
Abstract
Precise gene manipulation by gene editing approaches facilitates the potential to cure several debilitating genetic disorders. Gene modification stimulated by engineered nucleases induces a double-stranded break (DSB) in the target genomic locus, thereby activating DNA repair mechanisms. DSBs triggered by nucleases are repaired either by the nonhomologous end-joining or the homology-directed repair pathway, enabling efficient gene editing. While there are several ongoing ex vivo genome editing clinical trials, current research underscores the therapeutic potential of CRISPR/Cas-based (clustered regularly interspaced short palindrome repeats-associated Cas nuclease) in vivo gene editing. In this review, we provide an overview of the CRISPR/Cas-mediated in vivo genome therapy applications and explore their prospective clinical translatability to treat human monogenic disorders. In addition, we discuss the various challenges associated with in vivo genome editing technologies and strategies used to circumvent them. Despite the robust and precise nuclease-mediated gene editing, a promoterless, nuclease-independent gene targeting strategy has been utilized to evade the drawbacks of the nuclease-dependent system, such as off-target effects, immunogenicity, and cytotoxicity. Thus, the rapidly evolving paradigm of gene editing technologies will continue to foster the progress of gene therapy applications.
Introduction
Gene editing has emerged as one of the most revolutionary breakthroughs in the field of biomedical sciences over the past decade. The technological advancements developed by scientists have enabled precise and targeted manipulation of the genome. Gene editing approaches entail a site-specific modification of a gene by its deletion, replacement, or correction, thus producing the desired therapeutic effect.
Fundamentally, site-specific modification of genetic information at the DNA level requires two essential components: first, a sequence-specific DNA recognition and binding domain, and second, an effector domain that initiates DNA cleavage near the binding site. A double-stranded break (DSB) by a sequence-specific endonuclease activates the cell's endogenous DNA repair mechanisms, subsequently modifying the desired sequence. 1 –3 Two major repair pathways used by the cell to repair the nuclease-induced DSB are nonhomologous end-joining (NHEJ) and homology-directed repair (HDR). 4 –6 Depending on the DSB repair pathway, the outcome may be inactivation of the targeted locus by insertions or deletions (“indels”) introduced by NHEJ 7 or insertion of a new sequence by HDR from an exogenous DNA template. In the HDR pathway, the donor DNA has homology arms with sequences identical to the region surrounding the DSB, enabling precise correction or replacement of the original gene. 4 These alternations are triggered by engineered nucleases that induce a double-stranded break (DSB) in the desired genomic locus, leading to activation of efficient DNA repair mechanisms present in all organisms. Here we mention the different nuclease-mediated platforms, focusing on the CRISPR/Cas (clustered regularly interspaced short palindrome repeats-associated Cas nuclease) system for gene editing.
CRISPR has the potential to be used directly in patients, in vivo or ex vivo, for therapeutic gene editing. In vivo gene editing involves gene modification in situ by the direct delivery of CRISPR/Cas9 to target cells. Some parameters need to be considered for recognizing the efficacy and safety of therapeutic in vivo gene editing, discussed in the later sections of this article. Ideally, the carriers for delivering CRISPR/Cas directly to target cells should be nonimmunogenic, with minimal cytotoxicity. Only target cells harboring a mutated gene should be edited using CRISPR/Cas components; nonspecific targeting of the normal cells may adversely affect their physiological function. Off-target effects associated with CRISPR/Cas need to be restricted to prevent insertional tumorigenesis. Finally, a high editing efficiency may be required to obtain clinically relevant levels for therapeutic gene editing. Therefore, selection of an optimal CRISPR/Cas system and the appropriate delivery vector is imperative for precise and robust gene editing in vivo. In this review, we focus on the nuclease-dependent (CRISPR/Cas) HDR-based editing in vivo to treat human monogenic diseases, briefly evaluate the hurdles and mitigation strategies coupled with in vivo delivery, and discuss nuclease-free editing as an alternate gene targeting approach.
Nuclease-Dependent Platforms for Gene Editing
Nuclease-based platforms include meganucleases, zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the more recent CRISPR/Cas9 that can be engineered to target the genomic locus of interest (Fig. 1). Meganucleases or homing endonucleases recognize long DNA sequences to trigger a DSB. They were among the first to be reengineered for novel target site recognition using structure-based design and protein engineering approaches. 1,8 However, the process of designing meganucleases for therapeutic gene editing is laborious, thereby limiting its use. ZFNs consist of a zinc finger DNA binding domain that determines specificity and a nuclease domain derived from a restriction endonuclease, FokI. 9 A pair of ZFNs are designed for each target site for the FokI domains to dimerize, rendering the nuclease domain catalytically active. Using a wide array of approaches such as phage-based selection, bacterial-based selection, and modular assembly, ZFNs have been constructed to target a diverse range of sequences for gene editing. 10 –14 Engineered ZFNs have exhibited promise in enhancing targeted homologous recombination (HR) in human cells, 15 as well as therapeutic gene editing for cystic fibrosis, 16 sickle cell anemia, 17,18 and human immunodeficiency virus (HIV). 19 –21 SB-FIX by Sangamo Therapeutics is an in vivo gene therapy treatment that uses ZFN-based editing to deliver the correct copy of factor IX (FIX) gene for treating hemophilia B. Another ZFN-based in vivo gene editing therapy developed by Sangamo Therapeutics, SB-913, entered the first clinical trial for treating Hunter's syndrome. 22 A similar FokI nuclease-based editing platform, TALENs, derived from TAL effector proteins also demonstrated therapeutic gene editing potential. 23,24 This technology has been effectively used to mitigate the HIV coreceptor, CCR5 gene, 23 and manipulate immune cells for cancer treatment. 25 Despite the targeted gene editing efficacy of ZFNs and TALENs, the difficulty in cloning and reengineering them for each target site has limited their widespread use. The advent of the CRISPR/CRISPR-associated protein (Cas) technology, which is far more robust and flexible compared with the existing nucleases, paved the way for new possibilities in therapeutic gene editing.
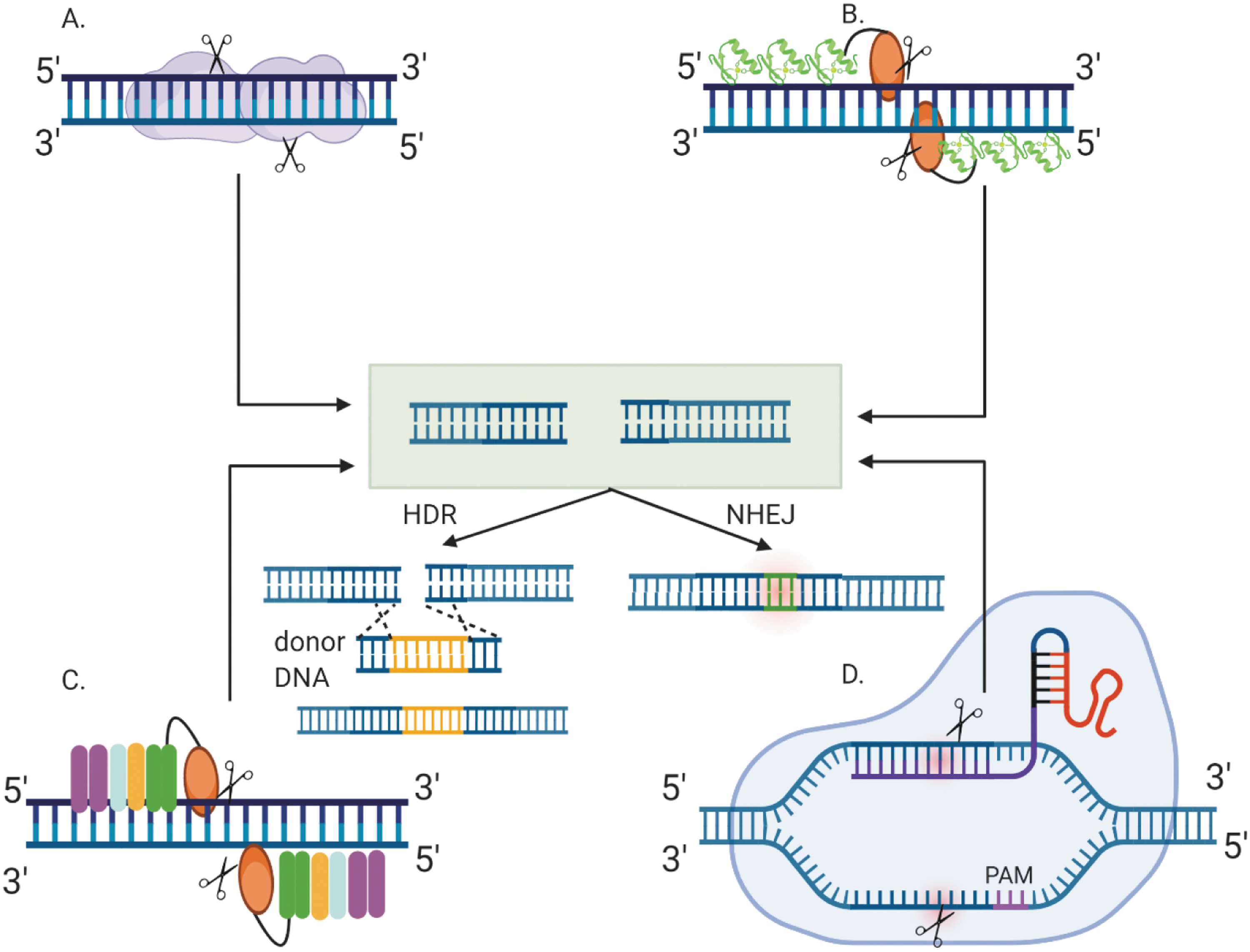
Different programmable nucleases for targeted gene editing.
CRISPR/Cas TECHNOLOGY
This powerful, multiplexed tool first studied as part of the bacterial adaptive immune system consists of a protein (Cas) and an RNA (crRNA and tracrRNA) component. A CRISPR/Cas locus is made up of Cas genes and a CRISPR array consisting of repetitive sequences interspaced by variable DNA bases, called spacers. These spacers serve as a “snapshot” of the invader's mobile genetic elements acquired during a previous infection. During future infections, this stored “snapshot” mediates recognition and protection against foreign cognate viruses or plasmids. 26,27 The CRISPR/Cas-mediated immune response, based on sequence-specific targeting of foreign nucleic acids, is divided into three main stages. The first stage of immune response elicited by the CRISPR system is termed as the acquisition stage, in which DNA fragments from invading viruses are introduced into the CRISPR locus of the host as spacers. The second stage, known as the expression stage, marks transcription of the CRISPR array containing spacers into pre-CRISPR RNA (pre-crRNA), followed by processing it by Cas proteins to mature crRNAs. A noncoding trans-activating CRISPR RNA (tracrRNA) is essential for crRNA processing and binding to Cas protein in type II CRISPR systems. The mature crRNA acts as a guide that can recognize invading foreign DNA and direct the Cas protein, thereby mediating target cleavage. During the final interference stage, the crRNA enables target recognition and Cas proteins cleave the foreign DNA, conferring protection to the host cell. 28 –30
Seminal work in the field has shown that this CRISPR/Cas system can be programmed to cleave host DNA in a diverse range of species, thereby enabling gene editing for a multitude of biomedical applications. A variety of CRISPR/Cas systems have rapidly evolved, resulting in their structural and functional diversity. CRISPR/Cas systems are divided into two classes that are each subdivided into three types and various subtypes. The class 1 CRISPR systems (type I, II and IV) present in bacteria and archaea are thought to be evolutionary ancestrally. Their effector complexes consist of multiple Cas protein subunits. Contrary to this, the class 2 CRISPR systems (type II, V, VI) are mostly restricted to bacteria and their effector complex is made up of a single multidomain Cas protein. 31,32 The type II CRISPR/Cas system has emerged as the most widely used and robust nuclease for genome editing studies. 32 This RNA-guided type II complex consists of a Cas9 endonuclease and a guide RNA (gRNA). The gRNA constitutes an ∼20-nucleotide crRNA complementary to the target DNA and a scaffold sequence required for Cas binding namely, tracrRNA. A breakthrough discovery showed that it is feasible to fuse the crRNA and tracrRNA into a single chimeric gRNA, which confers specificity to the CRISPR/Cas system for targeted gene editing. In addition, a protospacer-adjacent motif (PAM) sequence immediately downstream of the target site also determines the specificity of this system and serves as a binding signal for the Cas protein. Cas nucleases isolated from different bacteria recognize respective PAM sequences. The most commonly used and well-characterized Cas9 endonuclease is from the bacterium, Streptococcus pyogenes, which requires a 5′-NGG-3′ PAM sequence immediately downstream of the target site for binding. Cas9 and gRNA form a ribonucleoprotein (RNP) complex, facilitated by the gRNA scaffold (tracrRNA), while the spacer region (crRNA) is free to interact with the target DNA. Once the RNP complex binds to the putative target DNA, the gRNA anneals to the target and Cas9 undergoes a conformational change; its HNH nuclease domain cleaves the target strand at approximately three to four nucleotides upstream of the PAM sequence and the RuvC-like nuclease domain cleaves the nontarget strand resulting in a DSB at the desired genomic locus. 28,33 –35 The DSBs are repaired either by NHEJ or HDR pathways, as mentioned earlier. Thus, by altering the synthetic gRNA sequence to bind any desired target, the Cas9 protein can be utilized as a robust platform for precise genome targeting. Wild-type Cas9 nuclease variants generated by mutating either of the two nuclease domains function as a nickase Cas9 (nCas9) that cleaves a single strand of DNA. This feature helps in enhancing Cas9-based gene editing specificity. 36,37 When both the HNH- and RuvC-like nuclease domains are inactivated, dead Cas9 (dCas9) is formed, which only retains its DNA-binding ability. These engineered Cas9 mutants, fused to other functional effectors or ligands, can be extensively used for specific gene targeting, activation, silencing, epigenetic regulation, and base editing. 38 –40 Besides SpCas9, other Cas9 proteins have also been designed, such as Cas9 derived from Staphylococcus aureus (SaCas9) 41 and Neisseria meningitidis called Nme2Cas9. 42 These Cas9 proteins, discussed later in this review, have comparable editing potential such as SpCas9 but are better suited for in vivo delivery, owing to their smaller size.
CRISPR/Cas -MEDIATED IN VIVO GENE EDITING
A highly precise, robust, easily deliverable gene editing approach is required for safe ex vivo and in vivo clinical applications. During ex vivo editing, cells are first isolated, transfected with the appropriate gene editing toolbox, and then retransplanted into the patient. 43 On a clinical scale, this is a time-consuming, strenuous, and expensive process, thereby questioning its broad accessibility to patients particularly in underdeveloped nations. Furthermore, ex vivo editing is largely limited to cells that can be isolated from a patient's body, modified in vitro, and then reinfused back into the patient, such as hematopoietic stem cells (HSCs) and immune cells, for example, T cells and natural killer (NK) cells. CRISPR/Cas-mediated ex vivo therapeutic gene editing has been extensively used for genetic diseases, such as sickle cell anemia, β-thalassemia, and chimeric antigen receptor (CAR)-T therapy. 44,45 However, target cells implicated in the majority of the genetic diseases require in situ gene correction. Hence, in vivo gene editing is an ideal platform for treating various human genetic disorders. In the following section, we briefly highlight the rationale behind in vivo gene editing and CRISPR/Cas editing approaches devised for potential clinical use in monogenic diseases.
Ideal candidates for in vivo gene editing
First, in vivo gene editing involves local or systemic delivery of the gene editing components into a patient, avoiding the tedious process of cell isolation, expansion, editing, and reinfusion. 46 For example, the existing site-specific gene editing approaches to treat sickle cell disease include isolation of a patient's hematopoietic stem and progenitor cells, followed by either repairing the mutated hemoglobin gene (HBB) 47 or inducing fetal hemoglobin expression, 48,49 and finally, reinfusion of the corrected cells into the patient bone marrow. 50 Advancements in in vivo technologies might alleviate the need for bone marrow transplantation, making the process less painful and economical.
Second, in situ gene modification is preferred for certain cell types that might lose their properties and function when artificially cultured, such as neurons. Ex vivo editing techniques also affect the viability of cells and result in poor engraftment, which is evaded during in vivo editing.
Third, in some monogenic disorders where a single gene impairment causes defects in the entire cell lineage, such as severe combined immunodeficiency, 51 correcting HSCs generates healthy cells capable of differentiation, with a selective advantage over defective cells. As a result, a lower number of corrected cells are enough to attain therapeutic outcome. Since, the efficiency of in vivo editing is low, a selective advantage of the modified cells enhances the feasibility of this approach. Ideal candidates for in vivo gene editing are genetic disorders where allelic ablation of aberrant splice sites would help restore gene function, such as in β-thalassemia. 52
Moreover, some genetic disorders that affect small organs, such as the ear and retina, require localized injection of the genome editing toolbox to the target organ, with limited distribution to other tissues. A localized delivery achieved by the route of administration or using tissue-specific promoters improves the feasibility of organ/tissue-specific genome editing in vivo. 53 However, larger organs entail systemic injection for efficient targeting.
Finally, a conventional gene therapy approach involves replacement of the defective gene at the target locus. However, the low number of edited cells may not express adequate levels of the transgene necessary to alleviate the disease. This drawback can be resolved by delivery of the editing machinery to a native locus, “safe harbor” with high transcriptional activity, such as the serum albumin locus. 54 This strategy established a versatile platform for therapeutic levels of protein expression, substituting the donor for each transgene.
In vivo CRISPR/Cas applications
Most of the CRISPR/Cas-mediated therapeutic applications for monogenic disorders that are in clinical trials currently are ex vivo strategies. In recent years, in vivo gene editing studies that rely on both NHEJ and HDR pathways have emerged. Some of the recent applications of in vivo therapeutic genome engineering in preclinical and clinical studies are listed in Table 1. Here, we highlight some HDR-based precise gene modification studies in vivo using CRISPR/Cas that can be potentially translatable to human use in the future.
List of some of the recent therapeutic gene editing studies in in vivo preclinical and clinical models
AATD, alpha-1 antitrypsin deficiency; AAV, Adeno-associated vector; ABE, adenine base editing; Ad, adenovirus; ALS, amyotrophic lateral sclerosis; CAR, chimeric antigen receptor; CRISPR/Cas, clustered regularly interspaced short palindrome repeats-associated Cas nuclease; dCas9, dead Cas9; DMD, Duchenne muscular dystrophy; gRNA, guide RNA; HBV, hepatitis B virus; HDR, homology-directed repair; HIV, human immunodeficiency virus; HITI, homology-independent targeted integration; HR, homologous recombination; HTI, hereditary tyrosinemia; LCA, Leber's congenital amaurosis; LNP, lipid nanoparticles; NHEJ, nonhomologous end-joining; PNA, peptide nucleic acids; RNP, ribonucleoprotein; SCD, sickle cell disease; sgRNA, single-guide RNA; TALEN, transcription activator-like effector nuclease; ZFN, zinc-finger nuclease.
Genetic liver diseases
Alpha-1 antitrypsin deficiency (AATD) patients suffer from progressive lung disease due to loss-of-function of AAT antiprotease activity and some patients suffer from liver toxicity due to gain-of-function of the mutant allele. CRISPR/Cas9-mediated editing and NHEJ successfully impaired mutant AAT and effectively ameliorated liver fibrosis in a humanized mouse model, thus supporting a potential therapeutic possibility of treating AATD patients. 55 An additional study utilized coinjection of a dual adeno-associated vector (AAV): one encoding Cas9 and another expressing an AAT gRNA and an HDR donor template into the liver of a transgenic mouse model. This approach enabled precise AAT gene correction in vivo and partially restored wild-type AAT levels, 54 making it a probable therapeutic option upon further optimization for use in humans. Hereditary tyrosinemia type I (HTI) is another genetic liver disease, caused by loss-of-function of fumaryl acetoacetate hydrolase (FAH), a key enzyme of the tyrosine catabolic pathway. CRISPR/Cas9-mediated HDR has successfully corrected FAH mutation by two methods: (1) A hydrodynamic injection of the gene editing components, which yielded a low correction rate 56 and was tested in a clinical trial, 57 and (2) systemic delivery of Cas9 mRNA by lipid nanoparticles (LNPs) and a single-guide RNA (sgRNA)/HDR template by AAV, which resulted in an initial FAH correction in more than 6% of hepatocytes. 58 Moreover, a new-generation gene editing tool, base editing, which involves conjugating dCas9 with enzymes that catalyze direct conversion of A to G or C to T, ensues DNA base editing without causing any DNA breaks. 40,59,60 Using an adenosine base editing (ABE) strategy via an LNP delivery containing sgRNA and a codon-optimized base editor was shown to restore FAH point mutation in vivo, eliminating the need for any DNA donor template. 61 In addition, in a model of transthyretin amyloidosis, a single administration of LNP-mediated delivery of CRISPR/Cas9 along with chemically modified sgRNA facilitated efficient editing of the mouse transthyretin (Ttr) gene in the liver, and >90% reduction of TTR serum protein levels that persisted for at least 12 months. 62 This study achieving clinically relevant levels of editing in vivo may be extended to provide human data in future.
Duchenne muscular dystrophy
In vivo editing studies have been explored in genetic muscular diseases, for example, Duchenne muscular dystrophy (DMD), characterized by progressive muscle weakness and premature death due to mutation in the dystrophin gene. A DMD mouse model exhibiting a similar deletion in the Dmd gene (ΔEx50) occurring in DMD patients was generated using CRISPR/Cas9. CRISPR/Cas9-induced single cut in the dystrophin gene of these mice and a gRNA that enables exon 51 skipping restores up to 90% dystrophin gene expression in skeletal and cardiac muscles. 63 An important step toward clinical translation of therapeutic gene editing for DMD is using CRISPR/Cas9-mediated NHEJ to treat dogs with the ΔEx50 mutation, corresponding to a mutational “hotspot” in the human DMD gene. Systemic delivery of the gene editing apparatus in skeletal muscle provided 3–90% recovery, depending on the muscle type, and treated dogs revealed improved muscle histology. 64 Although this proof of principle study in the canine disease model has the potential to bridge the gap between mice and humans, there are some issues in large animal editing which needs further attention. Limited sample size, age of injection, treatment duration, characterization of the treatment results, safety and ethical concerns as discussed in earlier reports 65 –67 needs to be addressed in future.
Besides exon skipping, other groups have utilized AAV-based local and systemic delivery of CRISPR/Cas9 editing components to adult and neonatal DMD mouse models for removing the mutation in exon 23, resulting in partial recovery of functional dystrophin in skeletal myofibers and cardiac muscle. 68 –70 Moreover, local delivery of ABEs consisting of engineered adenine deaminase, and an SpCas9 nickase helped correct a nonsense mutation in a DMD mouse model. 71 An ideal therapy for a chronic disease such as DMD should ensure a lifelong, sustained restoration of dystrophin in the heart and skeletal muscle. To this end, a single-dose AAAV-CRISPR therapy that leads persistent alleviation of the disease phenotype is required. Successful results from short-term studies 68,70,72,73 prompted researchers to test the long-term restoration of the DMD gene. Systemic delivery of an AAV9 vector encoding SaCas9 and gRNA targeting introns 22 and 23 restored dystrophin expression, thereby improving skeletal and cardiac muscle function for 18 months in dystrophic mice. 74 Another approach for attaining enduring gene therapy for DMD would be editing muscle stem cells (MuSCs) using CRISPR. Since the self-renewing MuSCs, also known as satellite cells, regenerate skeletal muscle in response to tissue damage, correcting these cells would enable long-term therapeutic gene editing. CRISPR-edited MuSCs from dystrophic mice, when engrafted in a dystrophin null mouse, showed increased dystrophin expression and successful renewal. 75,76 These studies taken together demonstrate that with further development, in vivo gene editing approaches will be clinically useful for treating DMD.
Retinal disorders
A hallmark study that recently entered clinical trial uses in vivo CRISPR/Cas9 delivery for treating congenital blindness in patients. Leber congenital amaurosis (LCA) is a rare, debilitating monogenic disease resulting in vision loss in childhood, with no available treatment. A biallelic loss-of-function mutation in the CEP20 gene is responsible for this severe retinal dystrophy. Editas Medicine has developed a therapy named, EDIT-101, which delivers SaCas9 directly to remove the intronic IVS26 mutation in the CEP20 gene, implicated in aberrant splicing, thereby restoring functional CEP20 levels in human cells and humanized CEP20 mice. 77 A clinical trial of EDIT-101 by Allergan and Editas Medicine paves the way for a prospective curative strategy for treating congenital blindness using an in vivo approach.
Ideally, HDR-based precise gene correction can repair the genetic mutations implicated in inherited retinal disorders. Since HDR mainly occurs in mitotic cells, the postmitotic nature of most retinal cells limits the HDR efficiency. Hence, a majority of the in vivo gene therapy approaches for retinal dystrophies rely on the CRISPR-Cas-mediated NHEJ pathway. 78 –86 Another genome editing strategy, namely, homology-independent targeted integration (HITI), was utilized to successfully knock in exon 2 of Mertk (MER/AXL/TYRO3 receptor kinase) gene, thereby protecting from retinal degeneration. 87 HITI exploits the NHEJ repair mechanism and enables targeted transgene insertion without the need of an HR donor template in dividing and nondividing cells.
In vivo editing of stem cells and immune cells
Most of the HSC gene therapies involve removal of the patient's stem cells, their expansion followed by gene correction using editing machinery and then reintroduction to the patient's body. Although this approach has been used in ongoing clinical trials, they are associated with limitations, discussed earlier in this review. Another disadvantage of the ex vivo approach is that reinfusion of the edited cells into the bone marrow requires the patient to undergo chemotherapy. Recently, one of the ongoing clinical trials for sickle cell disease, initiated by Bluebird Bio, has come to a halt after two patients, who received the ex vivo gene therapy for SCD, were diagnosed with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). 88 Previously, in 2018, another patient in the same trial was diagnosed with MDS, likely due to the adverse effects of chemotherapy pretreatment. Whether these two new cases can be attributed to chemotherapy or insertional oncogenesis triggered by the lentiviral vector used in the trials is still elusive and needs further examination. In vivo editing bypasses the time-consuming, expensive, and laborious process of in vitro handling of HSCs as well as the DNA damaging chemotherapy during reinfusion. In vivo approaches either include a direct modification of HSCs in the bone marrow by an intraosseal injection, or a systemic injection of delivery vehicles that act on HSCs mobilized into peripheral blood, followed by their re-engraftment into bone marrow. Previous studies have reported successful lentiviral-mediated gene transfer in T cells 89 and in HSCs by direct intraosseal injection in mice, demonstrating high levels of transduction in bone marrow cells. 90 An in vivo HSC gene editing study was reported in thalassemic mice injected with nanoparticles containing triplex-forming peptide nucleic acids and a single-stranded homologous DNA donor, in combination with the stem cell factor. This editing strategy showed almost 7% editing frequency in the bone marrow, sufficient to ameliorate the disease phenotype. 91 An alternative in vivo gene therapy strategy involved mobilization of hematopoietic stem and progenitor cells (HSPCs) from the bone marrow into peripheral blood, followed by an intravenous injection of integrating, helper-dependent adenovirus (HDAd5/35++) vector system that targets human CD46 expressed on nascent HSCs. This transposase-based integration system achieved stable fetal γ-globin expression in CD46-transgenic and thalassemia mouse models. 92 –94 This method when tested in rhesus macaques demonstrated stable HSC transduction, thereby improving its feasibility in human HSC gene therapy. 95 Besides thalassemia, this approach has been recently used to correct the sickle cell phenotype. An HDAd5/35++ vector encoding two cassettes, one containing the CRISPR/Cas9 machinery and the other encoding the therapeutic fetal γ-globin transgene, was administered by an intravenous injection in an SCD mouse model. A combination of these two cassettes induced expression of the fetal γ-globin gene and ameliorated the disease phenotype. 96 Despite the promising results, the high titer of the immunogenic adenoviral vectors might hinder clinical trials. An AAV vector delivery system may be a safer and more efficient alternative for in vivo HSC gene editing. Recombinant tyrosine mutant AAV6 vectors displayed high transduction efficiency and robust transgene expression in human HSCs in vitro and in a mouse xenograft model in vivo. 97 –99 In addition, AAV8-mediated transduction of immune cells, such as T cells, B cells, macrophages, and dendritic cells, was achieved in vivo after systemic injection in mice, 100 thereby spurring the development of these vectors for in vivo immunotherapies. Another recent gene editing strategy using base editors delivered by HDAd5/35++ vectors revealed efficient HSPC transduction and stable γ-globin expression in transgenic mice, strengthening its immense potential for in vivo gene therapy for hemoglobinopathies. 101,102 Future studies exploring HDR-based in vivo HSC editing will enrich the field of hematopoietic gene therapy.
Brain disorders
NHEJ-based editing triggered by CRISPR/Cas9 system has been extensively studied in brain regions in vivo. 103 –107 Compared with NHEJ, the low efficiency of the HDR pathway in the postmitotic neurons makes precise gene correction difficult in these cells. To overcome this, HITI has been used to achieve targeted insertion of the desired donor sequence in situ. 87,108 This can be used to create knockin reporter systems for cell tracking in live animals, useful for studying neuronal circuits and brain functions. Moreover, some studies suggest that neuronal progenitors retain their ability to trigger HDR in vivo. 109 A rapid in utero electroporation method to deliver the editing components into neuronal progenitors in vivo enabled successful HDR editing in the mouse embryonic brain. 110 –112 HDR-facilitated gene editing has been shown in postmitotic neurons as well. A combination of CRISPR/Cas9 and AAV-mediated donor DNA delivery enabled HDR editing in vivo along with the insertion of a reporter tag in the brain regions. This strategy, known as vSLENDR (viral-single-cell labeling of endogenous proteins by CRISPR/Cas9-mediated HDR), was adapted to conduct precise gene modification by HDR in any regions of the brain. 113
Some of the existing in vivo therapeutic gene editing studies are summarized in Table 1. Despite these promising studies listed above, there still exists a lacuna between animal studies and applications in humans, further emphasizing the need for improved in vivo editing, discussed in the next section.
In vivo gene editing clinical trials
The gene editing landscape is evolving rapidly with the advancement of several therapeutic gene editing studies to clinical trials. Most of the ongoing trials are focused on gene modification ex vivo and have been reviewed. 114,115 Currently, the ex vivo gene editing preclinical and clinical trials primarily involve alteration of T cells to disrupt gene expression for treating HIV, 116 –120 engineering T cells for cancer immunotherapy, 121 –124 and modification of HSCs for treating hemoglobinopathies, such as β-thalassemia and sickle cell anemia. 18,48,125 –127 Clinical trials for β-thalassemia and sickle cell anemia using ZFN- and Cas9-mediated disruption of the fetal globin repressor BCL11A in HSCs ex vivo are ongoing (NCT03432364, NCT03653247, NCT03655678, and NCT03745287).
In vivo therapeutic gene editing approaches have also advanced into clinical trials, summarized in Table 2. ZFN-, TALEN-, and Cas9-based trials for treating cervical cancer have been registered. These approaches target the E6 and E7 genes of human papilloma virus (HPV), the causative agent of cervical cancer.
128
Although HPV vaccines are available now, they do not confer treatment for cervical cancer patients. Nonviral delivery of ZFNs, TALENs, or CRISPR/Cas9 achieved targeted disruption of the E7 oncogene, resulting in reduced tumor growth in mouse models.
129
–131
Besides nonviral delivery methods, AAV-dependent delivery of Cas9 targeting E6 and E7 viral genes showed encouraging results in xenograft models,
132,133
reflecting its therapeutic potential for cervical cancer. In addition to the in vivo trials on cervical cancer, ZFN-mediated gene editing has been used to treat hemophilia.
134
–137
ZFN-based gene correction of factor IX, α-
List of the in vivo gene editing clinical trials
U.S clinical trial data from
HPV, human papilloma virus; IDS, iduronate-2 sulfatase; IDUA, α-
LIMITATIONS ASSOCIATED WITH CRISPR/Cas- BASED IN VIVO THERAPEUTIC GENE EDITING
In this section, we briefly describe some of the unmet challenges and possible strategies to alleviate them, facilitating the clinical utility of therapeutic in vivo gene editing. A summary of the limitations associated with in vivo gene therapy is depicted in Fig. 2.
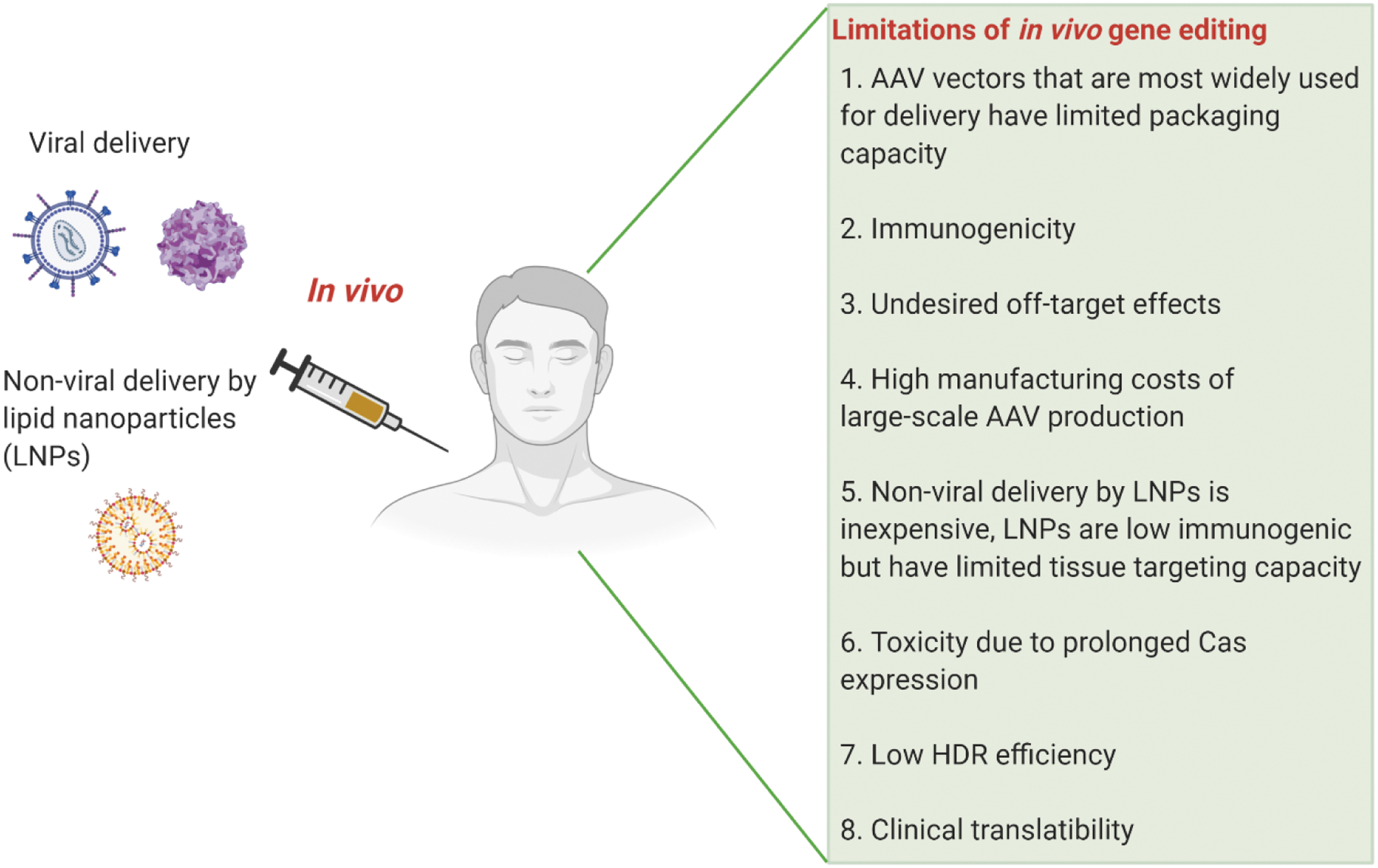
Schematic showing limitations of in vivo gene editing. In vivo gene therapy involves direct injection of the editing machinery into the patient by viral or nonviral delivery methods. Limitations associated with this approach are listed here. Created with
In vivo delivery
The key step that determines the clinical utility of genome editing is the efficient and safe delivery of the editing toolbox, including CRISPR/Cas enzymes, sgRNA, and repair template, to the target cells. Cas enzyme can be delivered to the cells in several formats: plasmid DNA encoding Cas gene, Cas mRNA or protein. These are coupled with the appropriate sgRNA. Electroporation of cells with a preformed Cas protein and sgRNA RNP complex is the preferred form of delivery in ex vivo gene editing. 139 Although electroporation has been used to deliver Cas9 to animal zygotes 140,141 and skeletal muscle in mice, 142 the high-voltage shock required to permeabilize cells is toxic and may not be favorable for a broad range of applications. In vivo delivery is more challenging and requires carriers that have high specificity, low cytotoxicity, and rapid clearance of Cas enzyme after gene editing. Overcoming these challenges to enhance the clinical prospects of in vivo gene editing has stimulated the development of viral and nonviral delivery systems. Among the viral delivery methods, which include lentiviruses, adenoviruses, and AAVs, the most widely used for in vivo delivery of CRISPR/Cas systems is AAVs.
AAVs are relatively nonimmunogenic, demonstrate capsid variant-dependent tissue specificity than other viral vectors, and long-term transgene expression without the necessity of genomic integration. AAV-mediated delivery of CRISPR/Cas9 has been used successfully in gene therapy for monogenic diseases, such as DMD, 68 –70,72 retinal impairments, 80,85,143,144 and liver, 41,58,145 heart, 146 –148 and lung disorders. 149 The single-stranded AAV genome and its unique inverted terminal repeats play an essential role in precise gene targeting. The ssDNA of rAAVs accommodates long homology arms, encodes selection markers, and has low NHEJ-based integration rates, thereby making it a bona fide HDR template. While the CRISPR/Cas components can be delivered by different methods that do not necessarily require AAVs, the HDR donor is often delivered by single-stranded AAVs. 150,151 Although AAVs serve as a favorable delivery vehicle for CRISPR/Cas, their packaging capacity is limited only to 4.7 kb. Using a dual-vector system, one expressing a gRNA and an HDR repair template and the second AAV encoding SpCas9 gene (4.2 kb), for HDR-mediated gene correction can avoid the packaging size limitation. 145,152 Alternate approaches for precise gene correction in vivo, such as base and prime editing, also need dual vectors to accommodate effectors fused to Cas9. 153 –157 However, the target cells need to uptake both the vectors together, thereby affecting editing efficiency. Also, a high AAV dose required in the dual-vector studies might raise safety concerns during clinical translation, considering the recent consequences of a high-dose AAV therapy in human trials. 158,159 Smaller Cas9 orthologs, such as SaCas9, NmeCas9, or Cas9, from Campylobacter jejuni can be combined with the gRNA and donor template in a single AAV vector to eliminate the packaging issue. 42,80,160,161 A novel all-in-one recombinant AAV vector encoding Nme2 Cas9 along with two sgRNAs was engineered to alleviate the disease phenotype in an HTI mouse model. To fulfill the need of a single-AAV for precise gene modification by HDR, this system was further updated. A self-inactivating single AAV vector, encoding Nme2 Cas9, a single sgRNA, and an HDR donor flanked by Nme2 Cas9 target sites, was designed. Self-cleavage during packaging was circumvented by including an anti-CRISPR protein (ACR). 162 Precise HDR-based therapeutic editing at clinically relevant levels were obtained in disease models of HTI and MPS I. 163 Newly discovered CRISPR/Cas systems, such as the hypercompact CasΦ, which is half the size of SpCas9, show similar efficiency and selectivity and have the potential of circumventing the size limitation of AAV-based delivery. 164 Second, the tissue tropism of AAVs needs further improvement to minimize any undesired side effects of CRISPR/Cas in other tissues. AAV capsids can be engineered to use tissue-specific promoters, 72,103,147 or with improved capsid variants 165 –167 or to increase target tissue specificity or transduction efficacy in vivo by incorporating ligands that bind to receptors on target cells. 168 Other constraints of AAVs such as delivery carriers include immunogenicity, 169 high viral titers beyond clinically accepted levels for obtaining therapeutic editing, and expensive manufacture and scalability for clinical use. 170
The administration route of AAV vectors also affects the efficiency and specificity of in vivo gene editing. Selection of an optimum injection route depends on the target tissue, tissue-specific promoter, and AAV capsid variant. For example, a systemic intravenous injection is the preferred delivery route for editing genes implicated in liver disorders since most of the AAVs accumulate in the liver. 145,171 However, a localized injection, such as subretinal or intravitreal injections, is favored while administering AAVs containing CRISPR/Cas components into the mouse retina. 85,143,144 Although most of the in vivo methods have the potential to be extended to human studies, the high dosage required to achieve clinically relevant editing levels questions their translatability to humans.
Despite the widespread use of AAV-mediated delivery systems, the limitations discussed above prompted the development of nonviral carriers for delivering CRISPR/Cas in vivo. Cationic LNPs have been used to deliver CRISPR/Cas9 RNP to mouse liver to obtain therapeutically relevant gene editing in vivo. 62 This study resulted in 70% editing efficiency with a single dose and yielded effective results in rats, validating its preclinical potential. 62 Other groups that have reported in vivo editing using nonviral delivery systems in the liver have efficiencies ranging from 3.5% of hepatocytes 172 to 35% editing after four systemic doses. 173 A recent study shows that LNPs were able to effectively deliver gRNA and Cas9 mRNA to splenic endothelial cells, thus identifying new accessible target cells in vivo. 174 Despite this method being inexpensive, rapid, and easy, 175 the LNPs show some evidence of toxicity. 58,176 A delivery system, consisting of gold nanoparticles conjugated to DNA and assembled with polymers that disrupt endosomes, can deliver Cas9 RNP and donor DNA to correct Dmd gene mutation in mice, with negligible off-target effects. 177 Further advancements in nanoparticles, nanowires, and cell-based delivery methods are crucial for therapeutic in vivo genome editing. 178,179
Off-target effects of CRISPR/Cas
Precise and accurate gene modification at the desired target site is imperative for therapeutic genome editing. Although the CRISPR/Cas system is known to be more precise in comparison with the other nucleases, it still exhibits off-target cleavage activity. Off-targeting occurs due to nonspecific CRISPR/Cas-induced DNA cleavage at sites other than the actual target and may result in deleterious effects, such as malignant transformation. 180 Some of the in vivo gene therapy studies revealed minimal or no off-target editing at the predicted sites, which is reassuring. 58,69,70,145 However, the possibility of off-target editing beyond the predicted sites requires the design of an unbiased genome-wide sequencing method. Several cell-based genome-wide sequencing tools, such as CHIP-seq 181 and Digenome-seq, 182 have aided in the identification of unpredictable off-target mutations in vitro. However, these in silico tools cannot be directly applied to identify undesirable genomic sites for in vivo editing. A two-step strategy, named “verification of in vivo targets” (VIVO), has been developed to first identify potential off-target locations using CIRCLE-Seq, and then confirm any alteration of these sites following CRISPR/Cas9 in vivo genome editing. 183 This powerful in silico tool allows identification of off-target mutation sites in vivo, vital for designing the most specific gRNA that acts on the desired genomic sites.
Besides optimizing gRNA design, reducing long-term expression of Cas9 is another way of minimizing off-target effects. Delivery of short-lived Cas9 protein instead of the Cas9 gene, 184 using a self-limiting CRISPR/Cas system for conditional genome editing, 143 or inducible Cas9 variants, 185 –187 diminishes duration of Cas9 exposure, thereby impeding its off-target effects. In addition, a self-inactivating AAV-CRISPR system containing a gRNA that cleaves Cas9 coding sequence can eliminate the Cas9 protein in vivo without affecting targeted editing efficiency, thereby alleviating the problems associated with long-term Cas9 expression. 163 Furthermore, LNP-mediated delivery of Cas9mRNA 58 and extracellular vesicle (EV)-mediated delivery of CRISPR-Cas9 RNPs minimize off-target cleavage by limiting prolonged Cas9 exposure. 188 –193 A recently developed all-in-one EV-based delivery system known as, NanoMEDIC (nanomembrane-derived EVs for the delivery of macromolecular cargo), promotes on-target gene editing both ex vivo and in vivo. 194 Alternate approaches to circumvent the off-target effects include editing methods that do not require double-stranded cleavage by CRISPR/Cas9. For example, dCas9 fused to transcriptional activators or repressors engaged in CRISPR activation and interference studies has higher specificity. 38,39 Base editors ensuing RNA-programmed DNA base editing without causing any DNA break also restrict undesirable off-target editing. 60,195 Another way to reduce off-target effects is using anti-CRISPR proteins that regulate dCas9 activity and generate cells resistant to nonspecific gene modifications. 196 –198 The robustness and specificity of these techniques in vivo still need to be studied comprehensively before their clinical use.
CRISPR/Cas immunogenicity
There are two predominant issues regarding the immunogenicity of CRISPR gene editing, one is the toxicity of Cas9 expression and the other is the preexisting immunity against Cas9. Toxicity associated with prolonged Cas9 expression and ways to alleviate them 58,143,163,184 –187 have been discussed in the previous section. A humoral and cellular immune response was elicited against SaCas9 only in adult mice receiving AAV-CRISPR based gene therapy for DMD. However, neonates did not exhibit any immune response against the bacteria derived SaCas9 proteins. 199 Humanized Cas9 protein might also be less immunogenic reducing its potential toxicity. Host immune responses against Cas9 may hinder in vivo therapeutic gene editing. Since the most widely used Cas9 orthologs, SpCas9 and SaCas9, are both derived from bacterial species that frequently infect humans, it is likely that humans will harbor preexisting immune responses against them. As expected, preexisting immunity of anti-Cas9 IgG antibodies was found against SaCas9 and SpCas9 in healthy human adults. 200 Reactive T cells against SpCas9 were also detected in humans. 201 Edited cells may be eliminated due to CRISPR/Cas-triggered immune response. In one study, preexisting immunity to Cas9 led to a high percentage of cytotoxic CD8+ T cells in mouse liver, resulting in removal of edited cells. 202 Development of methods for diminishing the immunogenicity of CRISPR/Cas toolbox requires further attention.
HDR efficiency
Precise gene correction for monogenic disorders is achieved by HDR. However, the efficiency of HDR-dependent precision gene modification is lower compared with other competing repair pathways, such as NHEJ. HDR occurs mostly in mitotic cells, making it difficult to improve its efficiency to match therapeutic levels. Although the editing efficiency for different diseases varies, a higher efficiency usually augments the therapeutic outcomes. Optimum and rational designing of HDR donors, 203 increasing sequence similarity between the donor template and target cleavage sites, 204 and inhibiting NHEJ pathways 205,206 are some of the advancements that enhance HDR efficacy. HITI strategies can also be used to obtain targeted integration of the desired transgene to facilitate in vivo gene therapy. 87,108,207 In addition, base editors 40,208 and prime editors 209 that allow precise gene editing, independent of DNA repair pathways, can potentially cure several genetic diseases.
NUCLEASE-INDEPENDENT GENE TARGETING AS AN ALTERNATE EDITING APPROACH
The risks associated with nuclease-dependent gene targeting, as discussed above, include the inadvertent prolonged expression of Cas9, resulting in potential off-target effects. To eliminate this problem, a nuclease-free gene targeting strategy based on HR was developed by Barzel et al. 210 In this method, a recombinant AAV8, containing a promoterless, codon optimized FIX coding sequence, flanked by sequences homologous to the mouse albumin locus, was designed. A porcine teschovirus-1 2A-peptide (P2A) encoding sequence preceding the F9 gene sequence was used for ribosomal skipping to ensure that the bicistronic Alb-FIX mRNA transcribed from the endogenous Alb promoter is translated into functional albumin and FIX proteins (Fig. 3). This alternative in vivo nuclease-free editing approach attained FIX expression at therapeutic levels to partially correct the spontaneous bleeding phenotype in hemophilic mice. 210 This forms the basis of LogicBio's proprietary GeneRide technology and utilizes HR-guided precise and targeted in vivo gene editing, eliminating the need for vector-driven promoters and engineered nucleases. 211 In addition, a versatile system for in vivo selection and expansion of gene-modified hepatocytes, irrespective of genetic background, has been established using GeneRide. 212 Another recent study published by Homology Medicines revealed the proficiency and specificity of HR-mediated, nuclease-free gene insertion in mouse liver containing human cells using AAVs derived from human HSCs. 213
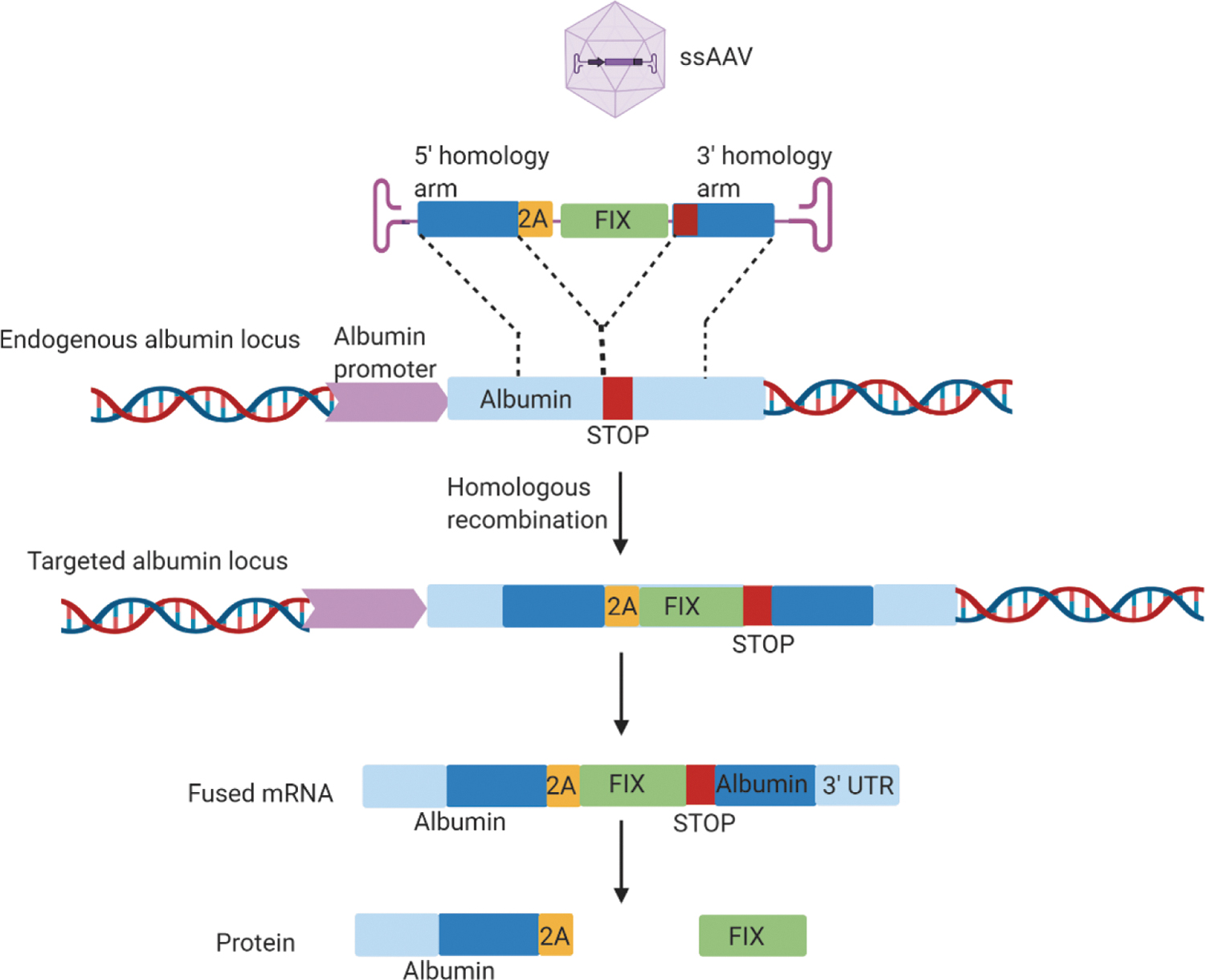
Schematic showing promoterless nuclease-free editing at the albumin locus. Recombinant AAV8 vector containing a promoterless codon-optimized human coagulation FIX sequence I (green), preceded by the 2A peptide (yellow) and flanked by albumin homology arms (dark blue) that covers the albumin stop codon (red), is designed. Homologous recombination results in integration of the rAAV8 vector into the endogenous albumin locus (light blue) and generates a chimeric bicistronic mRNA, which is translated into two distinct proteins, albumin and FIX, due to the ribosomal skipping. Adapted from Barzel et al.
210
AAV, adeno-associated vector; FIX, factor IX. Created with
Although this method is less efficient compared with nuclease-mediated editing, it can work well provided there is a selective advantage of the edited cells. 211 These examples using the nuclease-independent in vivo gene targeting strategy herald an overall safe, robust, and precise avenue for gene therapy.
Perspective
The advent of CRISPR/Cas technology has undoubtedly fostered the development of therapeutic gene editing for a multitude of genetic diseases. Currently, there are several ongoing clinical trials of nuclease-dependent gene therapy, with the hope to ameliorate monogenic disorders, such as hemoglobinopathies and retinal dystrophy, among others. Some examples include the ex vivo CRISPR-mediated gene therapy for β-thalassemia and sickle cell anemia, known as CTX001, currently in clinical phase 1/2 trials. SB-FIX by Sangamo Therapeutics is an in vivo gene therapy treatment that uses AAVs to deliver ZFNs to correct the factor IX gene for treatment of hemophilia B. Another milestone study is the first phase 1 clinical trial NCT02793856 on CRISPR/Cas9-based PD-1 gene knockout in T lymphocytes from metastatic nonsmall-cell lung cancer patients. Furthermore, Allergan and Editas Medicine are conducting a clinical trial of a candidate genome editing therapy, EDIT101, to cure LCA (Table 1). These studies reinforce the tremendous potential of engineered nucleases for treating genetic diseases.
In addition, gene editing has been applied to cancer immunotherapy, and one promising area that has garnered great interest is the development of allogeneic CAR-T therapy. ZFN- and TALEN-mediated gene editing has enabled the generation of allogeneic tumor-associated antigen-specific CAR-T cells, with negligible T cell immune response and graft-versus-host disease. 25,124,214,215 Furthermore, CRISPR/Cas9 triggered faster and easier multiplex gene editing in CAR-T cells, which exhibited CD19-specific antitumor activity in a lymphoma xenograft mouse model. 216 Allogeneic universal T cells were generated using a one-shot CRISPR technique with multiple sgRNAs in a CAR lentiviral vector that simultaneously depleted the endogenous T cell receptor and HLA 1, thereby eliminating rapid rejection from the host immune system. 217 Recent studies use CRISPR/Cas9 to specifically inhibit immune receptors 218,219 to enhance the generation of “universal” CAR-T cells, which might be an effective treatment for AML and other malignancies. The efficacy and safety of the CRISPR/Cas9-edited CAR-T cells in clinical studies need evaluation. The ongoing clinical trials of the modified universal CAR-T cells have been reviewed. 115,220,221 CRISPR/Cas editing machinery eliminated some of the limitations associated with CAR-T immunotherapies, thereby enhancing efficiency of off-the shelf CAR-T cells and minimizing their toxicity. 222 Overall, these findings reflect the immense prospective of gene editing as a robust platform to generate CAR-T cells as an off-the shelf therapy. Moreover, over 300 clinical trials are ongoing across the globe for improving CAR activity and broadening their clinical applications. 89 Yescarta for adult diffused B-cell lymphomas and Kymriah for pediatric acute lymphoblastic leukemia, approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) have hit the market. Generating human CAR-T cells directly in vivo will be very useful in circumventing the expensive and laborious ex vivo production of CAR T cells, rendering them more accessible to patients worldwide. In a recent study, a CD8 targeted lentiviral-based single systemic injection of CD19-CAR-T cells into humanized immunodeficient mice generated in vivo CAR-T cells, that successfully eliminated human B cells. This study resulted in a cytokine storm in humanized mice and further preclinical testing is required to test the feasibility of the approach. 223 Evaluation of the anti-tumoral activity of these in vivo generated human CAR-T cells was done in T cell engrafted immunodeficient mouse models for preclinical testing of CAR-T cells. 224 Next, successful in vivo generation of CD19 CAR-T cells in CD4+ T cells was reported that had the ability to eliminate the CD19 + cells and tumor cells in mice, highlighting the relevance of in vivo CAR-T cell therapy. 225 Although these results look promising, whether the in vivo generated CAR-T cells match the efficacy of the ex vivo-generated CAR-T cells needs further validation. Assessments in large animal models is required before the commencement of an in vivo CAR-T cell therapy a clinical trial in future.
Conclusion
Besides gene editing, the CRISPR/Cas toolbox has also been used for gene regulation, epigenetic modification, drug development, and precision medicine providing personalized therapies based on specific targets and diagnostics, extensively reviewed elsewhere. 226,227
In general, the CRISPR/Cas system provides a precise platform for ex vivo and in vivo therapeutic gene editing against debilitating genetic diseases. So far, ex vivo editing has been predominantly used to treat hemoglobinopathies, cancers, and immune cell disorders. Since a wide range of genetic diseases require in situ gene modification, in vivo gene editing has the tremendous potential to treat them. While the in vivo approach minimizes the risk of graft-versus-host disease and immunosuppression, there are existing barriers that hinder its clinical translatability. One of the primary bottlenecks of in vivo gene therapy is the targeted delivery of the editing machinery. Currently, AAV vectors are the most popular delivery tools for introducing the transgene and CRISPR/Cas system to target organs. However, the limited packaging capacity, off-target effects, and high production costs are some of the limitations of AAV vector delivery. Alternatively, nonviral delivery methods that allow flexible packaging ability, ease of manufacturing, and have low cytotoxicity have shown promise. Another concern that affects the efficacy and safety of the CRISPR/Cas-mediated in vivo editing is the off-target effects. Further progress in the delivery of viral and nonviral delivery vectors, and CRISPR/Cas components, is necessary to attain clinically relevant levels of gene editing in vivo.
Overall, the in vivo studies demonstrate the ability of both nuclease-mediated and nuclease-free editings as potent gene therapy tools. However, obstacles such as off-target effects, optimum delivery vehicles, HDR efficiency, and immunogenicity of the editing components have not been completely resolved. With innovative gene editing advancements in the future, these bottlenecks will be surmounted, thus bringing in vivo gene editing closer to human therapies.
Footnotes
Author Disclosure
No competing financial interests exist.
Funding Information
This study is supported by The Bill and Melinda Gates foundation. Grant ID is OPP1202116.