
Abstract
Duchenne muscular dystrophy (DMD) is a rare, X-linked, fatal, degenerative neuromuscular disease caused by mutations in the DMD gene. More than 2,000 mutations of the DMD gene are responsible for progressive loss of muscle strength, loss of ambulation, and generally respiratory and cardiac failure by age 30. Recently, gene transfer therapy has received widespread interest as a disease-modifying treatment for all patients with DMD. We designed an adeno-associated virus vector (rAAVrh74) containing a codon-optimized human micro-dystrophin transgene driven by a skeletal and cardiac muscle-specific promoter, MHCK7. To test the efficacy of rAAVrh74.MHCK7.micro-dystrophin, we evaluated systemic injections in mdx (dystrophin-null) mice at low (2 × 1012 vector genome [vg] total dose, 8 × 1013 vg/kg), intermediate (6 × 1012 vg total dose, 2 × 1014 vg/kg), and high doses (1.2 × 1013 vg total dose, 6 × 1014 vg/kg). Three months posttreatment, specific force increased in the diaphragm (DIA) and tibialis anterior muscle, with intermediate and high doses eliciting force outputs at wild-type (WT) levels. Histological improvement included reductions in fibrosis and normalization of myofiber size, specifically in the DIA, where results for low and intermediate doses were not significantly different from the WT. Significant reduction in central nucleation was also observed, although complete normalization to WT was not seen. No vector-associated toxicity was reported either by clinical or organ-specific laboratory assessments or following formal histopathology. The findings in this preclinical study provided proof of principle for safety and efficacy of systemic delivery of
Introduction
Duchenne muscular dystrophy (DMD) is a rare, X-linked, fatal, degenerative neuromuscular disease that occurs in ∼1 in every 3,500 to 5,000 males born worldwide. 1 –4 DMD and Becker muscular dystrophy (BMD) affect skeletal and cardiac muscle due to mutations in the 2.4 Mb dystrophin gene (DMD). Dystrophin is a 427 kDa cytoskeletal protein required for muscle fiber stability. Specifically, dystrophin links the sarcomere and the extracellular matrix. Loss of the protein results in destabilization of the dystrophin-associated protein complex (DAPC), repeated turnover of the muscle, satellite cell depletion, and replacement of fibers by fat and fibrosis. The most frequent mutations (65%) found in patients are deletions of one or more exons of the DMD gene, leading to shortened forms of nonfunctional dystrophin protein that undergo degradation. 5 Patients with DMD are characterized by progressive muscle weakness affecting skeletal and respiratory muscle and loss of ambulation typically by age 12, and generally death by age 30. 6
Corticosteroids are currently the standard-of-care treatment for most patients to modify symptoms. 7 However, while corticosteroids have shown benefit in delaying disease progression, long-term treatment is limited as it is associated with serious adverse effects such as bone fracture, infection, and gastrointestinal bleeding. 8 Moreover, once decline occurs, disease progression remains fatal as dilated cardiomyopathy and respiratory failure are often the major cause of premature death. 6 Eteplirsen is the first disease-modifying treatment (phosphorodiamidate morpholino oligomer) approved in the United States for DMD in patients with confirmed DMD mutations amenable to exon 51 skipping. 9 –11 Eteplirsen has been shown to produce functional dystrophin protein and slow the decline in ambulatory and pulmonary function after long-term use. 9 –12 Golodirsen was the first treatment approved by the U.S. Food and Drug Administration for DMD patients amenable to exon 53 skipping, 13 and a second antisense oligonucleotide has been approved for the same group of patients. 14 Recently there has been a significant focus on developing gene therapy approaches for DMD with the potential to become a disease-modifying treatment for all patients with DMD following single gene delivery.
Adeno-associated virus (AAV) systemic gene transfer therapy offers the possibility to ameliorate disease progression and normalize the muscle environment through restoration of dystrophin expression in cardiac and skeletal muscle. However, due to the enormous size of the dystrophin gene and the packaging limitation of AAV (∼5 kb), entire gene transfer is not feasible. Thus, investigators have developed micro-dystrophin genes that produce shortened functional variants of micro-dystrophin protein and demonstrated amelioration of muscle pathology and cardiomyopathy in various preclinical models. 15 –20
More than 30 different configurations of micro-dystrophin have been developed based upon the genotypes of patients with milder forms of DMD/BMD and preclinical structure–function studies. 21 Dystrophin is composed of four major domains: an N-terminus that contains an actin binding domain, a central rod domain of 24 spectrin repeats (R) that is flanked and interspersed with 4 hinge (H) subdomains, a cysteine-rich domain, and a C-terminal domain. 22 –24 The N-terminus and cysteine-rich domain are thought to be essential, as these contain an actin binding site 22 and the binding domain for β-dystroglycan, 25,26 respectively. In contrast, the C-terminus does not appear to be essential for muscle function or DAPC assembly. 23 The central rod domain is the largest and plays an important role in conferring protection and flexibility. 24,27 However, only a few spectrin repeats and hinges have been demonstrated to have functional relevance. 27,28 For example, spectrin repeats R2-3 have been shown to be necessary for optimized resistance to eccentric force loss, 28 and early structural studies have demonstrated that R1-3 may interact with the lipid membrane. 29,30 We have designed a micro-dystrophin transgene that would promote optimized functional efficacy upon delivery.
In addition to the micro-dystrophin transgene sequence, the efficacy and safety of AAV micro-dystrophin gene therapy are also dictated by the promoter sequence. Therefore, to maximize gene expression in cardiac and skeletal muscle, the cassette used for this study includes a skeletal and cardiac muscle-specific promoter, MHCK7, which provides assurance of safety and avoidance of off-target effects.
31,32
This promoter includes an α-myosin heavy chain complex (α-MHC) enhancer that leads to high levels of expression in cardiomyocytes and increased dystrophin expression in skeletal muscle versus MCK or CK7.
32
–34
In the present study, a dose-escalation study design was utilized to evaluate safety and efficacy in the mdx mouse model of DMD. This report provides proof of principle for the efficacy and safety of systemic delivery of
Materials and Methods
Animal models
Dystrophin-deficient mouse strain
All procedures were approved by The Research Institute at Nationwide Children's Hospital Institutional Animal Care and Use Committee (protocol AR08-00009; AR06-00054). Stocks of C57BL/6 and C57BL/10ScSn-Dmd mdx /J mice were bred and maintained as homozygous animals in standardized conditions in the Animal Resources Core at The Research Institute at Nationwide Children's Hospital. Mice were maintained on Teklad Global Rodent Diet (3.8% fiber, 18.8% protein, 5% fat chow) with a 12-h dark/12-h light cycle. All mice used in this study were male.
Micro-dystrophin gene construction
For all gene transfer studies, the human micro-dystrophin cassette contained the (R4–R23/Δ71–78) domains as previously described. 35 The complementary DNA was codon optimized for human usage and synthesized by GenScript (Piscataway, NJ). It includes a consensus Kozak sequence, an SV40 intron, and synthetic polyadenylation site (53 base pairs). The recombinant MHCK7 promoter used to drive transgene expression is a dual striated muscle-specific promoter and is based on the MCK promoter and the promoter described by Dr. Stephen Hauschka (University of Washington, Seattle, WA). 32 This MCK-based promoter utilizes an enhancer derived from the 5′ of the transcription start site within the endogenous muscle CK gene with a proximal promoter. 32 This enhancer, along with a modified CK7 cassette from the MCK family of genes, is ligated to an α-MHC enhancer 5′ of the CK portion to promote cardiac expression. 32 The MHCK7 micro-dystrophin expression cassette was cloned between AAV2 inverted terminal repeats (ITRs) using flanking XbaI restriction enzyme sites. MscI/SmaI restriction enzyme digestions, as well as Sanger sequencing through the ITRs, were used to confirm ITR integrity.
AAV vector production
In vivo gene delivery
The mice in this study were randomized in sequential order and dosed per mean body weight per group. Mdx mice were injected in the tail vein at 4 to 5 weeks of age with Lactated Ringer (LR) solution (untreated, mdx-LR, n = 6) or with
Hematology
Whole blood was obtained from cardiac puncture for serum chemistry panel analysis for C57BL/6 wild-type (WT), mdx-LR, intermediate dose (6 × 1012 vg) and high dose (1.2 × 1013 vg). Blood was collected into a serum separating tube and centrifuged for 10 min at 15,000 rpm. Supernatant was collected, frozen, and sent to Charles River Laboratories for chemistry testing. Due to limited sample volume, liver enzymes and glucose chemistries were prioritized.
Histopathology
Following euthanization, necropsy was performed, and organ samples were collected. Muscle tissue was fresh frozen in liquid nitrogen-cooled methylbutane, and all other organs were harvested, fixed in formalin, and embedded in paraffin. After processing, tissues were stained with hematoxylin and eosin, and slides and all tissues were sent to GEMPath, Inc., for blinded histopathology evaluation by a veterinary pathologist. The pathologist was subsequently unblinded for data analysis and report development.
Diaphragm tetanic contraction for functional assessment
Mice were euthanized, and the diaphragm (DIA) was dissected with rib attachments and central tendon intact and placed in Kreb's–Henseleit (K-H) buffer as previously described. 39 –43 One 2–4 mm wide section of DIA was isolated per animal per cohort (for one animal in the intermediate dose, two strips were analyzed). DIA strips were tied firmly with braided surgical silk (4-0; Surgical Specialties, Reading, PA) at the central tendon and sutured through a portion of rib bone affixed to the distal end of the strip. Each muscle was transferred to a water bath filled with oxygenated K-H solution that was maintained at 37°C. The muscles were aligned horizontally and tied directly between a fixed pin and a dual-mode force transducer-servomotor (305C; Aurora Scientific, Aurora, Ontario, Canada). Two platinum plate electrodes were positioned in the organ bath, to flank the length of the muscle. The muscle was stretched to an optimal resting tension of 1 g force (the current was set to 1A for duration of the analysis). The muscle was then allowed to rest for 5 min before initiation of the tetanic protocol.
Once the muscle was stabilized, it was subjected to a warm-up, which consisted of three 1-Hz twitches every 30 s, followed by three 150-Hz twitches every minute. After a 3-min rest period, the DIA was stimulated at 20, 50, 80, 120, 150, and 180 Hz, allowing a 2-min rest period between each stimulus, each with a duration of 250 ms to determine maximum tetanic force. Muscle length and weight were measured to determine cross-sectional area. The specific force was normalized to the cross-sectional area of the muscle.
Tibialis anterior tetanic contraction for functional assessment
The tibialis anterior (TA) procedure followed the protocol listed in Hakim et al. 44 Mice were anesthetized using a ketamine/xylazine mixture. A double square knot was tied around the patellar tendon with a 4-0 suture. The TA distal tendon was then dissected out, a double square knot was tied around the tendon with 4-0 suture as close to the muscle as possible, and then the tendon was cut. The exposed muscle was constantly dampened with saline. Mice were then transferred to a thermal-controlled platform and maintained at 37°C. The knee was secured by placing a metal pin behind the patellar tendon. The suture attached to the distal TA tendon was pulled to a resting tension of 3 g and adjusted to be level with the arm of the force transducer (Aurora Scientific, Aurora, Canada). An electrode was placed near the sciatic nerve to stimulate it.
Once the muscle was stabilized, the resting tension was set to a force (optimal length) where twitch contractions were maximal. To determine the optimal length resting tension was set at 2, 3, 4, 5, and 6 g force and stimulated at 1 Hz to determine the optimal tension to be used throughout the procedure protocol (the current was set to 200 mA for duration of the analysis). After a 3-min rest period, the TA was stimulated at 50, 100, 150, and 200 Hz, allowing a 1-min rest between each stimulus. Following a 5-min rest, the muscles were then subjected to a series of 10 isometric contractions, occurring at 1-min intervals with a 10% stretch–relengthening procedure. After the eccentric contractions, the mice were euthanized, and the TA muscle was dissected out and frozen for histology.
Immunofluorescence
Cryosections (12 μm) from the TA, gastrocnemius (GAS), quadriceps (QD), psoas major (PSO), gluteus (GLUT), triceps (TRI), and DIA muscles along with the heart were subjected to immunofluorescent staining for the dystrophin transgene through our previously used protocol. 39 Sections were incubated with a mouse monoclonal human dystrophin primary antibody (Santa Cruz Biotechnology, Dallas, TX) at a dilution of 1:50 and the human β-sarcoglycan primary antibody (Leica Biosystems, Buffalo Grove, IL) at a dilution of 1:100. Additional detail about antibodies used for immunofluorescence analysis is shown in Supplementary Table S1. Analysis for percentage of fibers positive for dystrophin staining included 4 random 20X images covering the four different quadrants of the muscle section per animal per tissue (n = 5 per cohort). Images were taken using a Zeiss AxioCam MRC5 camera (Germany). Percentage of fibers positive for dystrophin staining (>50% of muscle membrane staining) was determined for each image. Quantification data of immunofluorescent-positive fibers expressing dystrophin protein are reported as mean ± standard error of the mean of n = 5 per treatment group.
Western blot analysis
Western blots were performed according to our previously used protocol, with several modifications specific for each antibody used. 39 Samples from WT mice, mdx-LR mice, and vector-dosed mdx mice were used for each western blot. Protein (50 μg for muscle and organs) extracted from samples was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (3–8% Novex NuPAGE gradient gels, Invitrogen, Waltham, MA), blotted on polyvinylidene fluoride membrane, and probed with dystrophin primary antibody Dys1 for dystrophin detection and Dys3 for micro-dystrophin (Leica Biosystems) at a dilution of 1:50 and 1:20, respectively, or neuronal nitric oxide synthase (nNOS) primary antibody (Fisher Scientific) at a dilution of 1:1,000. Loading controls used included γ-tubulin antibody (Sigma) or α-actinin antibody (Sigma) at a dilution of 1:10,000 followed by Alexa Fluor 680 goat anti-mouse (1:5,000, LI-COR, Lincoln, Nebraska). Additional detail about antibodies used for western blot analysis is shown in Supplementary Table S1.
Morphometric analysis
Hematoxylin and eosin staining was performed on 12-μm cryosections of muscle from WT mice, mdx-LR mice, and
Biodistribution qPCR analysis
TaqMan qPCR was performed to quantify the number of vg copies present in targeted muscle, as well as nontargeted organs, as previously described.
34,45,46
Biodistribution analysis was performed on tissue samples collected from three vector-dosed mdx animals per dose level. Tissues were harvested at necropsy, and vector-specific primer probe sets specific for sequences of the MHCK7 promoter were utilized. A vector-specific primer probe set was used to amplify a sequence of the intronic region directly downstream from the MHCK7 promoter that is unique and located within the
Picrosirius red stain and collagen quantification
Frozen sections placed onto Fisherbrand Superfrost charged microscope slides were fixed in 10% neutral buffered formalin for 5 min and then rinsed in distilled water. Slides were then incubated in Solution B (Direct Red 80/2 4 6-Trinitrophenol) from the Picrosirius Red Stain Kit (Polysciences, Inc., Mount Arlington, NJ, Catalog no. 24901) for 15 min. After a thorough rinse in distilled water, the slides were placed in Solution C (0.1 N hydrochloride acid) for 2 min. Slides were counterstained for 5 min with 1% Fast Green in 1% glacial acetic acid from Poly Scientific (Bay Shore, NY, Catalog no. S2114) using a 1:10 dilution in deionized water. Finally, the slides were rinsed again in distilled water, dehydrated in graded ethanol, cleared in xylene, and mounted with coverslips using Cytoseal 60 media from Thermo Scientific (Catalog no. 8310). Images were taken using the AxioVision 4.9.1 software. Representative images of DIA tissue sections in mdx mice treated with
For analysis of Sirius red staining and percent collagen quantification, the contrast between the red and the green colors was enhanced using Adobe Photoshop. The color deconvolution plugin in the ImageJ software program was selected, and the RGB color deconvolution option was used. The red image includes all connective tissue from the Sirius red stain. The green image includes all muscle from the Fast Green counterstain. Only the red image and the original image were used. A threshold was applied to the images to obtain black and white images with areas positive for collagen in black and negative areas in white. Using the measure function, the area of collagen was calculated. The total tissue area was determined by converting the original image to “8-bit” and adjusting the threshold to 254 (one unit below completely saturating the image). The total tissue area was measured as done previously, and total area was recorded. Quantification of collagen accumulation in the DIA of WT mice, mdx-LR, and mice treated with low (2 × 1012 vg), intermediate (6 × 1012 vg), and high (1.2 × 1013 vg) doses was calculated by dividing the area of collagen by total tissue area and used to determine the mean percentage for each individual (n = 5, low dose; n = 8, intermediate and high dose; n = 6, mdx-LR; and n = 5, WT).
Statistical analysis
Data are expressed as the mean ± standard error of the mean (SEM; error bars) and were analyzed using a one-way analysis of variance (ANOVA) or two-way ANOVA with multiple comparisons between groups and statistical significance determined through Tukey's post hoc analysis test using GraphPad Prism 5 (GraphPad Software, La Jolla, CA), unless otherwise specified.
Results
Our micro-dystrophin transgene cassette was constructed by replacing the MCK promoter from previous studies with the MHCK7 promoter to enhance cardiac expression. 34 To establish the minimal efficacious dose in vivo, a dose-escalation study was performed. The dosing groups included a low dose of 2 × 1012 vg total, intermediate dose of 6 × 1012 vg total, and high dose of 1.2 × 1013 vg total. These doses were kept consistent throughout the study, and efficacy was determined through systemic delivery to the tail vein of mdx mice.
Histological improvements with systemic delivery in a dose-dependent manner
Four- to five-week-old mdx mice were treated with low, intermediate, or high total dose of
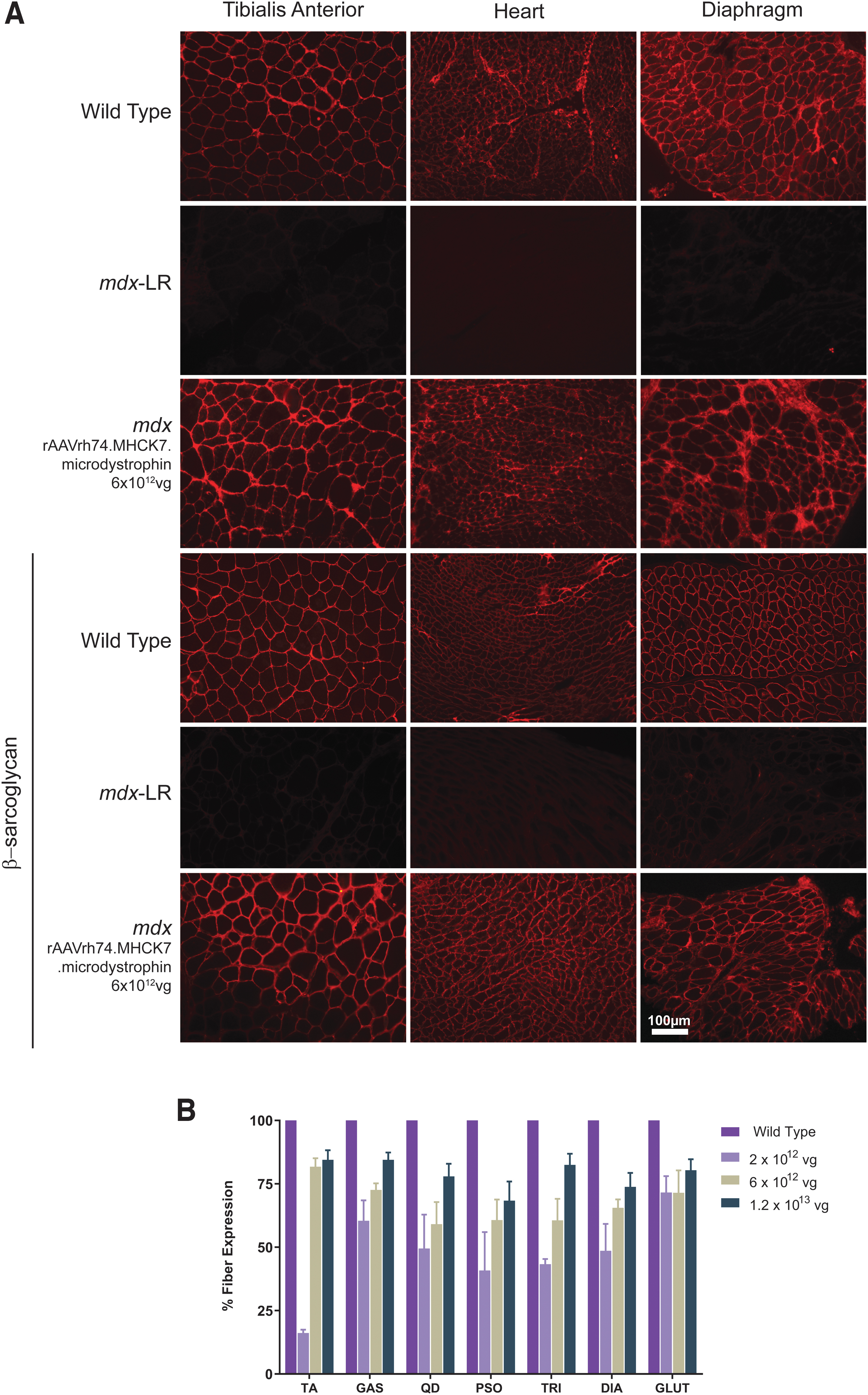
Robust micro-dystrophin expression at the sarcolemma membrane of skeletal and cardiac muscle after systemic delivery of
Fibrosis and inflammation have been previously reported in mdx muscle as part of the chronic dystrophic process. Therefore, fiber diameter, fibrosis, and central nucleation (CN) were evaluated postinjection to evaluate the muscle environment postvector delivery compared to mdx-LR mice. Gene transfer significantly reduced pathological severity at intermediate and high vector doses administered. Furthermore, gene transfer showed reduction of dystrophic features, including fibrosis (Fig. 2A and Supplementary Fig. S2A), and normalization of fiber size distribution with a significant increase in fiber diameter, indicating fiber size similar to WT fibers, specifically in the DIA, where results for low and intermediate doses were not significantly different from the WT. We also observed a normalization of fiber size in the TA for low and high doses and at the intermediate dose for the TRI (Fig. 2B and Supplementary Fig. S2B). Quantification of histological parameters demonstrated a significant reduction in CN in all skeletal muscles analyzed in a dose-dependent manner compared to mdx-LR mice. Gene transfer did not result in complete normalization to WT, and CN remains significantly higher in treated animals but we observed a significant improvement in the TA and GAS of the high dose cohort (Supplementary Fig. S2C).
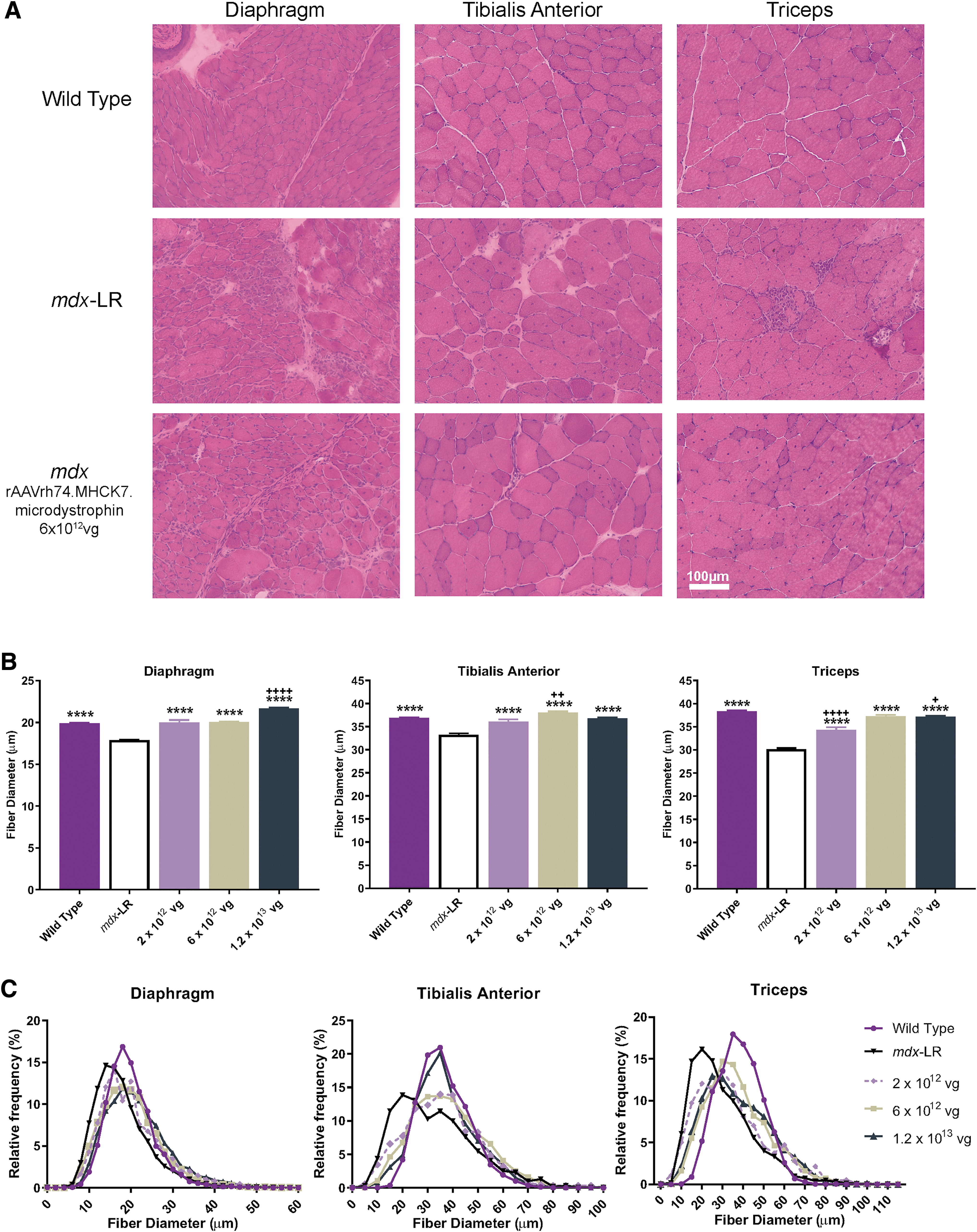
Systemic treatment with
In addition, Sirius red quantification, which represents the amount of collagen deposition in skeletal muscle, was measured to assess the effect of gene transfer on fibrosis (Fig. 3A). Gene transfer with
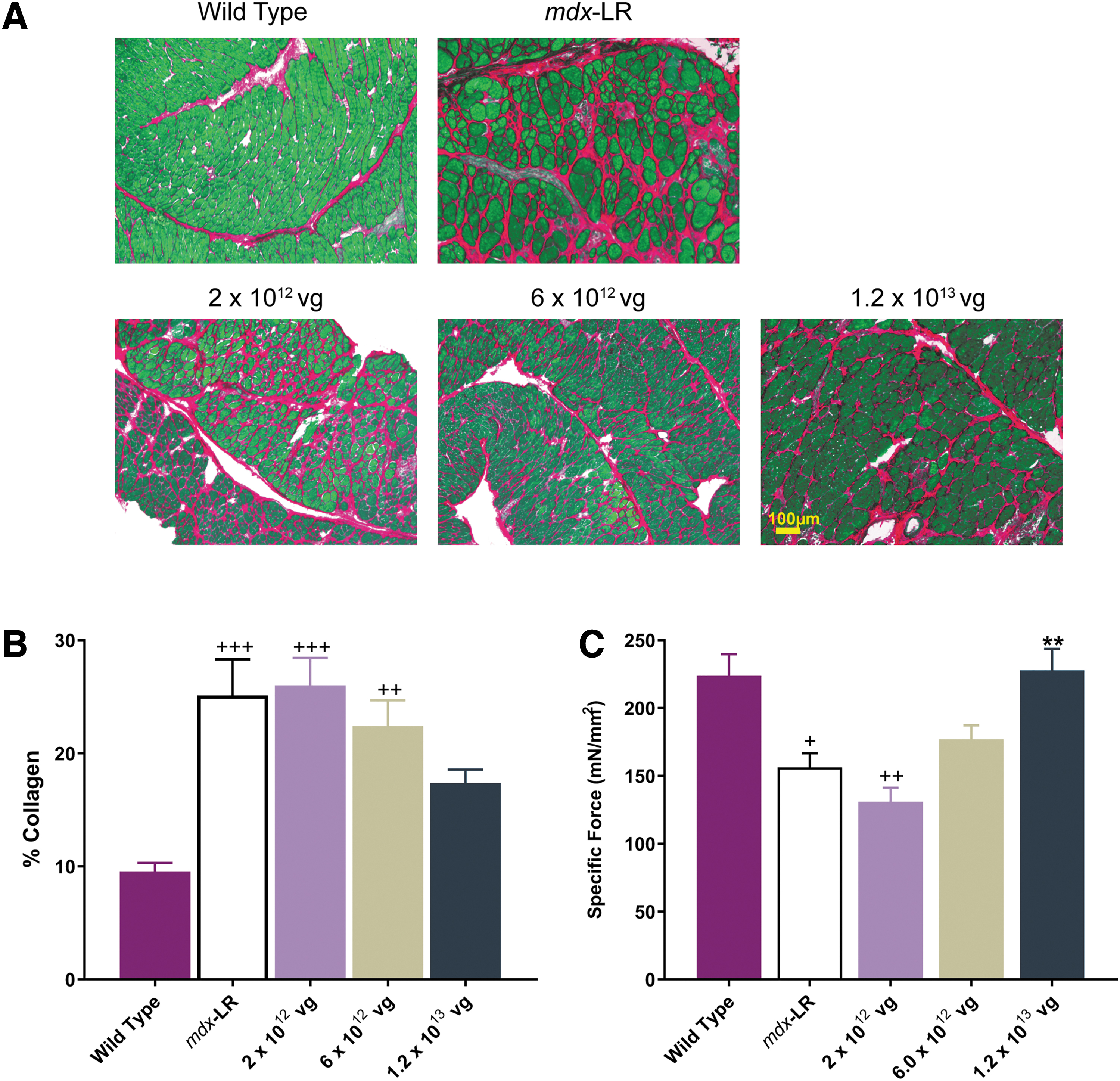
Reduction of fibrosis after systemic treatment with
Functional improvements with systemic delivery in dose-dependent manner
To determine whether micro-dystrophin gene transfer provides a functional benefit to diseased muscle, we assessed the properties of both the DIA (Fig. 3C) and TA (Fig. 4A, B) muscles from WT mice, mdx-LR mice, and
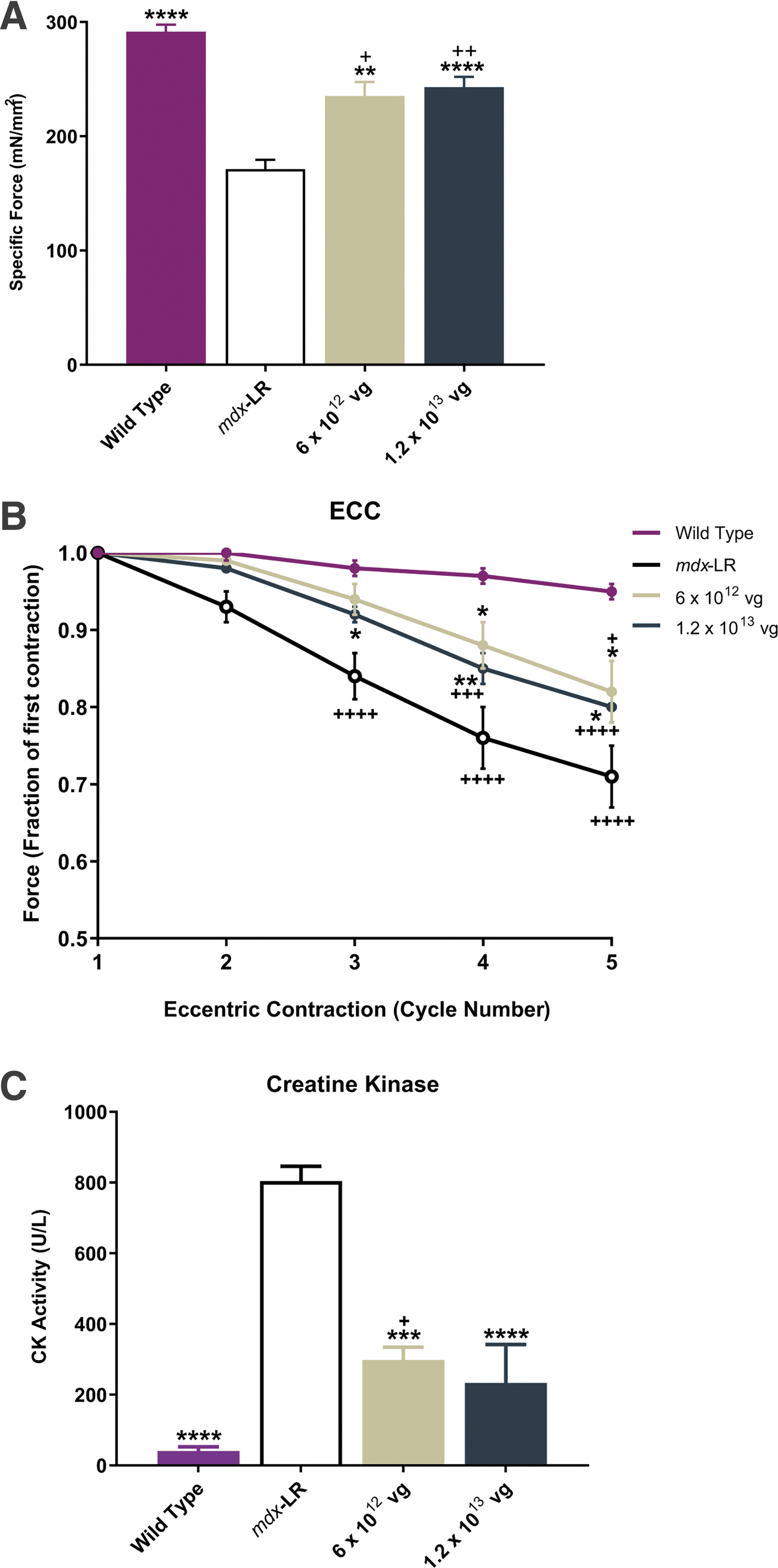
Functional benefits to skeletal muscle in intermediate- and high-dose cohorts.
In addition to sarcoglycan proteins, we also evaluated other DAPC-associated components, such as nNOS, using western blot analysis. Although the micro-dystrophin construct does not contain spectrin repeats 16 and 17, which have been identified as one of the sarcolemmal nNOS-specific sites, 49,50 when analyzing nNOS protein expression by western blot for all muscles analyzed in treated animals (n = 2 analyzed), we found the amount of nNOS protein in skeletal muscle to be equal to the amount found in WT TA tissues and increased compared to the untreated mdx mouse (Fig. 5A).
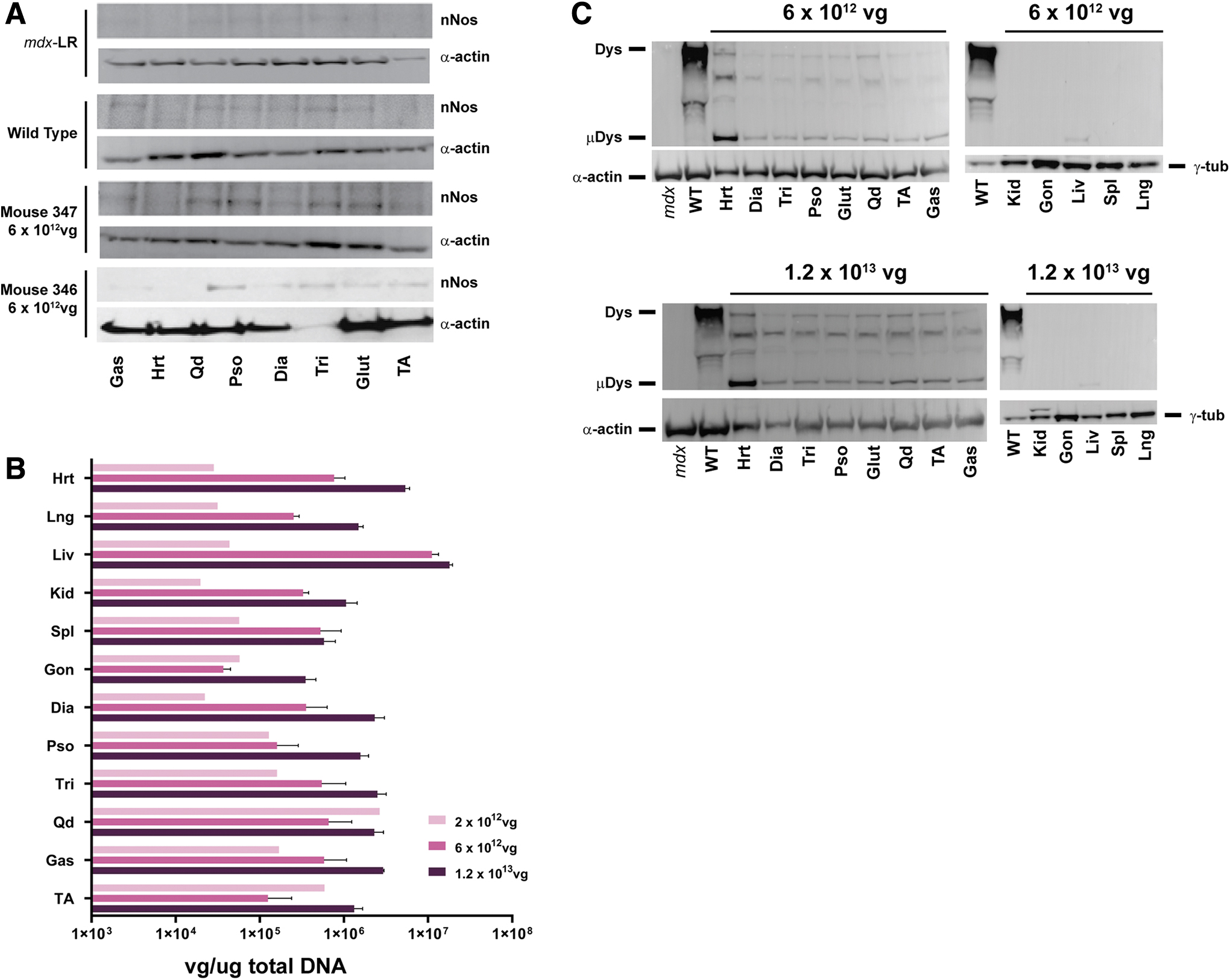
Biodistribution of vgs and protein expression in vector-treated mice.
Vector biodistribution following systemic delivery
To confirm the presence of test article-specific DNA sequences, biodistribution analysis was examined using real-time qPCR. Figure 5B depicts the vg copies detected in each tissue sample from
Durable improvements following a single systemic infusion
A cohort of mdx male mice were injected with an intermediate dose (6 × 1012 vg) at 4 weeks of age and necropsied at 6 months postinjection to evaluate long-term expression and functional benefit of a single intravenous treatment with
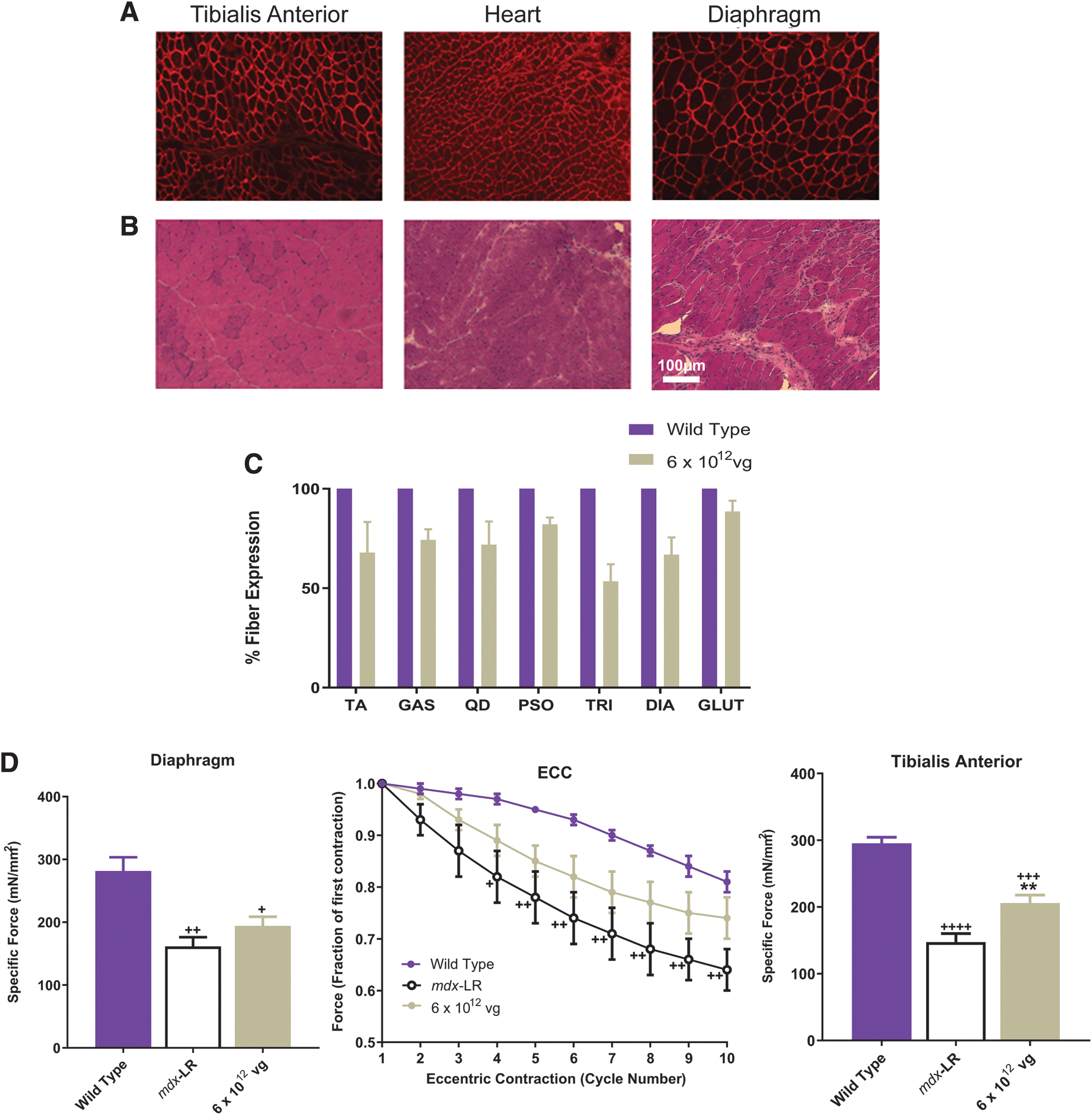
Long-term durability after a single administration of
Discussion
We have designed a therapeutic human micro-dystrophin cassette packaged into an rAAVrh74 vector, which includes the use of a dual striated muscle-specific promoter to selectively express transgene in skeletal and cardiac muscle. rAAVrh74 is a serotype recently isolated from lymph nodes of rhesus monkeys. rAAVrh74 displays high affinity for skeletal and cardiac muscle tissue, 51,52 and we have previously demonstrated robust biopotency using rAAVrh74 in biodistribution, delivery, and transduction of dysferlin, GALGT, and sarcoglycan proteins for other muscular dystrophies. 36,51 –54 In addition, rAAVrh74 seroprevalence is relatively low among other AAV serotypes, 55 which thus allows for application for more patients. Currently, the rAAVrh74 capsid is the vector that we have used in our gene therapy clinical trials in patients with DMD and with limb-girdle muscular dystrophy types 2E/R4, 2D/R3, and 2B/R2 (NCT02376816, NCT03769116, NCT03652259, NCT01976091, NCT02710500).
The safety and efficacy of AAV gene therapy for neuromuscular diseases are not only dictated by vector delivery (e.g., biodistribution and tissue uptake) but also by regulation of transgene expression in the targeted tissue (i.e., skeletal and cardiac muscle). Use of selective promoters is essential to maximize gene expression in targeted tissues and limit expression in nonrelevant tissues. In this study, we used MHCK7, which includes an α-MHC enhancer that has been shown to lead to high levels of expression in cardiomyocytes and increased dystrophin expression in skeletal muscle compared to muscle-specific promoters, MCK or CK7. 32 Indeed, we provide further support that MHCK7 limits off-target transgene expression, as demonstrated by widespread tissue and organ biodistribution of vector genomes, accompanied by micro-dystrophin protein expression limited to skeletal and cardiac muscle. Our findings presented here are consistent with our previous studies that demonstrated robust skeletal and cardiac muscle expression of β-sarcoglycan in mice after systemic delivery of the transgene using the rAAVrh74 capsid with expression driven by MHCK7. 39
In the present study we provide further evidence that demonstrates the translational relevance of systemic delivery of
It must be noted that given the limited DNA packaging capacity of AAV, numerous efforts to develop and evaluate shortened variations of dystrophin have been examined in preclinical animal models. While many constructs have shown success in delivery, varying degrees of expression and functional efficacy have been reported (for review, see Duan 56 ). We chose to use a transgene sequence designed to result in production of a protein with optimized functionality. Indeed, the transgene utilized here (ΔR4-23/ΔCT) has been shown to prevent and reverse dystrophic pathology associated in mdx mice and protect against damage due to eccentric contractions when delivered using AAV2, AAV6, or AAV8 with expression driven by the cytomegalovirus (CMV) or the muscle-specific promoter, MCK. 34,57,58 This variant of micro-dystrophin includes the N-terminus; spectrin-like repeats R1-3 and R24; hinges 1, 2, and 4; and the cysteine-rich domain. We believe that the ability of R1-3 to interact with the lipid membrane 29,30 may play an important role in maintaining structural integrity and protection of muscle tissue from damage.
In addition, inclusion of hinges is important for conferring flexibility by providing the structural mechanism for dystrophin to function as a shock absorber. 24 Prior studies reported that the presence of hinge 3 in ΔR4-23/ΔCT rescued muscle deterioration and structural deficits at neuromuscular junctions in mdx mice. 58 However, there were no differences in restoration of muscle functional capacity between hinge 2 and hinge 3 when configured in micro-dystrophin constructs ΔR4-23/ΔCT and ΔH2-23/ΔCT+H3 in mdx mice. 58 Banks et al. reported the formation of ringed fibers in mdx mice expressing ΔR4-23/ΔCT packaged into AAV6 with expression of the transgene driven by the constitutive CMV promoter. 58 In the present study, we did not observe any instance of ringed fibers in any vector-dosed tissues in mice. The reason for this discrepancy is unclear but could be due to differences in the constructs or technical differences between studies (e.g., age of mice, route of administration). Use of hinge 2 has the advantage of maintaining the natural linkage to the R1–3 domain, whereas use of hinge 3 would create a novel junction. Novel juxtapositions of protein domains may promote immune responses 59 or result in protein misfolding, which in turn may either promote degradation 60,61 or result in formation of protein inclusions. 34,62
It has been reported that nNOS binding domain R16–17 of dystrophin is essential to restore sarcolemmal nNOS expression in mdx mice and canine DMD models, as demonstrated by enhanced muscle perfusion, prevention of functional ischemia, and improvement in muscle force and exercise capacity. 19,28,48,63 –65 In contrast, several lines of evidence have also demonstrated that while lack of nNOS expression may be sufficient to promote deterioration, its presence is not required for muscle function or protection from overt dystrophy. For example, mice with genetic loss of nNOSμ did not exhibit skeletal muscle fatigue or loss of specific muscle force, 66 and forced expression of nNOSμ only partially rescued eccentric force loss in mouse models of DMD compared to WT animals. 67 Likewise, the presence of the R16–17 domains in micro-dystrophin constructs only partially protected the eccentric force loss observed in mdx mice. 28 Furthermore, transgenic mouse models that harbor genetic loss of nNOS expression do not overtly display a dystrophic phenotype. 68,69
In the present study, we analyzed nNOS expression for two treated mice from the intermediate-dose cohort, and the results show increased quantities of nNOS protein in all skeletal muscles compared with mdx-LR mice. These results are preliminary and further studies are needed, but suggest that nNOS protein presence in the muscle rather than increased nNOS sarcolemma-specific localization is beneficial for function, as has been previously suggested. 16,17
Altogether these studies, in conjunction with the present findings, demonstrate the importance in design and configuration of micro-dystrophin.
In addition to efficacy, this dose-escalation study demonstrated minimal toxicity when mice were treated with
In conclusion, the findings presented here provide proof of principle for safety and efficacy to support systemic delivery of
Footnotes
Acknowledgments
The authors thank the Nationwide Children's Viral Vector Core for Vector Production and Dr. Stephen Hauschka for his gift of the MHCK7 promoter. The authors also thank Terri Shaffer, MLAS, RLATG, for performing intravenous tail vein injections. Medical writing and editorial support were provided by Khampaseuth Thapa, PhD, and Lucia Quintana-Gallardo, PhD, of Sarepta Therapeutics, Inc., and Purvi Kobawala Smith, MS, MPH, of Health & Wellness Partners, LLC, Upper Saddle River, NJ, funded by Sarepta Therapeutics, Inc.
Authors' Contributions
Conceptualization, R.A.P., J.R.M., and L.R.R.-K.; Methodology, R.A.P., D.A.G., and L.R.R.-K.; Formal analysis, R.A.P; Investigation, R.A.P, D.A.G., K.N.H, E.L.P., and E.K.C.; Resources, R.A.P, D.A.G, L.R.R.-K.; Writing—original draft, R.A.P., and L.R.R.-K.; Writing—review & editing, R.A.P., D.A.G, K.N.H, J.R.M., and L.R.R.-K.; Visualization, R.A.P, D.A.G., J.R.M, and L.R.R.-K.; Supervision, L.R.R.-K.; Project administration, D.A.G.; Funding acquisition, J.R.M. and L.R.R.-K.
Author Disclosure
R.A.P. is an employee of Sarepta Therapeutics, Inc., D.A.G. is an employee of Sarepta Therapeutics, Inc., K.N.H. and E.K.C.: No competing financial interest exists. E.L.P. is an employee of Sarepta Therapeutics, Inc., J.R.M. is the coinventor of the rAAVrh74.micro-dystrophin technology. This technology has been exclusively licensed to Sarepta Therapeutics, Inc., L.R.R.-K. is the coinventor of the AAVrh74.micro-dystrophin technology and eligible to receive financial consideration as a result. L.R.R.-K. is an employee of Sarepta Therapeutics, Inc.
Funding Information
This study was funded by the Nationwide Children's Hospital Research Foundation, Parent Project Muscular Dystrophy, and Sarepta Therapeutics, Inc.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.