
Abstract
Recombinant adeno-associated virus (rAAV) vectors are increasingly popular gene delivery tools in biological systems. They are safe and lead to high-level, long-term transgene expression. rAAV are available in multiple serotypes, natural or engineered, which enable targeting to a wide array of tissues and cell types. In addition, rAAVs are relatively easily produced in a well-equipped lab or obtained from a viral vector core facility. Unfortunately, there is no standardization of quality control assays beyond titering and purity assessments. Next-generation sequencing (NGS) can be used to identify rAAV preparations. Because the rAAV genome is single stranded, previous studies have assumed that rAAV genomes must be converted to double strands before NGS. We demonstrate that rAAV DNA extracts exist primarily as double-stranded species. We hypothesize that these molecules form from the natural base pairing of complementary [+] and [−] strands after DNA extraction and show that rAAV DNA extracts are sufficient templates for downstream NGS without the labor-intensive double-stranding step. Here, we provide a detailed protocol for the simple and rapid NGS of rAAV genomes from DNA extracts. With this protocol, users can quickly confirm the identity of an rAAV preparation and detect the presence of contaminating rAAV DNA. In addition, we share custom Python scripts that allow users to accurately determine the serotype and detect Cre-independent DNA recombination events in rAAV containing Lox sites within minutes. We have used these scripts to analyze more than 100 rAAV preparations. Although we focused on the detection of cross-contaminating rAAV DNA and recombination events, our Python scripts can be customized to detect other sequences or events, such as reverse packaging of plasmid backbone or DNA from the packaging cell line. We find that the NGS of rAAV DNA extracts, termed viral genome sequencing, is a simple and powerful method for rAAV validation.
Introduction
The advent of next-generation sequencing (NGS) has reduced the cost of obtaining whole-genome sequence data 50,000-fold since the days of the Human Genome Project. 1 As the cost of NGS drops, the accessibility of this powerful method of data acquisition increases, and a method once considered too expensive for the average lab is becoming mainstream. One area that has been slow to adopt NGS as standard practice is in the quality control (QC) of research-grade recombinant adeno-associated virus (rAAV) vectors.
rAAVs are popular gene delivery tools used frequently in both basic research and drug development. rAAVs have several advantages over other viral gene delivery methods, including long-term expression of the transgene, reduced immunogenicity, and the ability to precisely target specific tissues and cell types by packaging in different serotypes. In addition, producing rAAV is fairly straightforward requiring a triple transfection of HEK293 cells with plasmids encoding the gene of interest, capsid and helper genes, and the isolation of the viral particles from cells, media, or both. Many research labs are able to produce rAAV on their own, and for those that cannot there are numerous viral vector cores that produce and assess the quality of rAAV.
Whether produced in house or by a vector core, rAAV QC tends to be sparse, usually consisting of titration by quantitative PCR (qPCR) and purity assessment by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The lack of comprehensive and standardized QC, especially among core facilities, is problematic for a variety of reasons.
First, scientists cannot be certain that the rAAV they are receiving is what they think it is. Most labs and core facilities titer by using primers targeting common features of the expression cassette such as inverted terminal repeats (ITRs), the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE), or the SV40 polyA. Although this approach streamlines production by allowing multiple lots of viral vectors to be titered in parallel, there is no certainty that the correct transgene was packaged since primers do not target the gene of interest. In situations where multiple lots of viral vectors are being produced at the same time, this is especially troubling. In addition, qPCR and protein staining fail to distinguish serotypes; therefore, even if transgene specific primers are used for titration there is no confirmation that the correct capsid was used during packaging.
Second, the common rAAV QC assays fail to ascertain the level of non-
Previous work using single molecule real-time sequencing (SMRT), ligation of thymine and adenine overhangs (TA-based ligation), and tagmentation-based library preparation methods have proved that NGS is an effective tool to confirm rAAV genome sequence and identify vector preparation anomalies such as truncated genomes, reverse packaging, and the presence of contaminating DNA. 12 –14 Unfortunately, at present, SMRT sequencing only works for self-complementary adeno-associated viral genomes precluding its widespread use. 12 Further, both TA-based ligation and Fast-seq, a tagmentation-based method, included a laborious double-stranding step. 13,14
Herein, we describe a rapid tagmentation-based library preparation method from viral DNA (vDNA) extracts termed viral genome sequencing (VGS). Using this approach, we are able to sequence to depths consistent with the Fast-seq method without a preliminary double-stranding step. Using these data, we are able to assess the identity and confirm the serotype of our rAAV preparations.
We show that VGS quickly and reliably confirms the identity of viral preparations, detects cross-contaminating rAAV DNA, and provides a simple platform for additional sequence-based analysis such as serotype confirmation, and detection of cre-independent recombination events.
In addition, herein we describe unique open-source Python scripts that confirm the serotype of our vector preparations and determine the rate of promiscuous recombination in Cre-dependent viral vectors. These scripts can be easily modified to detect other sequences or events, such as reverse packaging of plasmid backbone or the presence of specific DNA contaminants, and can analyze multiple samples within minutes. VGS is straightforward, requiring only a DNA extraction step before Illumina preparation and can be easily incorporated into the QC regimen of both independent laboratories and viral vector cores alike. Given the number of rAAV production services available and the widespread use of rAAV in both academia and industry, the standardization of AAV QC is a critical need for the research community.
Materials and Methods
AAV production
Vectors produced at Addgene were generated by using the helper-free triple plasmid transfection approach in adherent AAVPro-293T cells (Takara) in serum-containing conditions. Ninety-six hours after transfection, cells and media were harvested; cells were pelleted and resuspended in cold lysis buffer (50 mM Tris, 150 mM NaCl, 2 mM MgCl2) before sonication. After centrifugation, the clarified cell lysate was transferred to a clean tube. Viral particles present in the media were pelleted by addition of polyethylene glycol (PEG 8000) to a final concentration of 8%, stirred for 1 h, and incubated for 3 h at 4°C.
PEG-precipitated particles were pelleted by centrifugation, and the pellet was resuspended in cold lysis buffer. PEG-precipitated particles and clarified cell lysate were then combined and treated with Benzonase. The DNAse was added at a final concentration of 25 U/mL and incubated at 37°C for 45 min. Vectors were purified by iodixanol gradient ultracentrifugation at 350,000
The final formulation buffer was phosphate-buffered saline supplemented with 150 mM NaCl and 0.001% Pluronic F68. Purified AAVs were aliquoted, and a QC aliquot was stored at 4°C for all QC assays whereas the remainder of aliquots were stored at −80°C long term. All additional AAV vectors were obtained from the University of Pennsylvania Vector Core. Complete reagent information is listed in Supplementary Table S5.
DNA extraction for sequencing and restriction digests
vDNA was extracted from 20 μL of purified AAV by using PureLink Viral RNA/DNA purification kit (Thermo Fisher) following the manufacturer's instructions. DNA was eluted in 30 μL of nuclease-free water and transferred into a 2D barcoded micronic tube. DNA concentration and purity were determined by using the Nanodrop spectrophotometer, and the samples were stored at −20°C. rAAV DNA extracts or ΦX174 DNA (New England Biolabs) were digested with
qPCR of Sac II cleavage sites
Undigested and
Primer sequences:
37825 amplification
50465 amplification
Sample preparation for MiSeq and sequencing
vDNA was extracted from 20 μL of purified AAV by using PureLink Viral RNA/DNA purification kit (Thermo Fisher) following the manufacturer's instructions. DNA was eluted in 30 μL of nuclease-free water and transferred into a 2D barcoded micronic tube. DNA concentration and purity were determined by using the Nanodrop spectrophotometer, and the samples were stored at −20°C. Twenty microliters of vDNA extract was sent to Seqwell for library preparation by using a plexWell 96-well Library Preparation Kit and sequenced.
First, the average concentration of a 96-well plate was adjusted to 2.5 ng/μL by using a global dilution factor. Next, transposase complexes were added to tagment each sample in the 96-well plate with a unique P7 adaptor. After the first tagmentation step, the samples were pooled into a single tube and a second transposase was added to incorporate the p5 adaptor. Carrier DNA was included in the second tagmentation step to ensure a high enough DNA concentration of the pool to promote even incorporation of the adaptor. The prepared library was then sequenced on a MiSeq System in a 2 × 250 bp paired-end run.
Data analysis using Geneious
After sequencing, FASTQ files were imported into Geneious and paired as Paired End (inward pointing) with the insert size set at 500. The paired file was then trimmed by using BBDuk trimmer to remove poor-quality reads and adaptors. Adaptors were trimmed based on paired read overhangs with a minimum overlap set to 24. The minimum quality of reads was set at 20, and low-quality reads were trimmed from both ends. Reads under 50 bp were discarded.
Trimmed files were then aligned to a reference map of the
Data analysis using custom Python scripts
The serotype detection Python program was designed to analyze FASTQ sequencing files and determine serotype based on the presence of predefined signature sequences. The program takes, as configuration parameters, one or more signature sequences per serotype, as well as the directory where the FASTQ files containing the VGS data reside. When executed, the program loads each FASTQ file by using the BioPython library, extracts the sequence reads, and tallies the incidence of the different signature sequences by simply counting matches in both the reads and their reverse complement. The program then makes a serotype call based on the signature with the highest number of matches. The program outputs a spreadsheet summarizing the findings for all files in the input directory, as well as the detailed tallies for each file.
For the serotype determination script, unique signature sequences were identified by aligning the nucleotide sequences of the various
The following signature sequences were used for these studies:
AAV1-1: AGTGCTTCAACGGGGGCCAG
AAV1-2: GGGCGTGAATCCATCATCAACCCTGG
AAV1-3: CCGGAGCTTCAAACACTGCATTGGACAAT
AAV2-1: AGGCAACAGACAAGCAGCTACC
AAV2-2: AACAGACAAGCAGCTACCGCA
AAV5-1: TCCAAGCCTTCCACCTCGTCAGACGCCGAA
AAV5-2: CACCAACAACCAGAGCTCCACCACTG
AAV5-3: GCCCGTCAGCAGCTTCATC
AAV8-1: GCAAAACACGGCTCCTCAAAT
AAV8-2: CAGCAAGCGCTGGAACCCCGAGATCCAGTA
AAV8-3: AAATACCATCTGAATGGAAGAAATTCATTG
AAV8-4: CGTGGCAGATAACTTGCAGC
AAV8-5: ATCCTCCGACCACCTTCAACC
AAV9-1: AGTGCCCAAGCACAGGCGCA
AAV9-4: GGCGAGCAGTCTTCCAGGCA
AAV9-5: ATCTCTCAAAGACTATTAAC
PHPS: AGGCGGTTAGGACGTCTTTGGCACAGGCGCAGA
PHPeB: CTTTGGCGGTGCCTTTTAAGGCACAGGCGCAGA
AAVrg: ACCTAGCAGACCAAGACTACACAAAAACTGCT
The recombination calculation Python program was designed to analyze FASTQ sequencing files and determine the percent of recombination at specific recombinase sites. The program takes, as configuration parameters, the expected sequence of the predefined recombinase site, the number of base pairs before and after the recombinase site to consider for determining recombination (the head and tail), and the directory where the FASTQ files containing the VGS data reside.
The program analyzes each FASTQ file and produces a report per file. It reads each file by using the BioPython library, extracts the reads and their reverse complements, and compiles a list of all the sequences containing the recombinase site plus a head and a tail. The two most frequent sequences are assumed to be the nonrecombined sequences. The program then analyzes the remaining sequences to determine whether they contain recombination (
Transduction
Seven thousand AAVpro cells were transduced with 1 μL of AAV9-AAV-FLEX-rev-ChR2(H134R)-mCherry in the presence or absence AAVrg-pAAV-Cre-GFP in a 96-well plate. mCherry expression was assessed 3–9 days post-transduction by direct fluorescence. Wells were examined until 9 days post-transduction for promiscuous expression of mCherry in the non-Cre-containing wells.
Plasmids
Plasmids for transfection were purified by using Qiagen's endotoxin-free HiSpeed Gigaprep kit (Qiagen) and quantified by spectrophotometry. The integrity of the
The following plasmids were used to prepare the rAAV described in this study; pAAV-EF1a-double floxed-hChR2(H134R)-EYFP-WPRE-HGHpA (Addgene 20298, a gift of Karl Deisseroth, unpublished) AAV-Cre-GFP (Addgene 68544, a gift of Eric Nestler), AAV-EF1a-DIO-GCaMP6s-P2A-nls-dTomato (Addgene 51082, a gift of Jonathan Ting), AAV-FLEX-rev-ChR2(H134R)-mCherry (Addgene 18916, a gift from Scott Sternson), rAAV2-retro helper (Addgene 81070, a gift of Alla Karpova), pAAV-hSyn-DIO-hM3D(Gq)-mCherry (Addgene 44361, a gift of Bryan Roth), pAAV-hSyn-DIO-hM4D(Gi)-mCherry (Addgene 44362, a gift of Bryan Roth), pAAV-hSyn-hM3D(Gq)-mCherry (Addgene 50474, a gift of Bryan Roth, unpublished), pAAV-hSyn-EGFP(Addgene 50465, a gift of Bryan Roth, unpublished), pAAV-CAG-GFP (Addgene 37825, a gift from Edward Boyden, unpublished), pAAV-CAG-tdTomato (Addgene 59462, a gift of Edward Boyden, unpublished), and pUCmini-iCAP-PHP.eB (Addgene 103005, a gift of Viviana Gradinaru). 15 –20
Software
FASTQ data were analyzed by using Geneious Prime. The custom viral serotype determination and recombination Python scripts can be accessed at GitHub in Addgene's Open Toolkit (
Results and Discussion
To prepare for VGS, DNA is extracted from 20 μL of DNAse-treated purified rAAV by using commercially available vDNA extraction kits. The titers of the rAAV preparations range from 2 × 1012 to 2 × 1013 genome copies (GC)/mL, therefore each 20 μL aliquot contains between 4 × 1010 and 4 × 1011 GC.
Purified DNA is then sent to Seqwell for library preparation and NGS. At Seqwell, libraries are prepared via a tagmentation reaction in which transposons individually tag samples with sample-specific barcoded adaptors. Samples are pooled and tagged with pool-specific barcoded adaptors and then amplified with universal primers. The tagmentation library preparation method has been shown to work for circular plasmid DNA and, in addition to VGS, is used to sequence verify the plasmids used for AAV production. 21 After amplification, the library is normalized, sequenced and the data are provided to Addgene for analysis (Fig. 1).
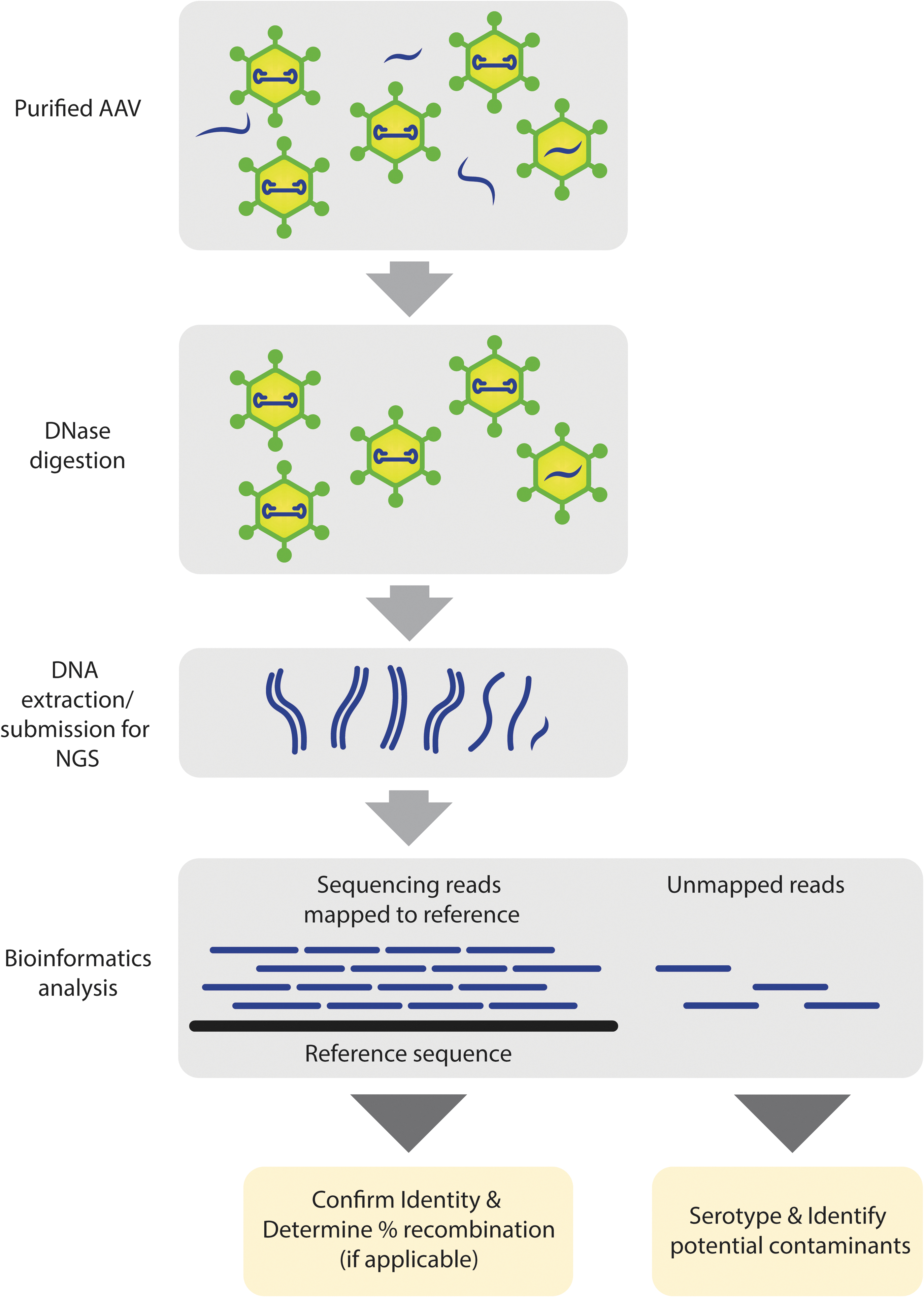
Simplified viral genome sequencing workflow. Packaged DNA is extracted from purified AAV and sequenced. Individual NGS reads are mapped to a reference sequence to confirm the identity of the viral genome. Unmapped reads are further analyzed to detect and identify potential contaminants. In addition to identifying confirmation and contaminant detection, the NGS data are used further to confirm serotype identity and determine relative recombination rate in Cre-dependent genomes (containing Lox sites). AAV, adeno-associated virus; NGS, next-generation sequencing. Color images are available online.
Previous work utilized TA-based ligation of adaptors for rAAV sequencing library preparation. 13 In addition, a recent method termed Fast-Seq successfully used a Tn5 tagmentation-based approach for rAAV library preparation. 14 A major drawback of these methods is that they require a laborious double-stranding step to generate a suitable ligation template. It has been demonstrated that rAAV package either a [+] or [−] strand DNA genome at equal rates. 22
Here, we hypothesize that the following DNA extraction complementary [+] and [−] strands, present in approximately equal numbers, hybridize
To determine whether [+] and [−] rAAV genomic extracts exist as single-stranded (ss) or ds species, DNA extracts were treated with the restriction enzyme
Unlike the rAAV extracts that contain equal proportions of [+] and [−] genomes, the ΦX174 genome is entirely [+] and therefore cannot hybridize.
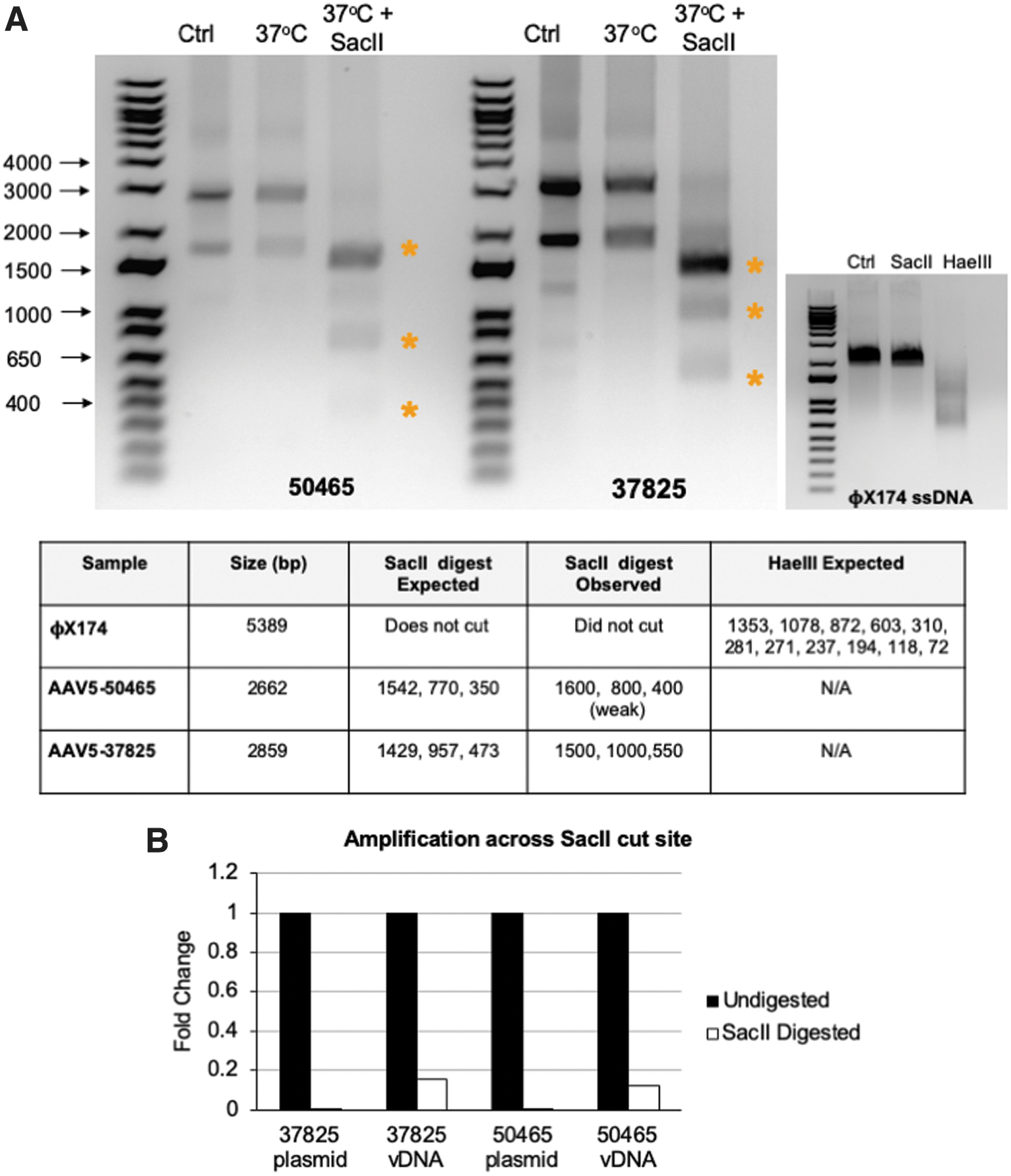
Analysis of DNA conformation of rAAV vectors.
Initially, we hypothesized that the bands might arise from distinct ss and ds species that migrate differently through the gel. To address this, the bands were extracted from the agarose gel, purified, and submitted for NGS. Both the high-molecular-weight and low-molecular-weight bands were sequenced with 2,730 and 1,478 total reads, respectively (Supplementary Table S1 and Supplementary Fig. S1). The majority of reads, 90% and 76%, from the high- and low-molecular-weight bands, respectively, aligned to the reference sequence. Given that both bands can be sequenced and are completely digested by
Although the data suggest that much of the DNA in the extract exists as a double-stranded species, it remains possible that the DNA extract is a heterogeneous mix of single- and double-stranded species. To address this, DNA extracts were left undigested or digested with
Of note, amplification of the cleavage site was not completely lost. To determine whether intact
To determine whether rAAV DNA extracts, shown to be present primarily as ds species, could serve as efficient tagmentation substrates without double stranding via random hexamer priming, DNA from several rAAV preparations was extracted and submitted directly to Seqwell for NGS. Seqwell was able to obtain tens of thousands of reads for the vast majority of rAAV samples (Supplementary Table S2).
As a negative control, the single-stranded ΦX174 genome was submitted for sequencing. Approximately 800 reads were obtained from the ΦX174 sample. Of these reads, only 89 aligned to the reference ΦX174 genome and those that did had very low (<10 × ) coverage and poor consensus to the reference (Supplementary Fig. S1, bottom panel). Of note, one study has shown that the Tn5 transposase is able to bind to certain conformations of ss DNA. 24 With this in mind, one cannot rule out the possibility that a low level of tagmentation may be occurring with ss DNA samples.
In the VGS analysis platform, rAAV NGS data are first analyzed by using Geneious Prime software to confirm identity, check packaging efficiency, and detect the presence of contamination. Briefly, using BBDuk (decontamination using kmers), low-quality reads and adaptors on paired read overhangs are trimmed and reads shorter than 50 base pairs are discarded. The reads are then aligned to the reference sequence of the
In a typical sample, the vast majority of reads, >90%, map to the reference sequence. The alignment is then checked to ensure an even distribution of reads over the entire expression cassette and consensus to the reference. The depth of coverage varies between samples but is typically between 500 and 1,000 × throughout most regions. This coverage is similar to the 1,400 × coverage observed by using the Fast-Seq method. 14
Of note, certain regions such as ITRs and GC-rich sequences such as the CAG and synapsin promoters are notoriously difficult to sequence. In addition, tagmentation-based methods have known sequence biases toward GC rich sequences and structural biases toward synapses. 25,26 Although a drop in coverage in these regions is expected and frequently observed, sequencing depths of >100 × are obtained in these regions, allowing for transgene identification and consistent with the Fast-Seq method. 14
Once the identity of the sample is confirmed, the files are analyzed for contamination via a Megablast search on all unmapped reads and a manual review of the hits. As previously mentioned, it is common for non-rAAV sequences such as plasmid backbones, elements from the capsid, and helper packaging plasmids, and genomic DNA from the packaging cells to be packaged with the rAAV genome. 2 –9 In confirmation of these findings, these sequences were often present at low levels in the Megablast search.
In a typical clean sample, the number of hits to a given non-
To validate this approach, rAAV preparations were prepared in which 0.1–20% of sample 2 was spiked into sample 1 (Fig. 3A). After preparing the spiked samples, DNA was extracted and the samples were sent to Seqwell for VGS. Sequencing results were blinded and analyzed following the standard VGS procedure. In the Megablast analysis of the unmapped reads, WPRE and the EF1alpha promoter—elements unique to the spiked DNA—were identified in samples spiked with as little as 0.1% of sample 2 (Fig. 3A, B).
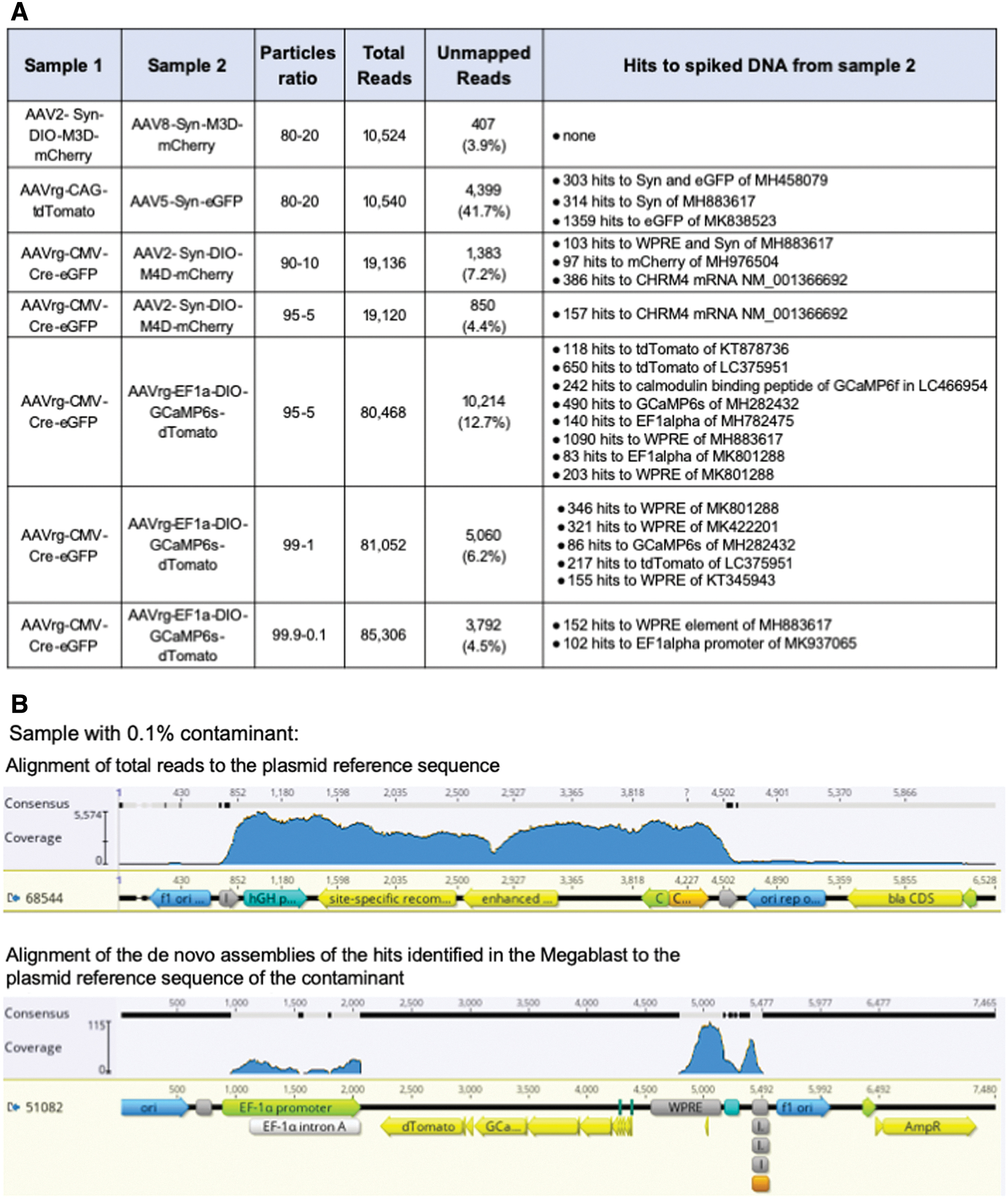
Validation of VGS workflow and analysis.
The only sample in which the spiked DNA sequences were unable to be detected was an instance in which the sequences of vector and contaminant differed only by the absence of two 34 bp lox sites in the spiked sample. In this case, the difference between samples would likely only be observed as a drop in coverage at the lox sites. This specific type of contamination was not able to be detected at levels up to 20% of the sample. Of note, since the spiked DNA lacks the lox sites, this would not be identified in the Megablast analysis stage.
Sequencing depth will influence the ability to detect contaminants using this method. For these analyses, depths ranged from 10,540 to 85,306 total reads, a range that is consistent with the recently reported Fast-Seq method for research-grade rAAV identification. 14 Overall, 19,120 reads were sufficient to detect a sample with 5% spiked DNA, and the sequencing depth was much higher for the sample in which 0.1% of spiked DNA was detected; in this case, of 85,306 reads only 102 and 150 hits to spiked DNA sequences were detected. Consequently, if adopting this method to identify low levels of contaminating DNA in research-grade rAAV, we would recommend sequencing to depths greater than 80,000 total reads.
In addition to confirming identity and detecting cross-contamination, VGS data can be used to examine the propensity of non-
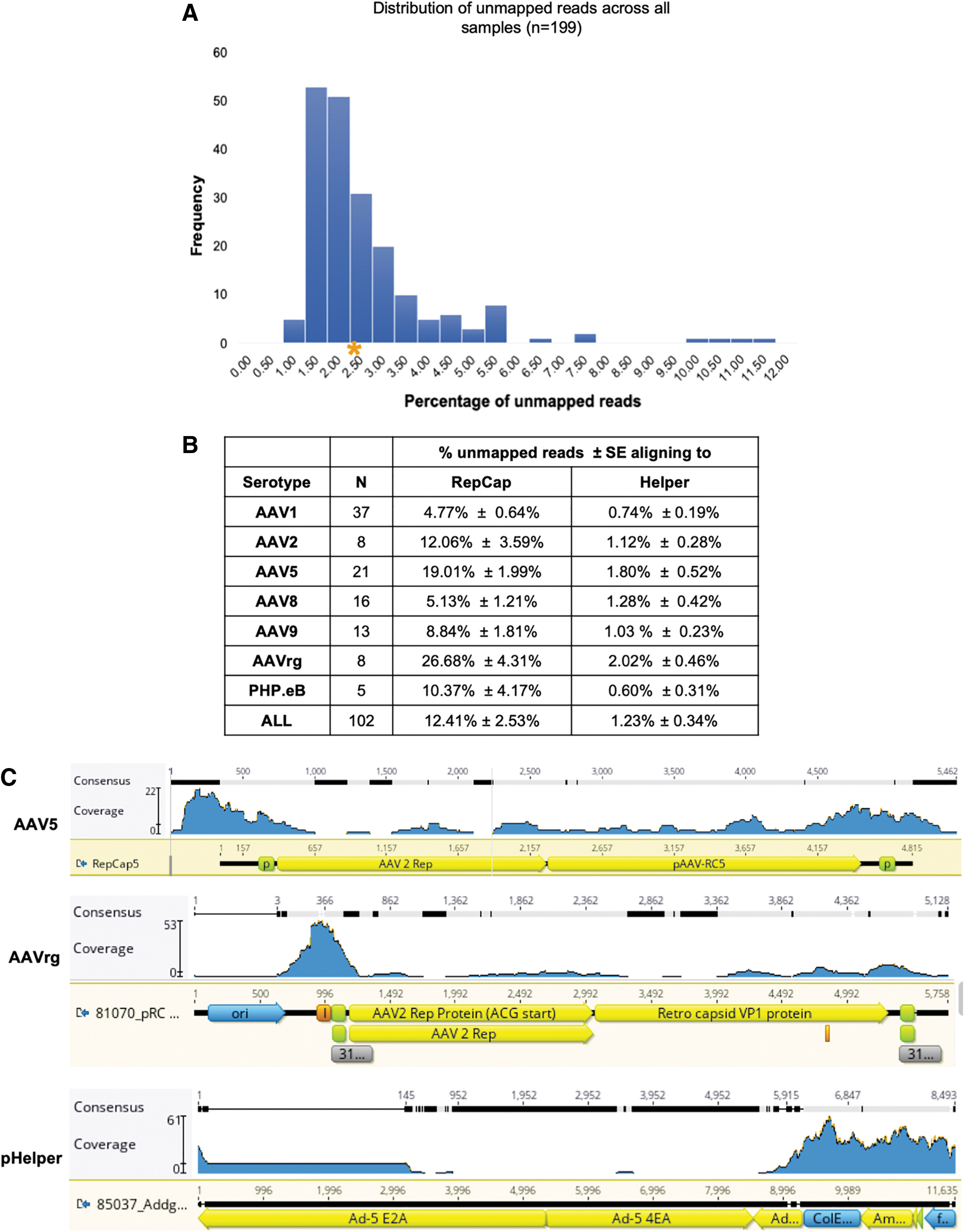
Analysis of unmapped reads.
The percentage of packaged capsid sequences was significantly higher than that of the helper sequences (
For both viral vectors, AAV2, AAV5, and AAVrg had the highest levels of capsid packaging. Previous work examining rAAV packaging demonstrated that the p5 promoter elements present in the capsid plasmids are prone to packaging. 12 Consistent with these findings, a clustering of reads around the p5 promoter region was observed in the samples (Fig. 4C, top panel). In addition, an adenoviral ITR element adjacent to one of the p5 promoters in the AAVrg capsid was frequently packaged (Fig. 4C, middle panel, peak at position 366). We speculate that this element may be structurally similar to the rAAV ITR elements, making it prone to packaging.
One concern, especially for those using AAV in the clinic, is the formation of a complete
We estimate that the DNA extracted for VGS contains between 4 × 1010 and 4 × 1011 GC. Despite the high number of genomes present, in the vast majority of cases, the
Although vector cores assess samples for titer and purity, verification of the serotype is not routinely done. Historically, serotype confirmation has required expensive and resource-intensive methods such as immunostaining and mass spectrophotometry precluding its incorporation into standard viral vector QC. Since DNA sequences from the capsid plasmids are often detected in the Megablast analysis stage, we hypothesized that we could use NGS reads to confirm the serotype of a sample. To address this, we developed a Python script that allows us to interrogate our VGS data for signature sequences that are unique to each serotype (Fig. 5A).
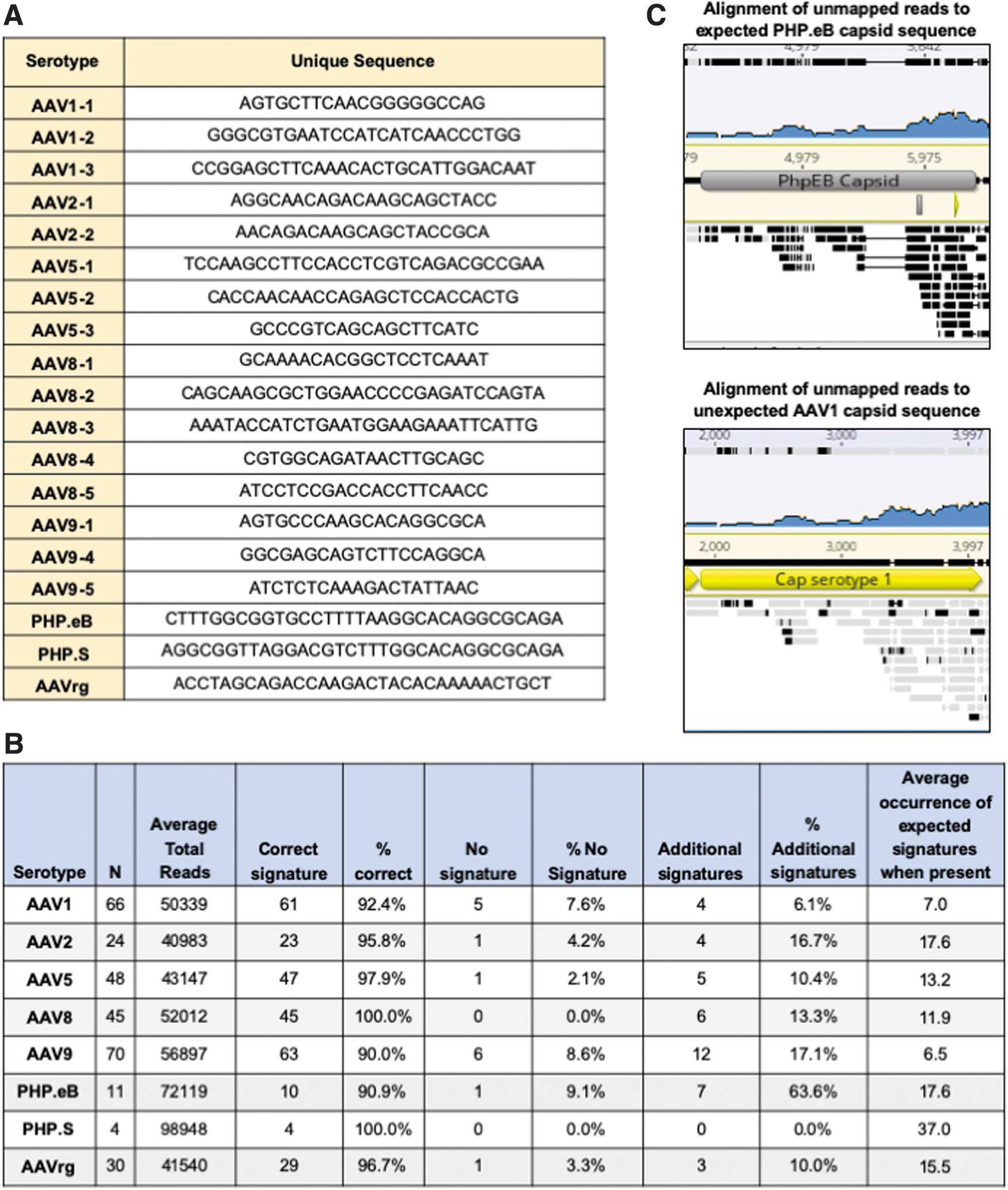
Serotype determination from NGS reads.
The Python script analyzes multiple samples within minutes without the need for expensive reagents. The program tallies the incidence of the signature sequences and makes a serotype call based on the signature with the highest number of hits. One limitation of the program is that it is designed to search for exact matches to the signature and will not tally divergent signatures. In addition, for highly homologous capsids it may be difficult to identify unique signatures. To date, this script has been used to analyze more than 250 vectors. The program accurately detects greater than 90% of samples across all serotypes tested (Fig. 5B). In the cases where the program fails to call the expected serotype, it is not due to miscalling but instead is due to the absence of the signature sequence in the rAAV DNA extract.
The presence of a serotype's signature sequences in the NGS data depends on several factors such as the sequencing depth, rate of signature sequence packaging, and number of signatures being interrogated. For this dataset, the percentage of unmapped reads ranged from 1% to 12% (Fig. 4A). The percentage of these unmapped reads aligning to the capsid gene varied across serotype, with AAV1 having the lowest at 4.8% (Fig. 4B). At a sequencing depth of 10,000 reads, if the sample were at the low end of the distribution, only 1% or 100 reads would align to the capsid.
In the case of AAV1 where less capsid elements are packaged, only 4.8% or 4.8 of the unmapped reads would align to the capsid. Assuming equal packaging of the capsid sequence, 4.8 reads at a read length of 250 bp would span 1,200 bp and would not provide complete coverage of the ∼2,200 bp capsid gene. At 20,000 reads, coverage would span 2,400 and provide roughly 1 × coverage of the capsid gene. Consequently, we recommend 20,000 reads as a minimum for this Python script, especially if only one single signature sequence is used.
It should be noted that this recommendation is based on the assumption that all sequences of the capsid gene are packaged with the same efficiency. Given that previous work and these data have demonstrated that particular regions are more prone to packaging than others, this is likely not the case. Consequently, some signatures may be more likely to be present or absent than others. We recommend using as many unique signatures as possible to increase the likelihood of the program returning a serotype call.
Of note, for some samples the program detects additional signatures from unexpected serotypes. On closer examination, when this occurs, the hits tend to be from capsids that have a high level of homology and the rate of unexpected signatures is significantly lower than that of the expected capsid. For example, in one sample there were 57 hits to the expected AAV2 signatures and 2 hits to AAVrg, a derivative of AAV2.
Although most serotypes have a relatively low level of additional signature calls, the average for PHP.eB is quite high at 64% (Fig. 5B). This is due to the high degree of homology between PHP.eB and its parental capsid, AAV9. Despite designing a signature sequence in the area of divergence, AAV9 signatures are consistently observed. Thus far, the number of hits to the PHP.eB signature has been high enough to easily make the correct call; of 11 samples, the average occurrence per sample of the PHP.eB signature is 17.8 whereas that of the AAV9 is only 1.6. Given that most labs do not routinely use PHP.eB and the large disparity in the number of PHP.eB and AAV9 calls, we do not think this will be a problem for users choosing to adopt this analysis method.
During this analysis, one sample was identified in which the serotype determination software provided an unexpected serotype call. The sample was labeled as PHP.eB, yet the software identified 13 AAV1 signatures and no PHP.eB signatures. Although it is common to see additional AAV9 signatures in PHP.eB samples, AAV1 is completely unexpected, especially to such a high degree. To confirm the program's findings, the FASTQ files from the sample were first aligned to the reference
As indicated by black bars in Fig. 5C, there was no consensus between the unmapped reads and the expected PHP.eB capsid sequence. In contrast, although the overall coverage is low, there was very good consensus with the AAV1 capsid, especially at the 3′ end of the sequence (see gray bars in Fig. 5C). This confirmed that the serotype detection program was, indeed, correct and the sample had been inadvertently swapped with an AAV1 of the same
More recently, a thermostability-based approach termed AAV-ID was developed that can distinguish AAV serotypes based on melting temperature. 27 AAV-ID can be done in a 96-well format and can yield results in as little as 6 h. Although AAV-ID is a marked improvement on previous methods, it does have some limitations. First, melting temperature will vary between different formulation buffers and can be affected by sample purity. In addition, some serotypes such as AAV6.2 and AAV9 have very similar melting temperatures despite limited homology in the VP3 protein and cannot be easily distinguished.
In cases such as this, where there is a high level of divergence of VP3 between serotypes, using a sequence-based approach for serotype identification would be beneficial. For viral cores producing large numbers of viral vectors, sequence-based identification and AAV-ID could be used as complementary methods of serotype confirmation. However, it is notable that if a core is already sequencing its rAAV preps, there is no additional cost to determine the serotype using the software, whereas additional lab work would be needed for the AAV-ID method.
The Cre-Lox system is a common tool used to control transgene expression. To obtain meaningful data when using the Cre-Lox system, it is critical that transgene expression is suppressed in the absence of the Cre recombinase. Consequently, scientists using Cre-dependent rAAV preparations are often concerned about promiscuous expression caused by random recombination events during plasmid propagation and viral packaging.
To address these concerns, we developed a Python script to interrogate VGS data for recombination events. Specifically, the program uses the Lox sequences as signatures and identifies which adjacent sequences are as expected and which could only exist if Cre-independent recombination occurred at those Lox sites. The color-coded cartoon in Fig. 6A shows the differences between the expected and recombined sequences. Using this program, we determined the relative recombination level in 198 rAAV preparations. Cre-independent recombination events range from 0% (below the limits of detection) to 2.70%, with most samples falling below a ∼0.2% recombination level (Fig. 6B).
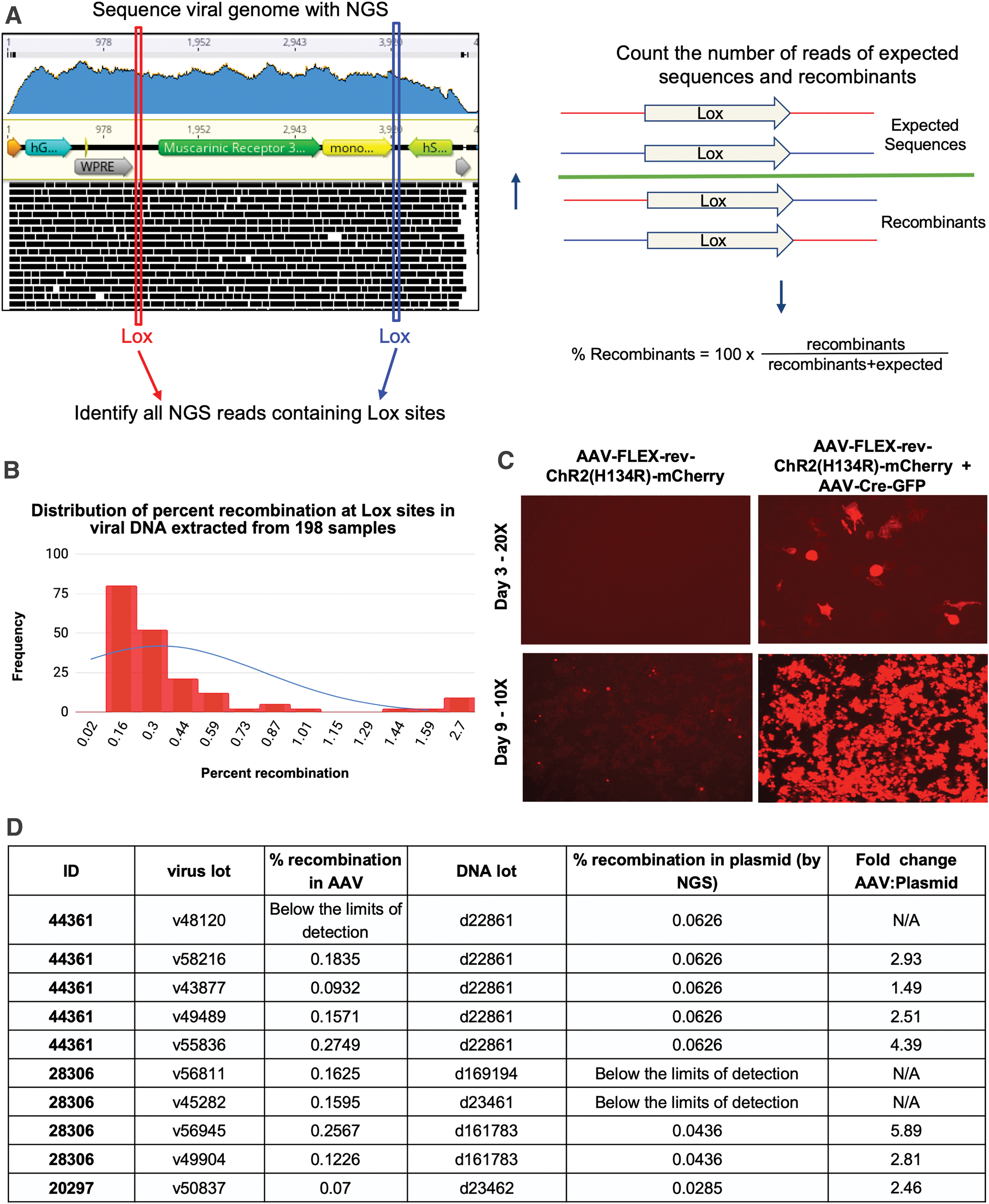
Analysis of promiscuous recombination at Lox sites in Cre-dependent viruses.
Although the overall level of recombination across samples is quite low, there were some samples with a high percentage of recombinants. To determine whether recombinants could be detected
mCherry expression was assessed by direct fluorescence up to 9 days after transduction. A high MOI was used as a worst-case scenario as to not limit the chances of observing Cre-independent expression. The lot of AAV-FLEX-rev-ChR2(H134R)-mCherry chosen had one of the highest recombination rates observed at 2.46%. Although mCherry expression was undetectable at 3 days, after incubating the sample for 9 days post-transduction, mCherry expression was present (Fig. 6C).
It should be stressed that this assay cannot be relied on as a detection method for leaky expression of Cre-dependent systems. To date, hundreds of samples have been analyzed and oftentimes, samples with high levels of recombinants are simply undetectable
Finally, the plasmid DNA used for viral production was compared with DNA extracts from the derived viral vector lots, to determine whether the Cre-independent recombinants were present in the
In one lot of 44361 plasmid DNA, d22861, the level of recombination was measured at 0.06%. This lot of DNA was used to produce five rAAV lots in which the rate of recombination ranged from undetectable to 0.27%, which was 4.4-fold higher than that observed in the plasmid DNA (Fig. 6D). Of note, the number of reads for the sample in which recombination was not detected was only ∼16,000, limiting the likelihood of signature-containing reads. Therefore, it is recommended that the program only be used to analyze samples with >20,000 reads. The broad range of recombination across viral lots demonstrates that first, the rate of recombination in the viral preparation cannot be inferred from that of the plasmid and second, recombination is occurring during the packaging step in mammalian cells.
Herein, we describe a powerful tool to rapidly characterize the identity of purified AAV from rAAV DNA extracts that we term “VGS.” VGS does not require a double-stranding step before tagmentation, reducing both the time and cost of sample preparation. Without the double-stranding step, it is likely that the DNA extract is a heterogeneous mix of ss and ds species, a theory supported by the low level of intact
The VGS method combined with our open-source custom Python scripts allows scientists to quickly and reliably confirm the identity and serotype of their AAV preparations and detect low levels of rAAV cross-contamination and recombination events. For scientists handling or producing multiple viral vectors, this allows for validation similar to plasmid sequencing and, because samples can be batched together on a single lane for sequencing, prices can be kept low.
In addition to providing a rapid method of rAAV identification, VGS can be used to create sequence datasets for the study of rAAV genomics. In this study, we used the data to identify non-
Footnotes
Acknowledgments
The authors thank Joanne Kamens, Lianna Swanson, Marcella Patrick, Will Arnold, and all other members of Addgene for their advice, helpful discussion, and support during the preparation of this article. The authors would also like to thank Benjie Chen for valuable advice on the Python scripts.
Author Disclosure
K.G., M.R., D.B., I.E., L.H., K.H.D., E.S., M.T., M.K., M.T., L.M.H., I.M., A.M., and M.F. are currently or have been employed by Addgene, a company that may be affected financially by the research reported in the enclosed article.
Funding Information
Addgene did not receive any funding for this work.
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.