
Abstract
T cells made with messenger RNA (mRNA) encoding chimeric antigen receptor (CAR) offer a safe alternative to those transduced with viral CARs by mitigating the side effects of constitutively active T cells. Previous studies have shown that mRNA CAR T cells are transiently effective but lack persistence and potency across tumor types. It was hypothesized that the efficacy of mRNA CARs could be improved by utilizing recent advancements in RNA technology, such as incorporating a modified nucleoside, 1-methylpseudouridine, into the mRNA and applying a novel purification method using RNase III to eliminate dsRNA contaminants. T cells electroporated with nucleoside-modified and purified mRNA encoding CD19 CAR showed an initial twofold increase in CAR surface expression, as well as a twofold improvement in cytotoxic killing of leukemia cells that persisted up to 5 days. T cells generated with nucleoside-modified and purified CAR mRNA also showed reduced expression of checkpoint regulators and a differential pattern of genetic activation compared to those made with conventional mRNA. In vivo studies using a leukemia mouse model revealed that the most robust 100-fold suppression of leukemic burden was achieved using T cells electroporated with purified mRNAs, regardless of their nucleoside modification. The results provide a novel approach to generate mRNA for clinical trials, and poise mRNA CAR T cells for increased efficacy during testing as new CAR targets emerge.
Introduction
H
One drawback of current CAR technology is that gene transfer into human T cells has primarily relied on a viral- or transposon-based vector. While this approach has proven successful, it also has several limitations due to the permanent genetic modification of the cells. These include the risk of insertional mutagenesis, as has been seen in trials for severe combined immunodeficiency and Wiskott–Aldrich syndrome using retroviral vectors in hematopoietic stem cells, 12,13 a time-consuming and expensive viral manufacture process difficult for iterative trials that incorporate evolving CAR design innovations, 14 and perhaps most importantly, the risk of persistent untoward side effects. The most often-reported side effect is cytokine release syndrome (CRS), which results from over-activation of the T cells and subsequent bystander activation of the innate immune system with increased cytokine production. 15 –17 CRS can be fatal, such as in the recent reports of cerebral edema. 18 In addition to CRS, severe side effects and death have also resulted from on-target off-tumor antigens of healthy cells that are being attacked by the constitutively active T cells. 19 –23 This last concern is particularly important, as many target antigens are not unique to the tumor itself.
To avoid viral vectors, several recent studies have used mRNA electroporation to express CAR molecules on T cells transiently, which has been successful in demonstrating antitumor activity. 24 –28 In vitro–transcribed (IVT) mRNA encoding CAR (mRNA CAR) offers a cost- and time-effective alternative to retroviral constructs, and its transient nature allows for dose escalation by multiple injections, without the risk of permanent T-cell activity. Mouse xenograft models of leukemia have been successfully treated with mRNA CAR therapy. 14,28 However, for solid tumors, there has been difficulty penetrating into the tumor, as the T cells become exhausted quickly and show limited ability to proliferate in the tumor microenvironment. 26,29,30 Despite these difficulties, clinical trials are currently underway utilizing mRNA CAR T cells, as their safety profile makes them the most attractive option for immunotherapy when side effects are unknown or pose a potential danger. Thus, mRNA CAR T cells need to be improved to enhance their ability for tumor penetration and cytotoxicity, as well as showing efficacy to give a reasonable estimate of target-specific toxicity.
Recent advancement of in vitro mRNA technology demonstrates strategies that can improve the translatability and stability of IVT mRNA, 31 which it was hypothesized would ultimately improve the antitumor activity of CAR T cells generated with mRNA. Naturally occurring modified nucleosides, such as pseudouridine (Ψ) and 1-methylpseudouridine (m1Ψ), allow the transfected mRNA to avoid immune stimulation and increase mRNA stability, leading to enhanced translational capacity. 32 –34 RNA purification to remove contaminating double-stranded RNA (dsRNA), which normally stalls translation, has been shown to result in further improvements, thus achieving maximal translation for longer duration. 35,36 The objective of this study was to combine these techniques to generate a more stable mRNA product, which would allow for a superior mRNA CAR T cell.
Methods
Generation of CAR constructs and mRNA
DNA of a third-generation CAR containing a scFv domain directed against CD19 linked to CD3ζ and 4-1BB intracellular signaling domains was generated, as previously described. 14,37 The CD19 CAR DNA was linearized, and then a MEGAscript T7 RNA transcription kit was used to synthesize the RNA. Four different mRNA isolates were generated. To synthesize the mRNA for the first group, the transcription reaction was supplemented with m1Ψ triphosphate (Trilink) in place of UTP. For the second group, the m1Ψ-containing mRNAs were purified by digesting with ribonuclease III (RNase III) (Epicentre), as described below. For the third group, mRNA was transcribed in the presence of UTP using standard methods followed by RNase III digestion. Finally, control mRNA was transcribed in the presence of UTP using standard methods without RNase III purification. MEGAscript T7 RNA transcription kit (Ambion, Thermo Fisher Scientific) was used to generate all RNA. To contain cap1, all mRNA was enzymatically capped with guanylyltransferase and 2′-O-methyltransferase (CellScript), and long polyadenylate tail was added using poly(A) polymerase (CellScript), according to protocols previously described. 38 RNA purification with RNase III for the two purified experimental arms was completed before capping and poly(A) tailing, using a protocol described below.
Purification of IVT mRNA using RNase III
To digest dsRNA contaminants present in the IVT mRNA sample, an aliquot of 2.5 μL of RNase III (Epicentre) diluted to 0.01 IU/μL in reaction buffer (33 mM of Tris, pH 8.0, 200 mM of potassium acetate, and 1 mM of magnesium acetate) was combined with 100 μg of mRNA in a final volume of 125 μL of reaction buffer and incubated at 37°C for 30 min. 39 Following a phenol-chloroform (pH 4.5; Thermo Fisher Scientific) and two chloroform extractions, the mRNA was precipitated from the aqueous phase by adding one tenth volume of 3 M sodium acetate, pH 5.5, and an equal volume of isopropanol. The centrifuged pellet was reconstituted in water and stored at −20°C. Verification of removal of dsRNA was completed using immunoblot assay, as previously described 35 and shown in Supplementary Fig. S1.
T-cell expansion and RNA electroporation
Human T cells were collected from de-identified healthy donors by the University of Pennsylvania Human Immunology Core and then stimulated with CD3/CD28 microbeads (Gibco), as previously described. 37 Stimulated and rested T cells were re-suspended at 5 × 106 cells in 100 μL of nucleofector reagent from the Nucleofector T-cell transfection kit (Lonza) prior to adding 10 μg of mRNA for electroporation. For in vitro studies, mRNA electroporation was performed using the Amaxa program T23 (Lonza). For in vivo studies, stimulated and rested T cells were electroporated with mRNA using ECM 830 Square Wave Electroporation System (BTX).
In vitro studies
A Nalm-6 leukemia cell line (obtained from DSMZ) permanently expressing firefly luciferase 40 was plated and incubated with the experimental CAR T cells at 10:1, 1:1, and 1:10 effector-to-target cell ratios. Tumor-specific T-cell cytotoxicity was measured using the Bright Glo Luciferase Assay System (Promega). Cytotoxic killing was measured as a decrease in luciferase activity and compared to killing of leukemia cells with triton detergent, which was set at 100%. T cells were simultaneously tested for CD19 CAR expression using a CD19 CAR-specific antibody (gift from Novartis) conjugated with R-Phycoerythrin AffiniPure F(ab′)2 Fragment Goat Anti-Human immunoglobulin G, Fcγ Fragment Specific, as well as the negative checkpoint regulators mouse monoclonal anti-human CD279 (PD-1) BV510 (BD Biosciences), mouse monoclonal anti-human CD223 (Lag-3) PerCP-eFluor 710 (eBioscience), and mouse monoclonal anti-human TIM-3 (CD366) BV421 (BD Biosciences). Flow cytometry acquisition was performed with a BD FACSVerse (BD Biosciences), and analysis was done with FlowJo (Treestar, Inc.). The luciferase assays and antibody testing were completed daily over 8 days to evaluate for persistence of CAR expression and T-cell viability among the different CAR T-cell constructs. T cells were counted throughout using the Moxi Z mini automated cell counter (Orflo). Experiments were repeated at least three times with different batches of mRNA and T-cell donors to ensure accuracy.
Mouse xenograft studies
Studies were performed in 6- to 10-week-old NOD-SCID-γc–/– (NSG) mice obtained from Jackson Laboratories or bred in-house under an approved protocol and maintained under pathogen-free conditions. Mice were injected via the tail vein with 1 × 106 primary leukemia cells transformed with luciferase (CHP101) in 0.1 mL of sterile phosphate-buffered saline (PBS), as previously described. 40 After achieving appropriate leukemic burden at 10 days, mice were injected via the tail vein with 2 × 107 CAR T cells in 0.1 mL of sterile PBS. T cells had been transfected with RNA 4 days prior and incubated at 37°C before injection. Mice were then evaluated frequently for persistent leukemia by optical imaging, as previously described. 40 Animals were closely monitored for the signs of xenogeneic graft-versus-host disease and other toxicity, and all efforts were made to minimize suffering. All animal studies were approved by the Institutional Animal Care and Use Committee of the Children's Hospital of Philadelphia (IAC 15-000232).
Gene expression analysis
To evaluate the CAR T-cell constructs for differences in immune activation following transfection with the various mRNAs, Nanostring RNA sequencing technology (Nanostring Technologies) was utilized to investigate gene expression. CAR T cells made from the experimental mRNA groups were lysed, and RNA was extracted using Quick-RNA Miniprep (Zymo Research) on days 1 and 4 post transfection. Isolated RNA (25–50 ng) was then hybridized with the nCounter PanCancer Immune Profiling Panel, which includes 770 genes involved in immune responses, cell cycle, and function. Gene expression data were then acquired on the nCounterTM Sprint Profiler v3.0 and evaluated using nSolver software (Nanostring Technologies). Gene expression studies were repeated twice with different mRNA batches and T-cell donors to ensure accuracy.
Statistical analysis
Statistical analysis was performed with Prism v7 (GraphPad Software). In vitro cytotoxic killing data represent means of triplicate, and comparisons of means were made via a t-test. Flow cytometry data were evaluated using mean fluorescence intensity, with confidence intervals generated by FlowJo, and statistical comparison was completed with a two-way analysis of variance (ANOVA), as well as Fisher's exact test for comparison of cell populations. For in vivo data, comparison of radiance was evaluated using a two-way ANOVA for each group on each day.
Results
Transfection of purified and m1Ψ-modified mRNA leads to increased CAR expression in T cells and cytotoxic killing in vitro
First, the various mRNA CARs were evaluated for CAR expression and cytotoxic killing efficacy in vitro over time. One day after transfection, the addition of purification and/or nucleoside-modification to mRNA induced a 1.5- to 2-fold increase in CAR expression compared to those mRNAs prepared by conventional techniques (Fig. 1A). CAR mean fluorescence intensity (MFI) for mRNA prepared by conventional techniques was 53,563 for day 1 compared to 88,981 for nucleoside modified m1Ψ-mRNA (p < 0.0001), 76,296 for purified m1Ψ-mRNA (p < 0.0001), and 95,535 for purified U-mRNA (p < 0.0001). CAR expression decreased rapidly over the subsequent days, with the purified U-mRNA and purified m1Ψ-mRNA falling to similar expression levels as the original mRNA CAR by day 2, and a slower decline for m1Ψ-mRNA, which normalized with U-mRNA at day 4. All groups showed a small amount of CAR expression persisting until day 6 post-transfection.
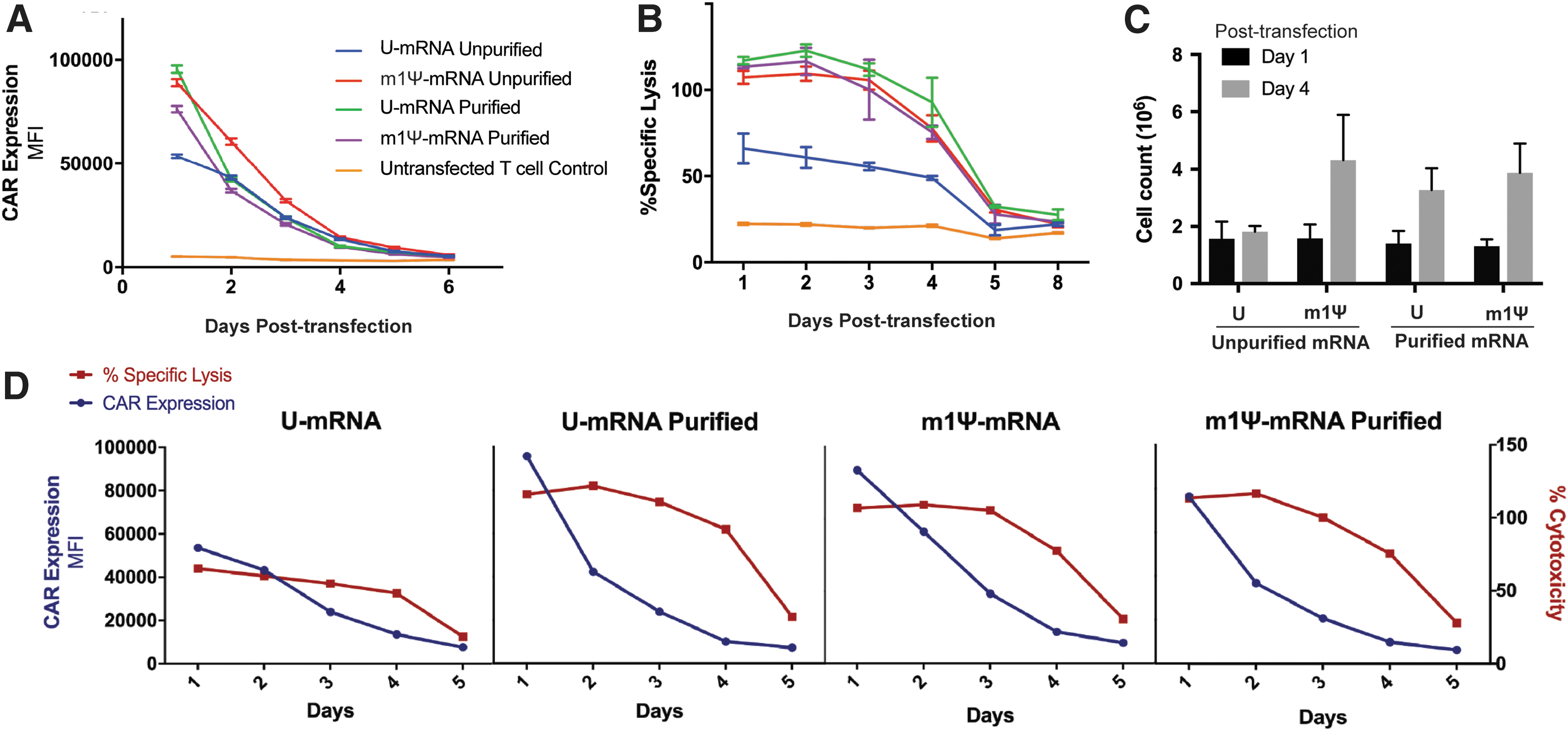
High expression of chimeric antigen receptor (CAR) and improved cytotoxicity of T cells transfected with nucleoside-modified and purified mRNA CAR.
In parallel, the cytotoxic killing potential of the various mRNA CARs was evaluated against Nalm 6 leukemia cell lines in vitro. T cells electroporated with purified U-mRNA, m1Ψ-mRNA, and purified m1Ψ-mRNA encoding CARs all showed a twofold increase in cytotoxic killing compared to the conventional, unpurified U-mRNA CAR, with improved efficacy that persisted up to 5 days (Fig. 1B). When evaluated together, the novel CAR mRNAs showed a significant and persistent increase in cytotoxic killing efficacy, despite rapidly decreasing CAR expression (Fig. 1D), suggesting that the improved cytotoxic effect was mediated by additional factors other than improved CAR expression alone. Investigation of proliferative capacity showed the experimental groups containing purified and/or m1Ψ-mRNA CAR T cells doubled in growth (p < 0.05) compared to U-mRNA CAR T cells that remained overall stagnant (Fig. 1C).
T cells transfected with m1Ψ-modified or purified mRNA encoding CAR express lower levels of checkpoint regulators
As T cells undergo activation, they express a series of negative checkpoint receptors that by binding to their ligands inhibit cell growth and decrease the proliferative capacity of T cells, leading to exhaustion. The first of these receptors to appear is programmed cell death protein 1 (PD-1), which is expressed when the T cell is initially activated. 41 Subsequently, additional markers such as lymphocyte activation gene 3 (Lag-3) and T-cell immunoglobulin mucin-3 (Tim-3) are expressed, and when T cells exhibit two or more of these markers, they are highly susceptible to inhibition and reach the end of their life-span. 41,42 PD-1 and Lag-3 expression was measured in the mRNA CAR T cells over time after transfection (Fig. 2). Initially, PD-1 and Lag-3 were co-expressed at similar levels on all T-cell groups on day 1, as the shock of electroporation itself activated a proportion of T cells likely via type I interferon responses 43 and eIF2alpha phosphorylation, 44 resulting in expression of multiple negative checkpoint regulators. On day 4, after the effect of electric shock subsided, T cells that had been transfected with purified or m1Ψ-modified CAR mRNA exhibited less dual expression of PD-1 and Lag-3 compared to those transfected with the unpurified U-mRNA (Fig. 2). The proportion of cells with dual expression of PD-1 and Lag-3 was 56.97% when unpurified U-mRNA was used compared to 8.29–15.63% when the purified and/or m1Ψ-modified mRNA was transfected (p < 0.0001). Measurement of Tim-3 showed minimal staining across all groups with similar expression compared to untransfected cells (p = 0.4391).
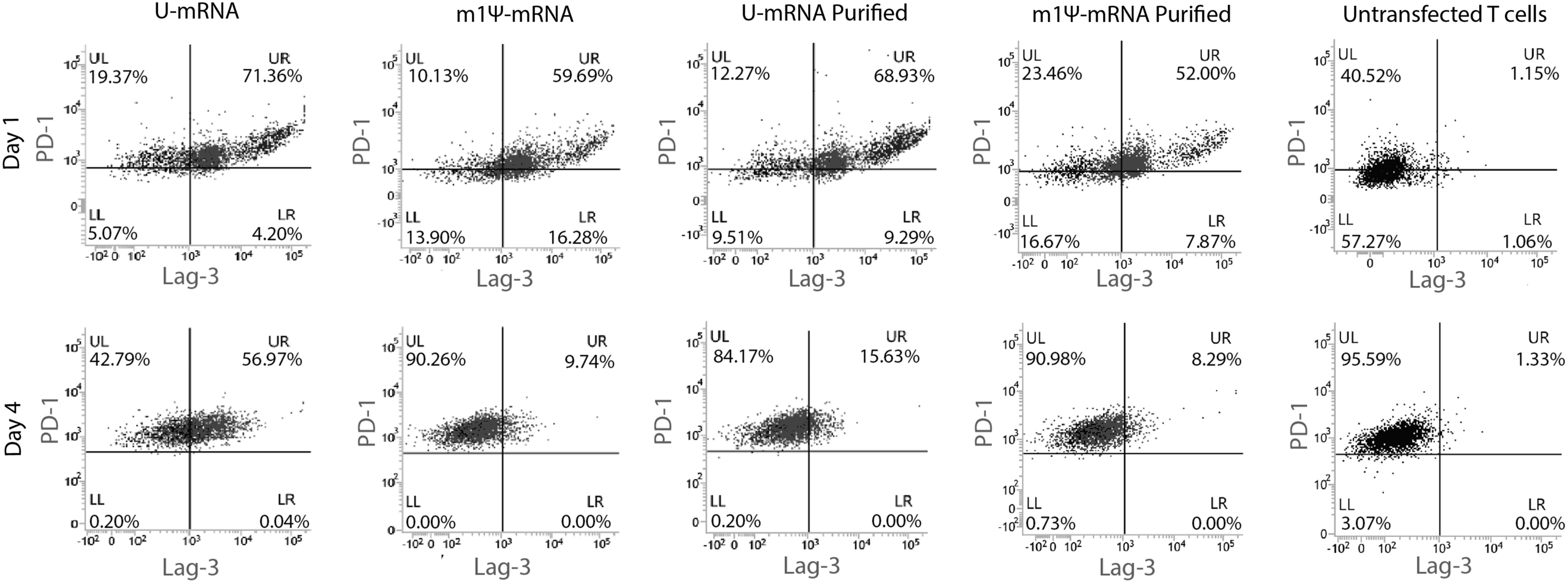
Low levels of negative checkpoint regulators on T cells transfected with nucleoside-modified and purified mRNA CAR T cells. Flow cytometry with gating on CD3 and CAR expressing T cells only. Day 1 shows similar values of programmed cell death protein 1 (PD-1) and lymphocyte activation gene 3 (Lag-3) double-positivity, as the transduction process activates and exhausts a portion of the T-cell population across all transfected groups. On day 4, there are less double-positive T cells in the cohort transfected with m1Ψ-modified and purified mRNA CAR, with T cells showing PD-1 positivity only.
Improved cytotoxic killing in vivo by T cells when generated with purified CAR mRNA
To assess anti-leukemic activity in vivo, mice injected with primary leukemia were subsequently injected with a single dose of 2 × 107 mRNA CAR T cells, saline, or untransfected T cells as a control. RNA CAR T cells had been transfected 4 days prior to injection and kept incubated at 37°C. Following a single dose of mRNA CAR T cells, leukemia progressed rapidly over the course of 1 month for control animals (Fig. 3A). Mice treated with CAR mRNA generated with unpurified U-mRNA had slightly reduced leukemic growth compared to controls, but they still had a two-log increase in leukemic burden over the course of the month (Fig. 3A). Among the improved CAR mRNA constructs, all three groups showed improvement in leukemic burden compared to both the control groups and U-mRNA CAR T cell–treated group (p < 0.05), with the two purified mRNA groups showing the most improvement. The U-mRNA purified CAR T cells as well as the m1Ψ-mRNA purified CAR T cells were successful in keeping the leukemic burden at its baseline, two orders of magnitude improved from the unpurified U-mRNA CAR T cell and control groups (p = 0.0138 and 0.0106, respectively; Fig. 3A and B).
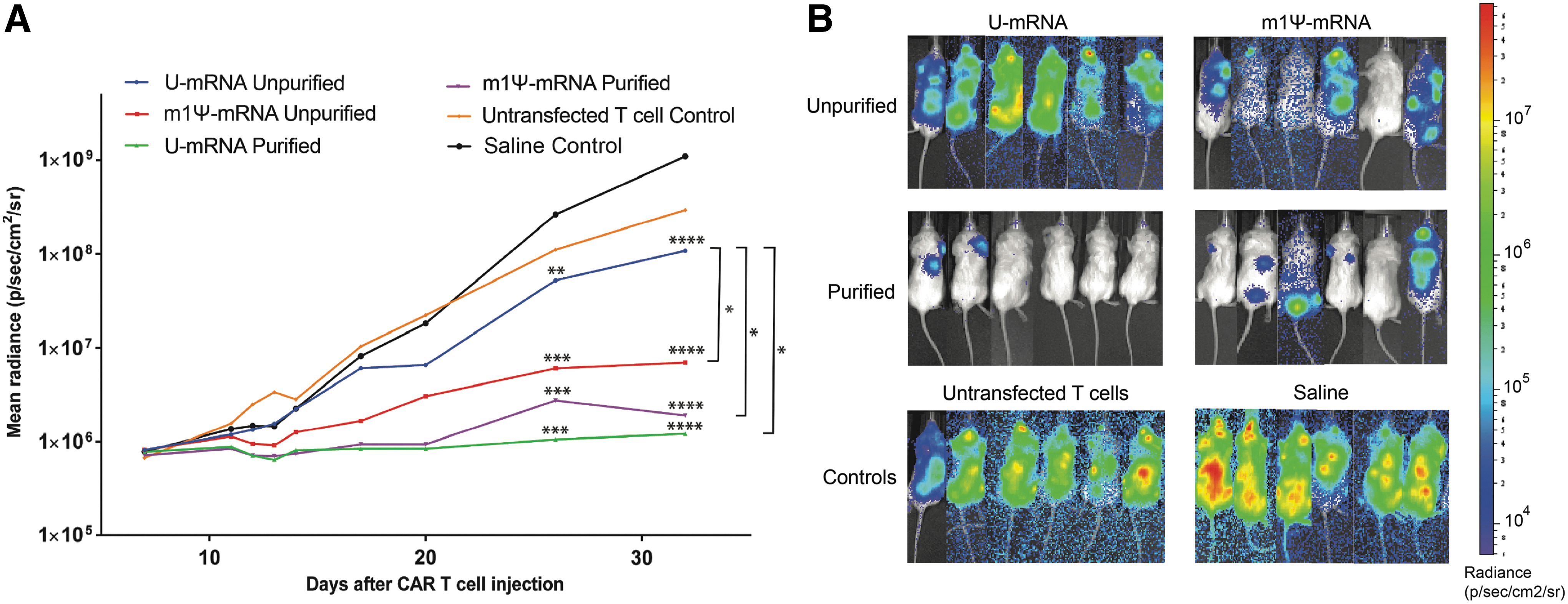
T cells electroporated with purified mRNA encoding CAR show the most efficient killing in vivo.
Distinct activation profile for purified and m1Ψ-modified mRNA CAR T cells
To investigate the underlying mechanism for the differences in cytotoxicity and negative checkpoint regulator expression in the CAR T cells, genomic expression results were evaluated across a panel of immune and cell function genes after transfection with selected mRNA constructs. Using the nCounter PanCancer Immune Profiling Panel, a heat map representation was created for multiple grouped genes such as those involved in overall cell function, T cell–specific function, encoding cytokines and chemokines (Supplementary Table S1). A pattern emerged showing overall increased expression of numerous genes in the unpurified U-mRNA, with generally decreased expression in both the purified and m1Ψ-mRNA cohorts (Fig. 4). While the CAR T cells containing m1Ψ-mRNA segregated with untransfected cells in their pattern of gene activation based on unsupervised clustering analysis, the purified U-mRNA CAR T cells appeared to have a distinct expression profile at a moderate level of gene expression between unpurified U-mRNA and untransfected cells. Type I interferons were downregulated in both the m1Ψ-mRNA-containing and purified groups compared to U-mRNA-containing T cells (Fig. 4). Among the groups of genes with increased activation in the unpurified U-mRNA T cells included Toll-like receptors (TLR) and the tumor necrosis factor superfamily (TNFSF; Fig. 4). Genes induced in this family in the U-mRNA T cells included pro-apoptotic proteins Fas, TNSF12, Death receptor 5 (DR5), and tumor necrosis factor receptor superfamily member 10B (TNFRSF10B), as well as genes normally not expressed in T cells including TNFSF15 (Fig. 4). This clustering pattern of over-activation in the unpurified U-mRNA T cells was persistent across profile panels (Fig. 4).
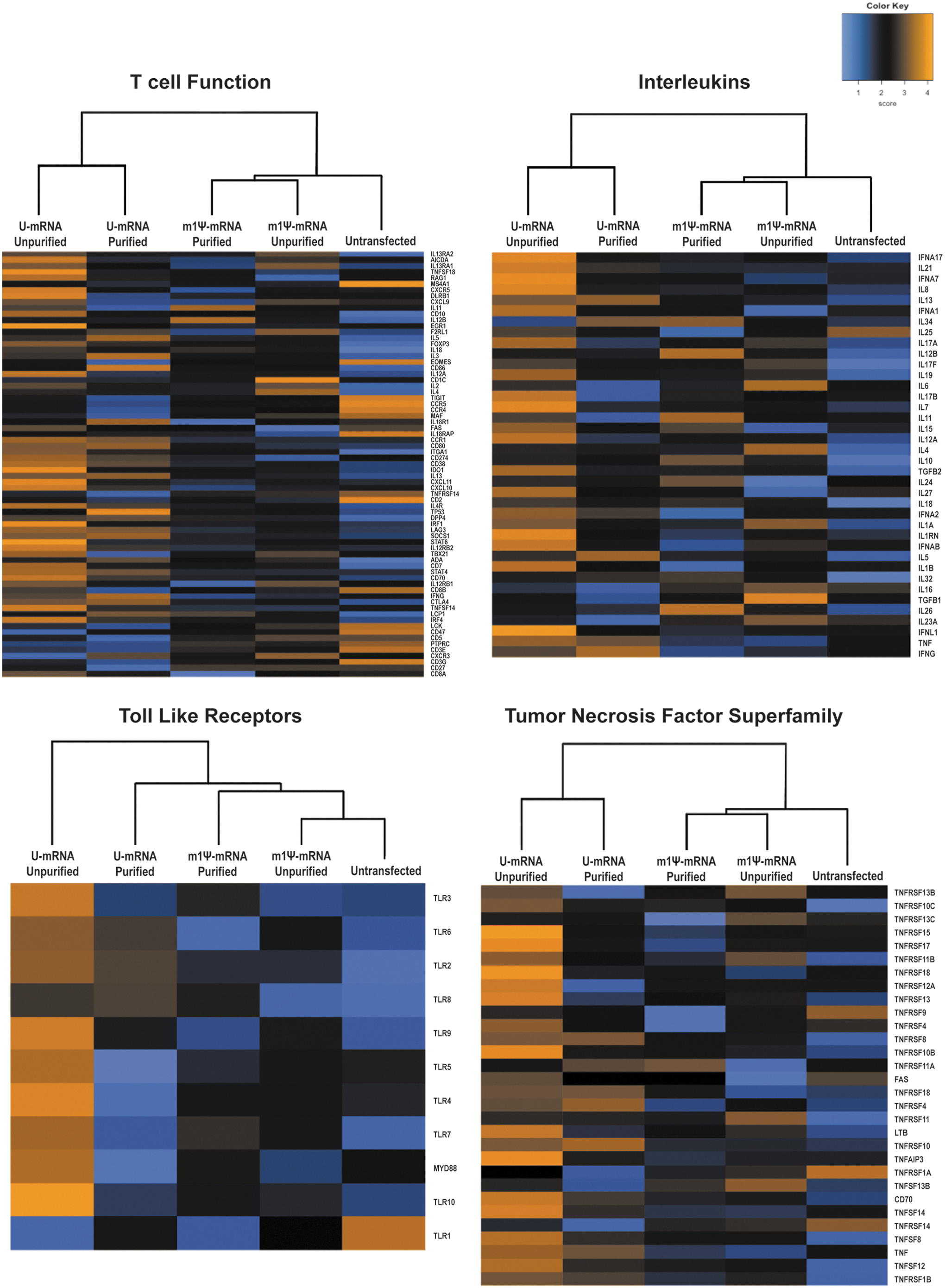
Gene expression profiles of T cells reveal differential pattern of activation by mRNA CAR constructs. T-cell function, interleukins, toll-like receptors, and tumor necrosis factor superfamily RNA sequencing data are displayed at day 4 after transfection with CAR mRNA. Unpurified U-mRNA transfected cells show the highest expression across all profiles. Heat maps include normalized RNA sequencing data and are scaled to give all genes equal variance. Orange represents high expression; blue represents low expression. The experiment was repeated with a different batch of mRNA and T-cell donor with identical clustering.
Discussion
While immunotherapy has emerged as a major breakthrough in the treatment of cancer, there remains a continued need for improvement and advancement of the current techniques. In particular, CAR T-cell therapy investigators are actively searching for novel tumor antigens, each with as yet undetermined potential toxicities related to normal tissue expression, which magnifies the need for a transiently expressed CAR that can provide both an evaluation for efficacy against tumor, as well as a transient mechanism to evaluate for toxicity. Several groups have investigated the use of alternate means to create a transient CAR, such as incorporating the inducible Caspase 9 suicide gene. However, prior work using this model in T cells has shown re-expansion of the T-cell population over time 45 and failure of the suicide mechanism, 46 as it is difficult to ensure that all T cells have the incorporated gene or that the agent given to activate the suicide gene reaches all cells. As a direct consequence, it is difficult to assure that all CAR T cells would truly be eliminated using this approach. mRNA is unique in its ability to offer a truly transient expression of CAR. However, prior work has shown only modest efficacy with clear limitations when applied across multiple tumor types. 29,30,47 This report demonstrates that elimination of dsRNA contaminants by introducing a digestion with RNase III resulted in pure single-stranded CAR mRNA, which when transfected to T cells led to robust improvement in the efficacy of tumor killing, both in vivo and in vitro. With continued transient expression, this new CAR mRNA is an improved platform for novel investigations in CAR therapy as well as other therapeutic applications of mRNA.
The improved efficacy that was found in vivo and in vitro throughout these experiments exceeded that expected in the context of the CAR expression profile. While it was possible to show initial improved CAR expression on the surface of T cells using these new mRNA techniques, CAR expression rapidly declined to a level comparable to the conventional CAR construct, as the T cells proliferated and mRNA was diluted and degraded. However, despite similar CAR expression over time, the new mRNA constructs offered robust improved potency. This increase in potency was attributable to two findings. First, the updated mRNA resulted in an increased number of T cells due to increased proliferative capacity, resulting in rapid decrease of CAR expression and a sheer increase in volume of effector-to-target cells. In this context, cytotoxicity was enhanced. In addition, a decrease in the level of negative checkpoint receptors was also observed in T cells transfected with the improved mRNA constructs. Those T cells appeared to be activated without overstimulation to the point of expression of multiple negative checkpoint receptors, enabling the T cells to proliferate and extending their cytotoxicity through cytokine release and directed killing compared to those generated by prior methods. This result is supported by the genomic expression profiling, which reveals an over-activation of U-mRNA containing T cells compared to m1Ψ and purified mRNA containing T cells. Interestingly, a different gene expression profile emerged for the two purified constructs, U and m1Ψ- mRNA, which was surprising, as the two purified constructs performed similarly in vivo. Most likely, this difference in gene expression is related to the broad array of genes investigated in the panel, with only a select few genes being most important in determining T-cell cytotoxic function. Pattern recognition receptors (PRR) activation including TLR (Fig. 4) appeared to be similar among the purified constructs, making these a likely candidate for the enhanced efficacy. Future studies will continue to investigate these genes and opportunities for maximizing T-cell cytotoxicity, such as the use of co-delivery of PRR inhibitors.
The in vitro and in vivo model showed that purification alone of the CAR mRNA was sufficient to achieve maximal T-cell cytotoxicity. During purification, dsRNA, the major contaminant in IVT mRNA, was eliminated by dsRNA-specific RNase III digestion. DsRNA, which activates innate immune receptors, can lead to type I interferon secretion and inhibition of translation. 35 Similarly, unmodified mRNA also activates the innate immune system, as immune cells detect unmodified nucleosides in RNA. 34 The gene expression data showed decreased type I interferon expression across all of the purified mRNA constructs. In the in vitro studies presented here, as well as in a prior investigation where purified, dsRNA-free IVT RNAs were tested, removal of contaminants led to improved translation, but with an additional improvement when the purified mRNA contained modified nucleosides. 35 In the present study, both purified U-mRNA and purified m1Ψ-mRNA performed equally well in vivo compared to slightly decreased efficacy of m1Ψ-mRNA alone. This discrepancy is likely due to the use of electroporation for delivery of the mRNA into T cells. Kirschman et al. 48 recently showed that when using electroporation for mRNA delivery, the nucleoside-modified and unmodified mRNAs translated equally, which is in contrast to transfection of formulated mRNA where modifications in mRNAs play a significant role in improving their translation. Unlike formulated mRNA, the electroporated naked mRNA enters directly into the cytoplasm, thus avoiding endosomally located TLR7 and TLR8, both of which are activated by uridine-containing ssRNA or uridine, respectively. 49,50 Although the dsRNA contaminants present in the electroporated mRNA also bypass the endosome and its receptor, TLR3, there are many different types of dsRNA sensors and effectors in the cytoplasm such as RIG-I, MDA5, PKR, and OAS. 51 Activation of these dsRNA-dependent proteins induces interferon response and inhibition of protein synthesis. Removal of dsRNA with RNaseIII treatment thus contributed to the superior performance of the T cells electroporated with purified mRNA CAR.
The impact of purification with RNase III on the translational performance of IVT mRNA has never been reported. To the authors' knowledge, this is the first report in which T cells were transfected ex vivo with purified and nucleoside-modified IVT mRNA and then characterized in vivo. Prior studies have used pseudouridine-containing IVT mRNA to generate induced pluripotent stem cells 52 and to transfect ex vivo into mesenchymal stem cells that were subsequently evaluated in vivo. 53 In these studies, Ψ-mRNA was superior to U-mRNA, 52,53 most likely because the IVT mRNAs were formulated prior to transfection. In recent studies, m1Ψ-modified and purified IVT mRNA was formulated and then injected into mice directly to generate T cell-engaging bi-specific antibodies, and no adverse reactions were detected. 54 This report is the first to apply purified and nucleoside-modified IVT mRNA in chimeric antigen receptor technology.
As new mRNA CAR clinical trials are currently underway for both hematologic and solid tumor malignancies (NCT01837602, NCT03060356, and NCT02624258), incorporation of the present findings into the manufacturing of mRNA CAR T cells becomes even more paramount to achieve the aims of the trials. This work demonstrates that utilizing purification can vastly improve the efficacy of mRNA CAR T cells and reduce the overstimulation of T cells during the electroporation process. It is anticipated that this purification technique will be rapidly translatable to clinical trials and large-scale applications, as well as enhancing the platform for evaluating novel chimeric antigens in both preclinical and clinical testing.
Footnotes
Acknowledgments
Research Supported by St. Baldrick's Stand Up To Cancer Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT-27-17; J.B.F. and A.C.R.). Stand Up to Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. This work was also supported by the St. Baldrick's Foundation and the Gabrielle's Angels Foundation for Cancer Research (D.M.B.).
Author Disclosure
S.A.G. has a consulting agreement with and receives research funding from Novartis. K.K. is currently employed by BioNTech RNA Pharmaceuticals. None of the other authors have any conflicts of interest to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.