of switch constructs with a focus on ITSM containing inhibitory domains. To this purpose, we assembled a library of all ITSM containing inhibitory domains within the human proteome 21 with a total length of <200 amino acids. To ensure utility also in CAR-modified T cells, this new panel of CRASH-IT variants, plus the variants from the first screen, were first evaluated with respect to their capacity to control signaling through a second-generation CD19-specific CAR in primary human T cells (Supplementary Fig. S4), and the activity of well-performing switch designs was subsequently verified in TCR-T cells (Supplementary Fig. S5). T cell activation upon culture with CD19+ Daudi cells in the absence or presence of 10 μM asunaprevir, was used to quantify and rank the degree of T cell inhibition provided by the various signaling domains (Supplementary Fig. S4B). Furthermore, to be able to identify those switches that already mediate efficient suppression at low expression levels, we focused on T cell inhibition as a function of switch expression level. Notably, analysis of suppression of T cell activity in cells with intermediate switch expression levels revealed that switch systems that were based on the myeloid cell inhibitory receptors SIGLEC5, SIGLEC9, and SIGLEC11 all yielded significantly stronger inhibition of T cell activity, as compared with the PD-1-based switch (p ≤ 0.000001, p = 0.000003, p ≤ 0.000001, respectively), and a SIRPA-based switch trended toward higher activity, whereas there was no statistically significant difference between inhibition provided by PECAM1 and PD-1-based switches. Substantial, but lower, control over T cell activity was obtained with BTLA, SIGLEC6, and LY9 containing CRASH-IT variants, whereas SLAMF1, SLAMF6, SLAMF7, LAG3, and TIM3 containing switches inhibited T cell activation only weakly. CD22, FCGR2B, CTLA4, TIGIT, CD84, CXADR, and CD244 containing switches inhibited T cell activation even less than the switch without inhibitory tail, possibly because of impaired folding or stability of these fusion proteins (Supplementary Fig. S4B). Notably, in cells with high-level switch expression, asunaprevir-induced degradation of SIRPA and SIGLEC5, 9, or 11 containing switches failed to efficiently restore T cell effector functions, further suggesting the strong inhibitory activity of these signaling domains (Supplementary Fig. S6), and indicating a need for improved control over switch protein levels for such second-generation switch systems. Importantly, efficient suppression of T cell activity by SIGLEC5, SIGLEC9, SIGLEC11, and SIRPA-based CRASH-IT switches was also observed in primary human T cells modified with a tumor-reactive TCR, indicating that strong T cell signaling inhibitory domains efficiently downregulate T cell functions independent of the type of antigen receptor (Supplementary Fig. S5).
Efficient control over TCR-modified T cell function with optimized FKBP(V)-based CRASH-IT switches
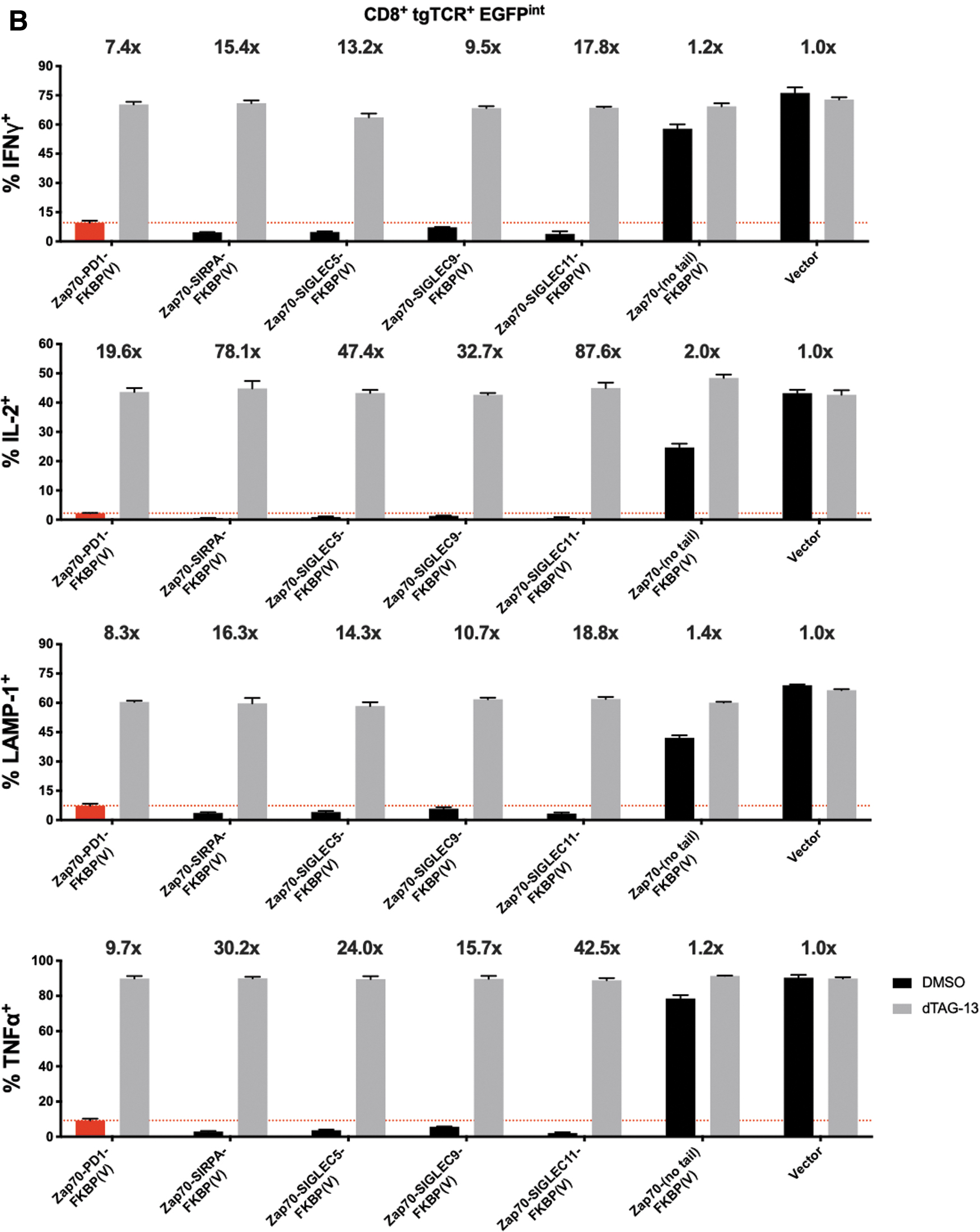
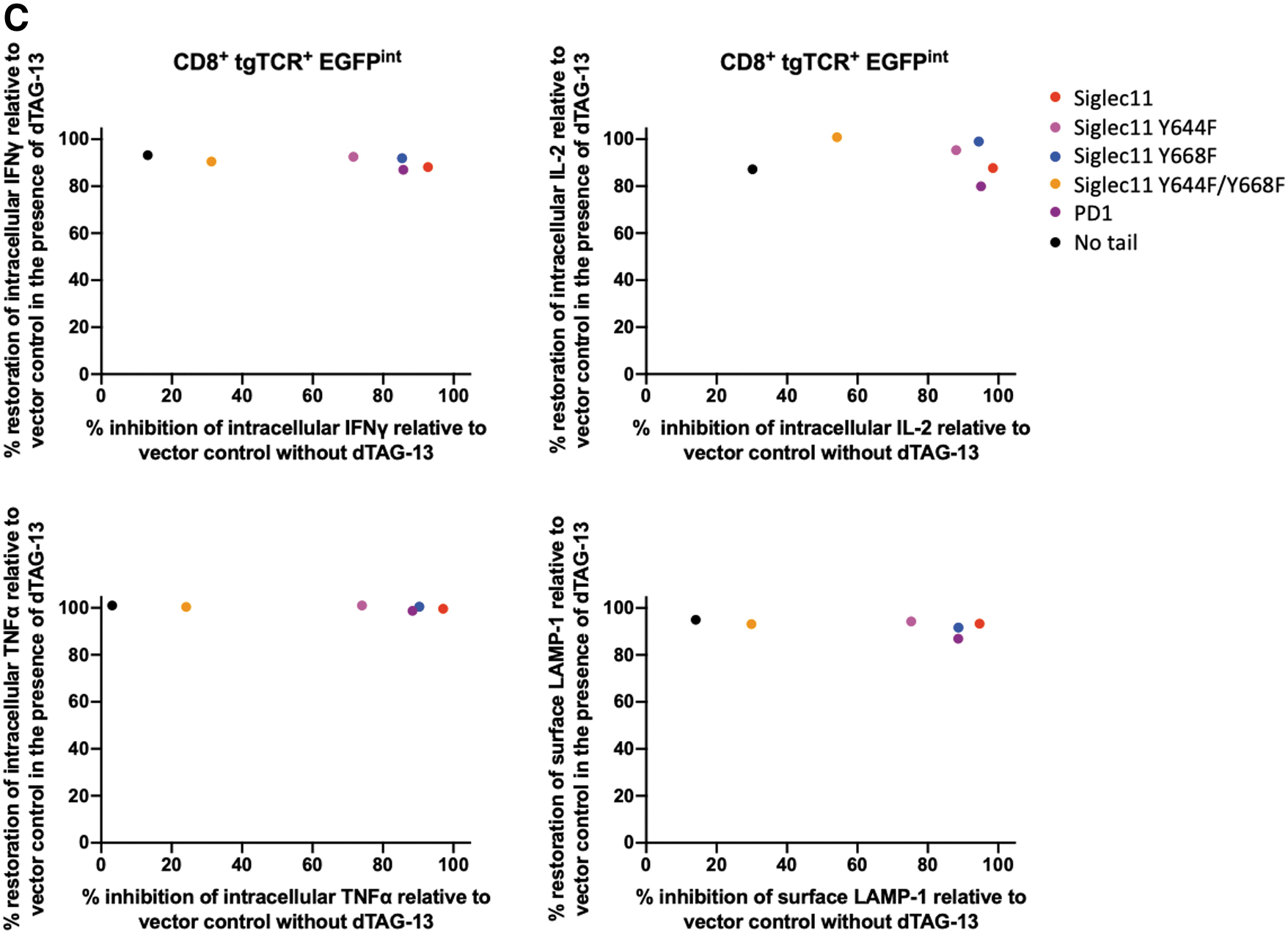
In T cells modified with SIRPA/SIGLEC5/SIGLEC9/SIGLEC11 inhibitory domain-based CRASH-IT switches that employ the SMASh domain to regulate switch protein levels, asunaprevir addition only results in a partial restoration of T cell activity, suggesting that residual switch protein levels still result in substantial inhibition of TCR signaling (Supplementary Figs. S3–S6). To determine whether induced protein degradation systems with a higher dynamic range could increase the utility of such highly active designs, we created a panel of dTAG-13 inducible FKBP(V)-based CRASH-IT variants. Importantly, despite the very potent inhibitory potential of these four myeloid inhibitory receptor domains in the absence of the dTAG-13 PROTAC, cytokine production, and surface LAMP-1 levels were largely restored in the presence of 0.5 μM dTAG-13, both in cells with high and intermediate levels of switch expression (Fig. 3). Particularly tight regulation of T cell output signals was observed with the SIGLEC11-based switch system, with PROTAC addition boosting the fraction of intracellular IFNγ, IL2, TNFα, and surface LAMP-1-positive cells in the presence antigen-positive tumor cells by 61-, 422-, 235-, and 29-fold in cells with high level switch expression. Furthermore, relative to PD-1 switch expressing cells, a 2.3-, 4.5-, 4.2-, and 2.5-times greater fold induction of intracellular IFNγ, IL2, TNFα, and surface LAMP-1-positive cells was observed in SIGLEC11-based switch expressing cells, with the activity of T cells in the presence of 0.5 μM dTAG-13 closely approaching the activity of control vector modified cells. These data identify second-generation CRASH-IT-based switch designs with improved properties over PD-1-based switches and indicate a particular utility of a number of members of the SIGLEC family. Site-directed mutagenesis of the ITIM and/or ITSM motifs in the SIGLEC11-based CRASH-IT switch revealed that both the SIGLEC11-ITIM and SIGLEC11-ITSM contributed substantially to the high degree with T cell inhibition, but with a larger contribution of the former (Fig. 4). Together with the prior data revealing a dominant role for the PD-1 ITSM in T cell inhibition (Fig. 1C–G), these data suggest that the relative contribution of ITIM and ITSM motifs to T cell inhibition is context dependent.
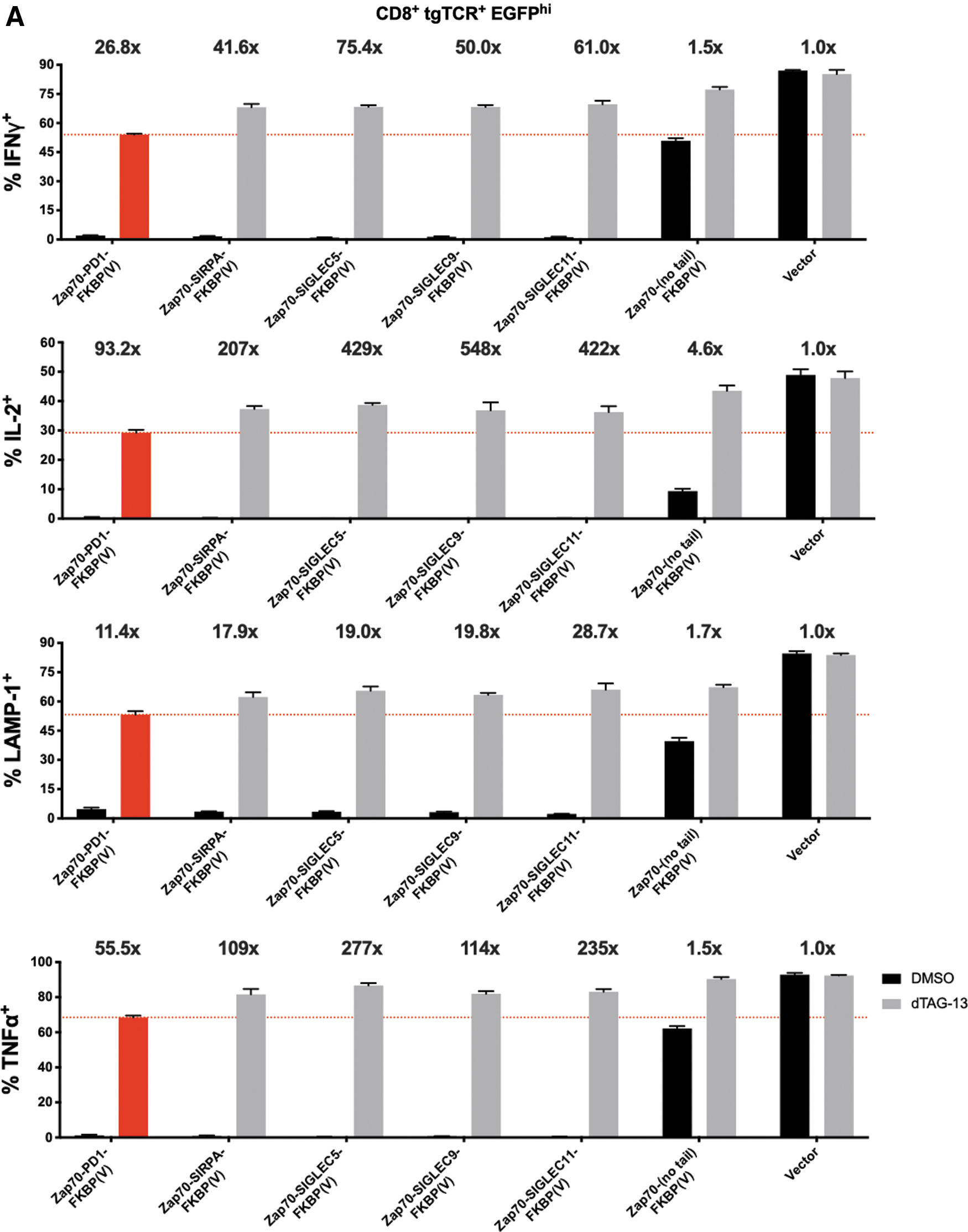
Improved dynamic range of PROTAC-based highly active CRASH-IT switch designs. Primary human T cells modified with the CDK4 TCR plus vector control, plus a switch without inhibitory domain, or plus a Zap70 (2xSH2)-inhibitory domain-FKBP(V) switch in which the inhibitory domain was chosen from PD-1, SIRPA, SIGLEC5, SIGLEC9, or SIGLEC11 signaling domains, were pretreated with 0.5 μM dTAG-13 or DMSO control. Data depict intracellular IFNγ, IL2, TNFα, and cell surface LAMP1 expression of CDK4 TCR+, CD8+, EGFP high
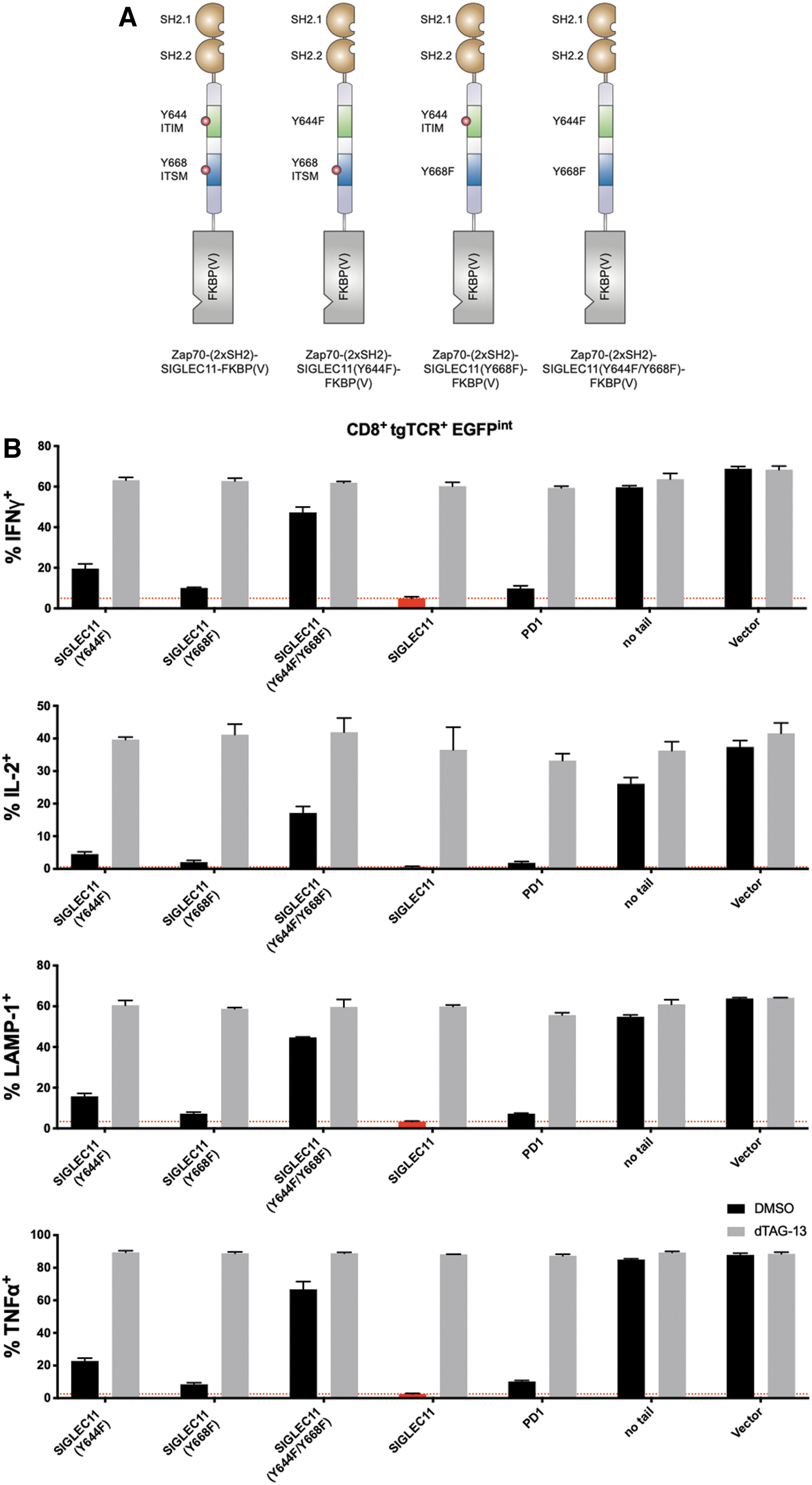
Context-dependent contribution of ITIM/ITSM inhibitory motifs to T cell inhibition.
Efficient regulation of a diverse panel of ITAM receptors with a SIGLEC11-based CRASH-IT switch
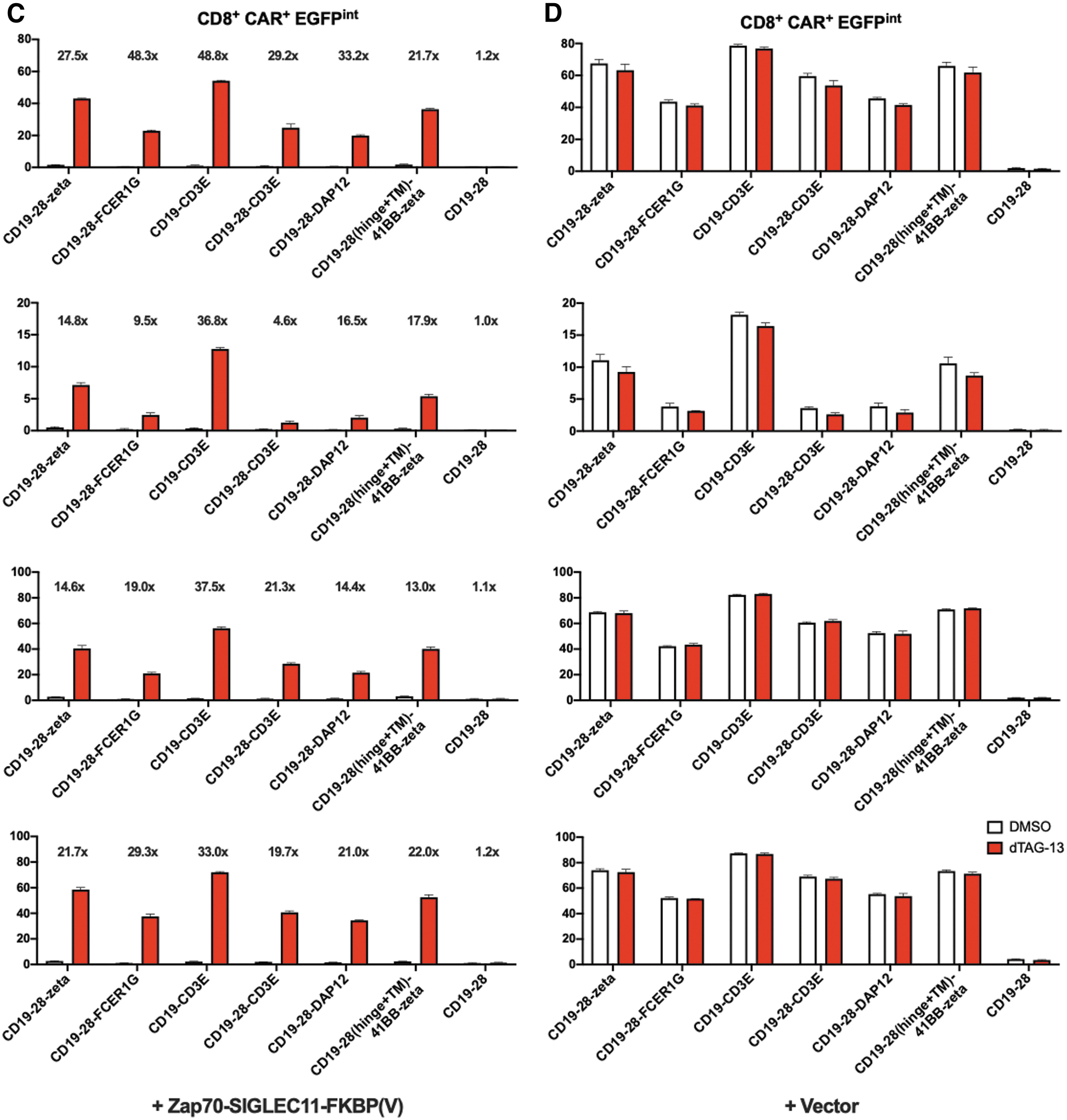
The aforementioned data indicate a substantial flexibility in the capacity of different inhibitory domains to regulate signaling through the CD3 ITAM motifs in TCR-modified cells and the CD3zeta ITAM motifs in CAR-modified T cells. To explore whether a given inhibitory domain that can be used to control T cell signaling through the CD3 complex can also be used to control T cell activation through other ITAM containing domains, we generated primary human T cells modified to express CD19-specific CARs that contained the ITAM containing domains of either CD3zeta, the gamma chain of the immunoglobulin E receptor (FCER1G), CD3epsilon, or DAP12. Exposure of resulting CAR T cells to CD19+ Daudi cells in the absence or presence of 0.5 μM dTAG-13 revealed tight control over T cell activation in all four cases (Fig. 5).
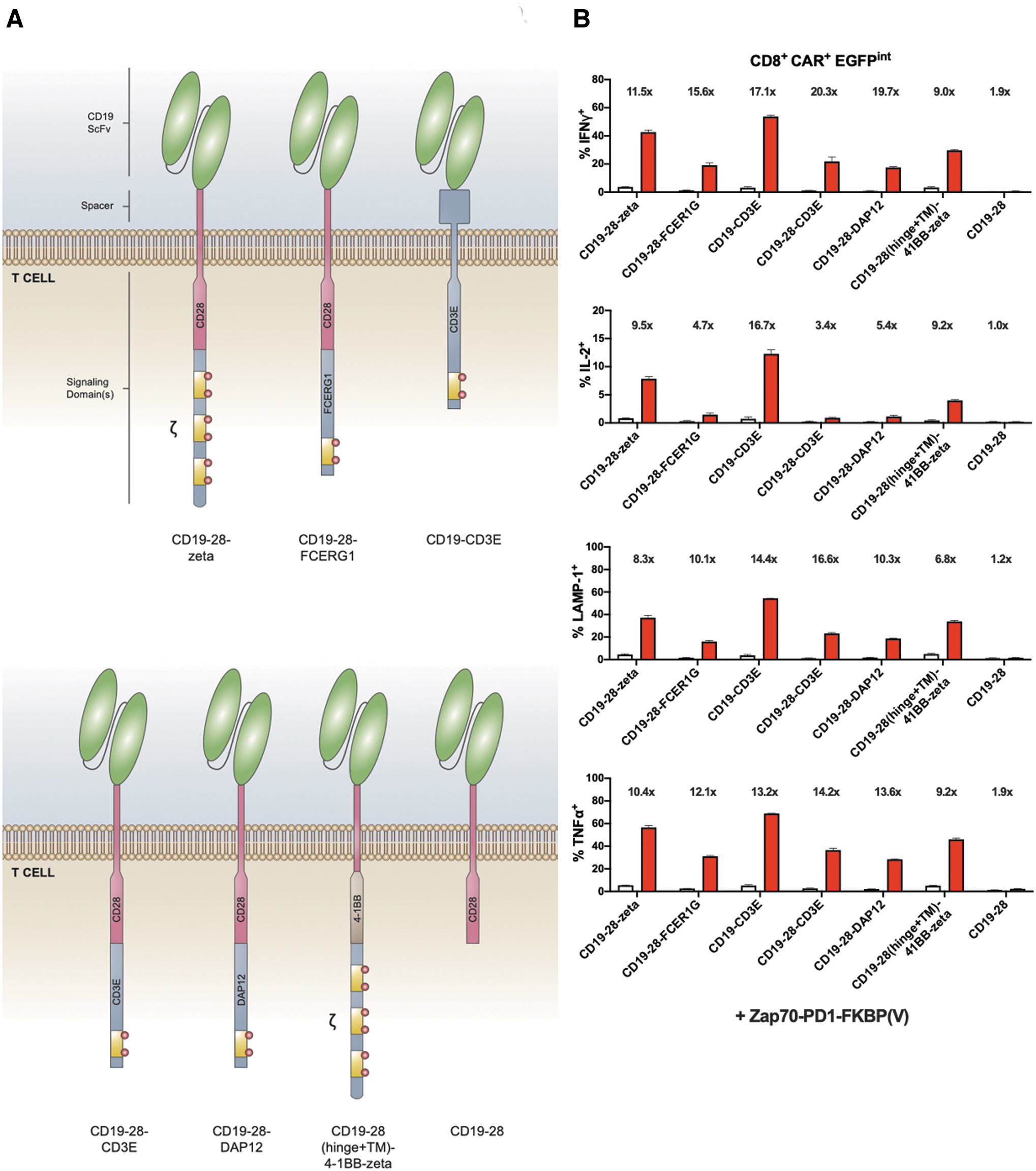
Efficient regulation of different CAR designs by second-generation CRASH-IT switches.
To explore the capacity of CRASH-IT-based systems to also control activity of ITAM containing NK cell receptor complexes in their physiological context, we modified the KHYG-1 NK cell line with first- or second-generation CRASH-IT switches. Coculture of resulting cells with HLA class I deficient K562 cells showed efficient inhibition of cytokine production in the absence of small molecule for both PD-1 and SIGLEC11-based designs, but with superior restoration of cytokine production in the presence of 0.5 μM dTAG-13 for the second-generation switch (Fig. 6). Thus, whether antigen recognition is provided by the TCR, CD3 zeta chain-containing classical CAR designs, a range of other CAR designs with alternative ITAM containing domains, or by NK cell receptors, SIGLEC11-based switch systems provide superior control over immune cell activity.
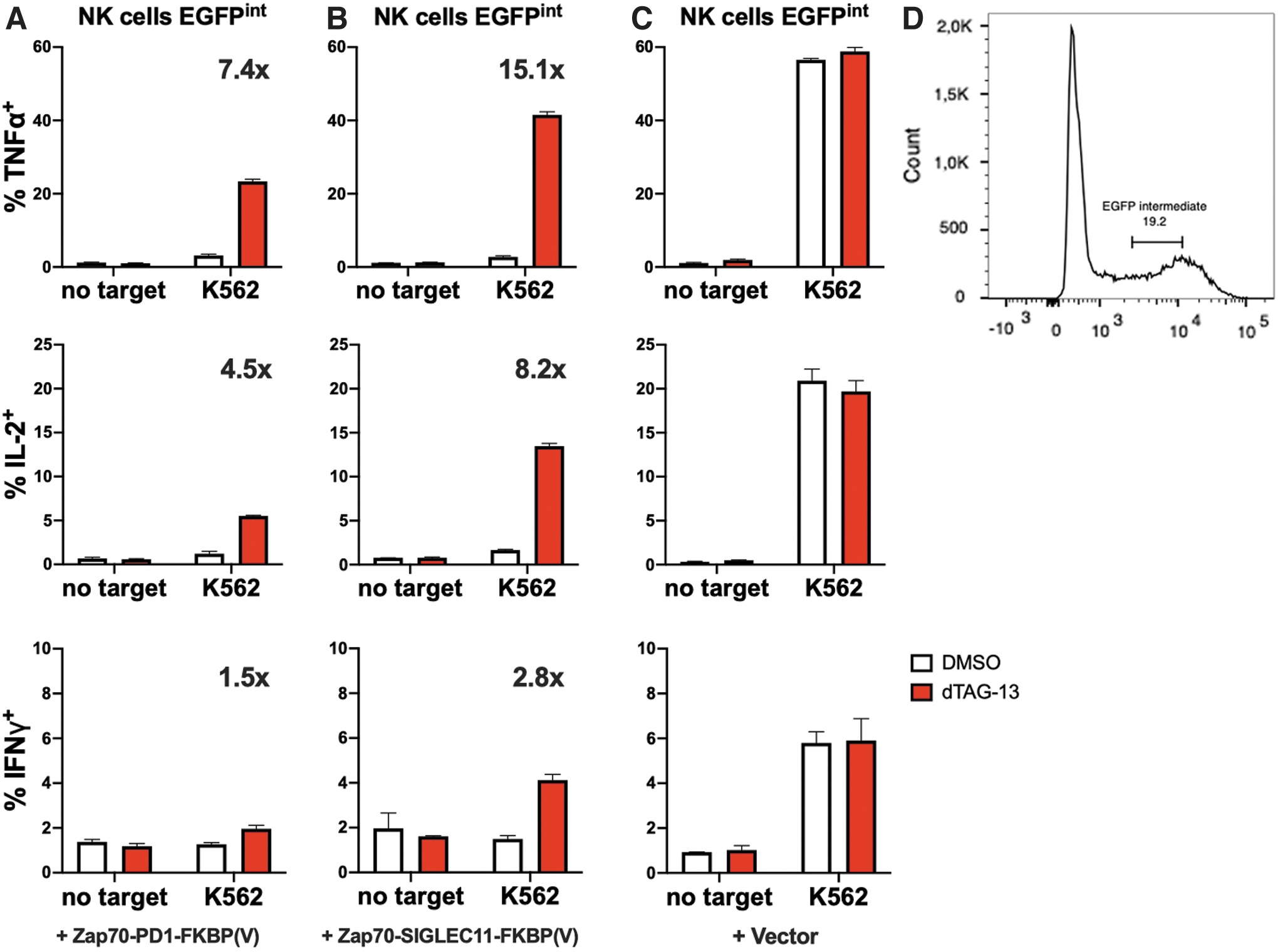
Control of NK cell function by second-generation CRASH-IT switches. The human NK cell line KHYG-1 modified with Zap70-PD1-FKBP(V) switch
Conclusion
The efforts to optimize the different structural domains of CRASH-IT switches described in this study yielded second-generation switch designs with improved capacity to control T cell activity. In particular, SIGLEC11-based switches allow an extremely tight control over T cell activity, with an on average 189-fold induction in the fraction responding cells by dTAG-13 addition over the four T cell functions tested. As demonstrated using the dTAG-13 regulated FKBP(V)-based designs, such switch systems can be assembled fully from human protein domains, thereby significantly reducing the probability of immune-mediated rejection. Furthermore, the fact that control over T cell activity is achieved in cells with both low and high switch expression also significantly facilitates clinical implementation and may, for instance, enable engineering of compact vector designs that combine antigen receptor and CRASH-IT in a single vector.
CRASH-IT-based switches may potentially be used for the clinical testing of gene-modified T cells in settings in which on-target off-tumor toxicity forms a concern, or to reduce the likelihood of cell therapy-induced CRS or NT. In addition, a recent study has suggested that CAR T cells may benefit from pulsatile—rather than constitutive—antigen receptor signaling, 32 and CRASH-IT triggering could provide a means to achieve this. On a more general note, the ability of ITIM/ITSM containing inhibitory signaling domains derived from different lineages of the blood system to control signaling through a variety of ITAM containing receptors provides clear evidence of the modular nature of immune receptor signaling. This malleability of immune receptor systems makes the use of components of this system in other synthetic biology approaches that aim to develop novel signaling networks particularly attractive.
Footnotes
Authors' Contributions
A.C.S. and T.N.S. conceived the project, analyzed the data, and wrote the article. A.C.S. performed experiments and visualized the data. T.N.S. supervised the project.
Acknowledgments
We would like to thank R.N. Ozdeslik (UMC Utrecht, The Netherlands) for illustrations.
Author Disclosure
A.C.S and T.N.S. are inventors on the patent application PCT/NL2020/050655 that covers the CRASH-IT switch system. T.N.S. is advisor for Adaptive Biotechnologies, Asher Biotherapeutics, Allogene Therapeutics, Merus, and Neogene Therapeutics; is a recipient of grant or research support from Merck KgaA; is a stockholder in Allogene Therapeutics, Asher Biotherapeutics, Merus, and Neogene Therapeutics; and is venture partner at Third Rock Ventures, all outside of this study.
Funding Information
This study was supported by Oncode Institute core funding.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8
Supplementary Table S9
Supplementary Table S10
Supplementary Table S11
Supplementary Table S12
Supplementary Table S13
Supplementary Table S14
Supplementary Table S15
Supplementary Table S16
Supplementary Table S17
Supplementary Table S18
Supplementary Table S19
Supplementary Table S20
Supplementary Table S21
Supplementary Table S22
Supplementary Table S23
Supplementary Table S24
Supplementary Table S25
Supplementary Table S26
Supplementary Table S27
Supplementary Table S28
Supplementary Table S29
Supplementary Table S30
Supplementary Table S31
Supplementary Table S32
Supplementary Table S33
Supplementary Table S34
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.