
Abstract
B-cell maturation antigen (BCMA) expression has been proposed as a marker for the identification of malignant plasma cells in patients with multiple myeloma (MM). Nearly all MM tumor cells express BCMA, while normal tissue expression is restricted to plasma cells and a subset of mature B cells. Consistent BCMA expression was confirmed on MM biopsies (29/29 BCMA+), and it was further demonstrated that BCMA is expressed in a substantial number of lymphoma samples, as well as primary chronic lymphocytic leukemia B cells. To target BCMA using redirected autologous T cells, lentiviral vectors (LVV) encoding chimeric antigen receptors (CARs) were constructed with four unique anti-BCMA single-chain variable fragments, fused to the CD137 (4-1BB) co-stimulatory and CD3ζ signaling domains. One LVV, BB2121, was studied in detail, and BB2121 CAR-transduced T cells (bb2121) exhibited a high frequency of CAR + T cells and robust in vitro activity against MM cell lines, lymphoma cell lines, and primary chronic lymphocytic leukemia peripheral blood. Based on receptor quantification, bb2121 recognized tumor cells expressing as little as 222 BCMA molecules per cell. The in vivo pharmacology of anti-BCMA CAR T cells was studied in NSG mouse models of human MM, Burkitt lymphoma, and mantle cell lymphoma, where mice received a single intravenous administration of vehicle, control vector–transduced T cells, or anti-BCMA CAR-transduced T cells. In all models, the vehicle and control CAR T cells failed to inhibit tumor growth. In contrast, treatment with bb2121 resulted in rapid and sustained elimination of the tumors and 100% survival in all treatment models. Together, these data support the further development of anti-BCMA CAR T cells as a potential treatment for not only MM but also some lymphomas.
Introduction
T
CAR T cells targeting CD19 in B-cell malignancies have demonstrated remarkable efficacy in several clinical studies. In acute lymphoblastic leukemia (ALL), response rates of up to 90% were reported, and in diffuse large B-cell lymphoma (DLBCL), response rates were similarly impressive at 70–80%. 14,15 These results were reported by multiple groups using different CAR constructs, delivery vectors, and cell manufacturing methods, indicating the robustness of the application. Promising initial results have also been reported in CLL and follicular lymphoma (FL), suggesting that most CD19-expressing B-cell malignancies could be targeted by this approach. Furthermore, results with anti-CD19 engineered T cells were superior to results reported with monoclonal antibody-based approaches, including bi-specific antibodies targeting CD19-CD3. 16
Disseminating CAR T therapies beyond patients with B-cell lymphoma and leukemia requires the identification of additional therapeutic molecular targets. In the case of multiple myeloma (MM), only a small fraction of tumor cells would be expected to express CD19 and thus be eligible for CD19-directed CAR T-cell therapy. 17,18 MM is characterized by the clonal expansion of malignant plasma cells in the bone marrow and remains an incurable disease, despite recent therapeutic advancements. BCMA (CD269, TNFSFR17) has been proposed as a potential target for CAR T cells in MM. 19 One feature of BCMA that makes it an attractive CAR T-cell target is its limited distribution in normal tissue. Several reports have documented BCMA expression on differentiated plasma B cells in normal lymphoid tissue (e.g., bone marrow, spleen, lymph nodes, and tonsil) and lack of expression on naïve B cells and other hematopoietic cells, including neutrophils, macrophages, and T cells, as well as any other normal tissue. 20 –23 Most significantly, BCMA is consistently expressed on malignant plasma cells from MM patients, and Carpenter et al. reported that anti-BCMA CAR T cells demonstrated specific activity restricted to BCMA-expressing targets (BCMA-expressing MM cell lines and patient MM samples). 23 –26
In addition to MM, BCMA expression has been identified in the Raji Burkitt lymphoma (BL) cell line 22,27 and primary lymphomas. 21,28 Therefore, it is possible that BCMA-specific CAR T cells could be used as a therapy in lymphomas as well as MM. This report demonstrates that anti-BCMA CAR T cells recognized low levels of BCMA expressed on lymphoma and leukemia cell lines (<250 BCMA molecules per cell), and demonstrated significant in vivo antitumor activity in both MM and lymphoma xenograft models. The implication of these data to the development of novel T cell–based therapeutics is discussed.
Materials and Methods
Cell lines and primary cells
The MM cell lines NCI-H929, U266-B1, and RPMI-8226 were obtained from American Type Culture Collection (ATCC; CRL-9068, TIB-196, and CCL-155, respectively). K562 is a chronic myelogenous leukemia cell line (ML; ATCC; CCL-243). K562.BCMA are K562 cells transduced with the gene for full-length BCMA, sorted by flow cytometry for high expression, and expanded from a single-cell clone in the authors' laboratory. Daudi and Ramos are BL cell lines (ATCC; CCL-213 and CRL-1596, respectively). NALM-6 and NALM-16 are ALL obtained from Deutsche Sammlung von Miroorganismen und Zellkulturer, GmbH (DSMZ; ACC-128 and ACC-680, respectively). REC-1 and JeKo-1 are mantle cell lymphoma (MCL) cell lines (ATCC; CRL-3004 and CRL-3006, respectively). HDLM-2 and RPMI-6666 are Hodgkin lymphomas (HL; DSMZ; ACC-17, and ATCC CCL-113, respectively). Leukapheresis product from healthy donors was obtained from Key Biologics, LLC. Peripheral blood mononuclear cells (PBMCs) were isolated and cryopreserved in the authors' laboratory. Whole blood from two CLL patients was obtained from Conversant Biologics, Inc.
Immunohistochemistry
Twenty-nine MM and 35 lymphoma biopsies were obtained as formalin-fixed, paraffin-embedded (FFPE) blocks (Cambridge Bio). Lymphoma samples (n = 7/subtype) were comprised of HL or NHL, including MCL, marginal zone (MZL), DLBCL, and FL. MM biopsies were from bone marrow; lymphoma biopsies were from various tissues. Cell pellets from 14 different tumor cell lines for which BCMA expression was determined by flow cytometry were processed similarly. Tissues and pellets were sectioned at 5 μm, deparaffinized, rehydrated through graded alcohols, and antigen retrieved in heated citrate buffer (Ventana). Following a protein blockade step (Dako), serial sections were incubated with polyclonal goat-anti-human BCMA (R&D Systems) or irrelevant goat immunoglobulin G (IgG; Jackson Immunoresearch). Following a wash step, sections were incubated with rabbit-anti-goat F(ab′)2 fragments (Jackson Immunoresearch), washed, and then developed with the Optiview DAB detection kit (Ventana). Slides were subsequently counterstained with hematoxylin, dehydrated through graded alcohols and xylene, and cover-slipped. BCMA staining was scored categorically, which was defined as the BCMA relative strength of staining (0–4 scale) on a typical, positively stained cell, and frequency, which was defined as the prevalence (0–4 scale) of BCMA+ cells within a tissue section. For MM and lymphoma biopsies, the approximate percentage (0–100%) of the total section that stained positively for BCMA was also estimated.
BCMA receptor density
BCMA receptor density was assessed on various tumor cell lines by flow cytometry. Mean fluorescent intensity (MFI) of cells stained with saturating concentrations of PE anti-human CD269 (BCMA) antibody, clone 19F2 (BioLegend) was used to calculate antibody binding capacity (ABC) using Quantum™ Simply Cellular® anti-Mouse IgG microsphere kit per the manufacturer's instructions (Bangs Laboratories, Inc.). ABC values were normalized to values obtained using PE Mouse IgG2a,κ, clone MOPC-173 isotype control (BioLegend).
Quantitation of BCMA mRNA levels in tumors
Lymphoma cDNA Array (LYRT301) plates were purchased from the TissueScan™ Cancer cDNA Array (OriGene Technologies). Quantitative polymerase chain reactions (qPCR) were run on the StepOnePlus™ Real-Time PCR System (Applied Biosystems) using TaqMan® Gene Expression Master Mix (Applied Biosystems) and assay primer/probe set (Hs03045080_m1 for TNFRSF17 [BCMA]; Applied Biosystems), according to the manufacturer's instructions. The ΔCt was calculated for each sample against the endogenous control gene (Hs99999903_m1 for ACTB; Applied Biosystems), which provides normalization for each cDNA sample.
Lentiviral vectors, T-cell transduction, culture, and CAR detection
Anti-BCMA CAR lentiviral vectors (LVV) were replication defective, self-inactivating (SIN), third-generation human immunodeficiency virus type 1 (HIV-1)–based LVV, pseudotyped with the vesicular stomatitis virus–glycoprotein (VSV-G) envelope protein. The vectors used the murine leukemia virus–derived MND promoter 29 to drive expression of the chimeric antigen receptor. The anti-BCMA CARs contain an anti-BCMA single-chain variable fragment (scFv) coupled to the CD8α hinge and transmembrane domains, and the intracellular CD137 co-stimulatory (4-1BB) and CD3ζ chain signaling domains using design principles previously reported. 30,31 It was not possible to obtain the anti-BCMA hybridomas or antibodies to perform independent analysis to determine their affinity. Anti-BCMA CAR LVVs were produced by transient transfection of HEK293T cells with the plasmid transfer vector and packaging plasmids encoding GAG/POL, REV, and VSV-G. Anti-BCMA CAR LVV was purified via chromatography and formulated before storage at ≤−65°C. Per internal nomenclature, capital letters (i.e., BB2121) refer to the LVV, while T cells transduced with a given LVV are designated with lower-case letters (i.e., bb2121).
To initiate T-cell cultures, bulk PBMCs were activated with soluble human anti-CD3, clone OKT3 (Miltenyi Biotec), and human anti-CD28, clone 15E8 (Miltenyi Biotec), in T-cell growth media (TCGM) consisting of X-VIVO™ 15 media (Lonza) supplemented with 5% human serum, type AB (Valley Biomedical), 2 mM of GlutaMAX™-I (Gibco), 10 mM of HEPES buffer solution (Gibco), and 250 IU/mL of recombinant human interleukin-2 (rhIL-2; CellGenix GmbH) with culture at 37°C in a 5% CO2 incubator. The next day, cells were transduced with LVV (multiplicity of infection [MOI] = 20), and T cells were expanded for 7–15 days at a concentration of 0.3–0.5 × 106 cells/mL. T-cell controls compared in each individual study were from donor-matched parallel cultures.
Cell surface detection of CAR molecules was performed as follows. T cells were labelled with LIVE/DEAD® Fixable Near-IR Dead Cell Stain Kit (Molecular Probes) according to the manufacturer's instructions to exclude dead cells. The cells were then incubated with Biotin-Goat Anti-Mouse IgG (H+L; Molecular Probes) for 20 min at 4°C. Following a wash step, the cells were incubated with R-phycoerythrin (PE) streptavidin and CD3 PerCP-Cy5.5, clone SK7 (BD Pharmingen), for 20 min at 4°C and fixed. Samples were acquired on an LSRFortessa™ Cell Analyzer (BD Biosciences) and analyzed using FlowJo Single Cell Analysis Software v9.0 (FlowJo, LLC).
Vector copy number (VCN) was determined using qPCR, as described previously. 32 Briefly, gDNA was isolated from transduced T cells using Quick-gDNA™ MiniPrep kit (Zymo Research Corp). qPCR reactions were run on the StepOnePlus™ Real-Time PCR System (Applied Biosystems) using TaqMan® Universal Master Mix II, no UNG (Applied Biosystems), according to the manufacturer's instructions. TaqMan® Copy Number Reference Assay, human RNase P (Applied Biosystems), and custom psi gag primer/probe set (Life Technologies) were run in multiplex. Analysis was performed using StepOne Software v2.2 (Applied Biosystems) using the ΔΔCt method. The average VCN was calculated by normalizing psi gag copies to RNaseP copies in diploid cells.
Tumor cell co-culture and interferon-gamma enzyme-linked immunosorbent assay
A total of 100,000 T cells (effector, E) and tumor cells (target, T) were co-incubated at a 1:1 effector:target (E:T) ratio overnight in a 96-well plate (0.1 mL/well) in TCGM (not supplemented with IL-2). Cell culture supernatants were harvested for use in human interferon-gamma (IFN-γ) enzyme-linked immunosorbent assay Ready-SET-Go!® (Thermo Fisher Scientific), according to the manufacturer's instructions. Samples were acquired on a SpectraMax® M Series Multi-Mode Microplate Reader (model M3; Molecular Devices) at a wavelength of 450 nm. Data were analyzed using SoftMax Pro 6.4 software (Molecular Devices).
T-cell proliferation assay
T cells were labeled with a CellTrace™ Violet Cell Proliferation Kit for flow cytometry (Molecular Probes), according to the manufacturer's instructions. Following labeling, T cells were co-incubated with tumor target cells for 48 h at a 1:1 ratio with antigen-negative or antigen-positive tumor targets in TCGM without IL-2 supplementation. After incubation, co-cultured cells were labelled with LIVE/DEAD® Fixable Near-IR Dead Cell Stain Kit (Molecular Probes), according to the manufacturer's instructions. Cells were then stained with CD3 fluorescein isothiocyanate (FITC), clone SK7 (BD Pharmingen). Following staining, samples were acquired on a LSRFortessa™ Cell Analyzer (BD Biosciences). Dilution of CellTrace™ Violet was assessed on viable CD3+ cells using FlowJo Single Cell Analysis Software v9.0 (FlowJo, LLC).
Cytotoxicity
The ability of anti-BCMA CAR effector T cells to lyse BCMA-expressing tumor target cells specifically was assessed in a fluorescence-based cytotoxicity assay and was adapted from Carpenter et al. 23 Briefly, BCMA+ tumor cells were pre-labeled with CellTrace™ carboxyfluorescein succinimidyl ester (CFSE) Cell Proliferation Kit, for flow cytometry (Molecular Probes), according to the manufacturer's instructions. Antigen-negative tumor cells were pre-labeled with a CellTrace™ Violet Cell (CTV) Proliferation Kit for flow cytometry (Molecular Probes), according to the manufacturer's instructions. Both antigen-positive and antigen-negative tumors were combined at a theoretical 1:1 ratio and co-cultured with CAR T cells at the indicated E:T ratios in triplicate wells of a 96-well round-bottom plate. The cultures were incubated for 4 h at 37°C. Immediately after the incubation, samples were labeled with a LIVE/DEAD® Fixable Near-IR Dead Cell Stain Kit (Molecular Probes) and acquired on a LSRFortessa™ Cell Analyzer (BD Biosciences). For each T cell plus target cell culture, cytotoxicity was determined as follows: percent cytolysis = 100 – (effective ratio of CFSE+ cells to CTV+ cells/the baseline ratio of CFSE+ cells to CTV+ cells). The baseline ratio of CFSE+ cells to CTV+ cells was the measured ratio of antigen-negative and antigen-positive tumor targets without T cells. The effective ratio of CFSE+ cells to CTV+ cells was the measured ratio of antigen-negative and antigen-positive tumor targets with T cells.
Xenograft models
MM
Female NOD-Cg-Prkdc scid IL2rg tm1Wjl /SzJ (NSG; Jackson Laboratories) mice received subcutaneous (s.c.) injections of 0.2 mL of a 5 × 107 cells/mL suspension containing 1 × 107 BCMA+ RPMI-8226 MM tumor cells to establish s.c. xenografts. At 18 days post tumor implantation, mice with xenografts were randomized to four groups of 10 mice, with mean tumor volumes of ∼96 mm3. On the same day (day 1), groups received a single intravenous (i.v.) injection of 0.2 mL of a cell suspension containing either the vehicle alone (vehicle control) or an identical total T-cell dose of an anti-CD19 CAR deleted for internal signaling domains (anti-CD19Δ CAR) T cells (negative control) or anti-BCMA CAR T cells. An additional group received 1 mg/kg of bortezomib (Velcade®; positive control) i.v. twice weekly for 4 weeks. Mice were monitored until day 85.
Lymphoma
Female NSG mice received i.v. injections of 0.1 mL of a 2 × 107 cells/mL suspension containing 2 × 106 BCMA+/CD19+ Daudi (BL) or 106 BCMA+/CD19+ JeKo (MCL) transduced with firefly luciferase (FFLuc) to establish systemic xenografts. Mice with established xenografts were randomized to four groups of five mice each, with a mean tumor size (bioluminescence or total flux values) of ∼3 × 108 photons/s/cm2/sr (Daudi) or ∼1 × 107 photons/s/cm2/sr (JeKo). On the same day, animals received a single i.v. injection of 0.2 mL of a cell suspension containing either the vehicle alone (vehicle control) or anti-CD19Δ CAR T cells (negative control), anti-CD19 CAR T cells (positive control), or anti-BCMA CAR T cells. Mice were monitored until around day 50 post tumor injection. In all studies, the activity of the anti-BCMA CAR T cells were evaluated by determining the tumor size twice weekly (calipers for MM, IVIS® Spectrum in vivo imaging system for lymphomas). General safety was evaluated by observing the animals daily and recording their body weights twice weekly. All in-life personnel were blinded to the identity of the test and control articles.
Results
Expression of BCMA on MM and lymphoma cell lines and tumor biopsies
Prior investigators demonstrated high and restricted BCMA RNA and cell surface protein expression on MM plasma cells, 23,25,33 but little has been published on BCMA expression in B-cell malignancies. To investigate BCMA protein expression, an immunohistochemistry (IHC) assay was established to determine BCMA expression in archival tumor samples (most readily available as FFPE slides). First, this procedure was verified on myeloma and lymphoma cell lines (Fig. 1A, representative staining shown for BCMA- K562, BCMA+ HL line RPMI-6666, and BCMA+ MM line RPMI-8226). To determine the number of BCMA molecules per cell, next, a flow cytometry-based BCMA receptor density assay was developed using fluorescent microspheres to quantitate BCMA surface expression accurately (Table 1 and Fig. 1B). MM cell line RPMI-8226 and K562 cells engineered to express BCMA showed the highest BCMA expression (>12,000 BCMA molecules), while a low but detectable amount of BCMA was observed on a variety of lymphoma cell lines (222–3,173 BCMA molecules/cell). Within most MM and lymphoma cell lines examined, BCMA IHC staining intensity was highly correlated to the number of expressed BCMA molecules, as determined by flow cytometry (Table 1 and Fig. 1B). One notable exception was a BL cell line (Daudi), which had surface expression of 1,173 BCMA molecules, but it was not possible to generate BCMA+ staining readily by this IHC assay. Multiple factors such as fixation or epitope retrieval could have influenced IHC reactivity of this specific cell line.
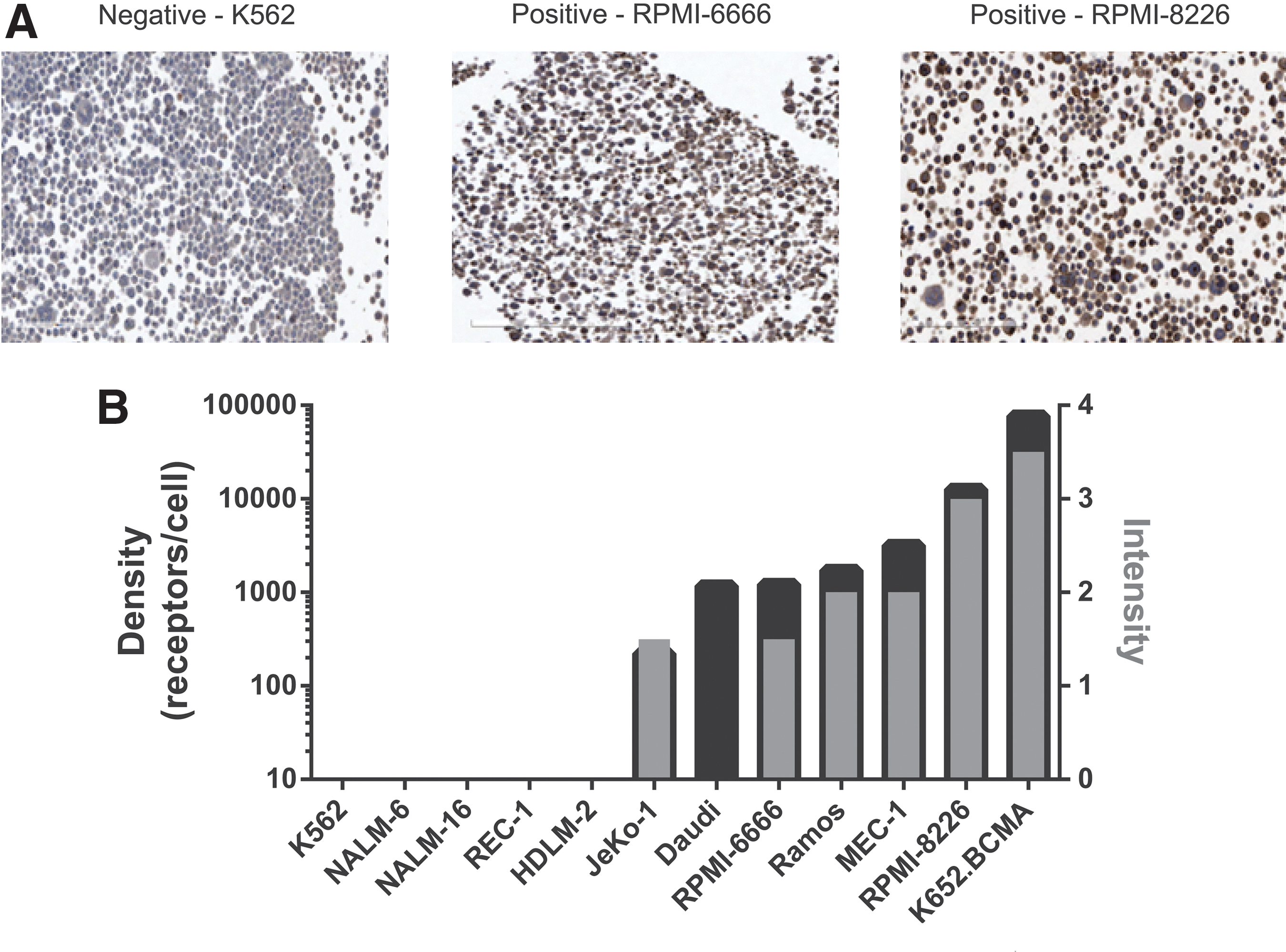
B-cell maturation antigen (BCMA) expression on human lymphoma and multiple myeloma (MM) cell lines by immunohistochemistry (IHC) correlated with receptor density.
BCMA immunohistochemistry and receptor density for human MM and lymphoma cell lines
BCMA receptor numbers determined by flow cytometry using antibody binding capacity (ABC) assay.
IHC intensity: 0 = negative to equivocal; 1 = weak; 1.5 = weak to moderate; 2 = moderate; 2.5 = moderate to strong; 3 = strong; 3.5 = strong to intense; 4 = intense.
IHC frequency: 0 = negative; 1 = very rare; 1.5 = very rare to rare; 2 = rare; 2.5 = rare to occasional; 3 = occasional; 3.5 = occasional to frequent; 4 = frequent.
Mixed results: negative by IHC, yet positive by flow cytometry.
MM, multiple myeloma; ND, not determined; td, BCMA transductant; NHL, non-Hodgins's lymphoma; HL, Hodgkin's lymphoma; ML, myelogenous leukemia; ALL, acute lymphoblastic leukemia; MCL, mantle cell lymphoma; BL, Burkitt lymphoma; B-CLL, B-cell chronic lymphocytic leukemia.
These observations were extended to MM biopsies from 29 patients (FFPE bone-marrow biopsies). IHC demonstrated that BCMA was expressed on all MM samples tested, but the frequency was variable (Fig. 2A, representative examples shown). The staining intensity of archival MM biopsies was not as pronounced as a freshly prepared MM cell line (RPMI-8226), but was easily discernable and was similar to MM line U226-B (Table 1). BCMA+ cells represented >50% of the tumor tissue area in 41% of the biopsies.
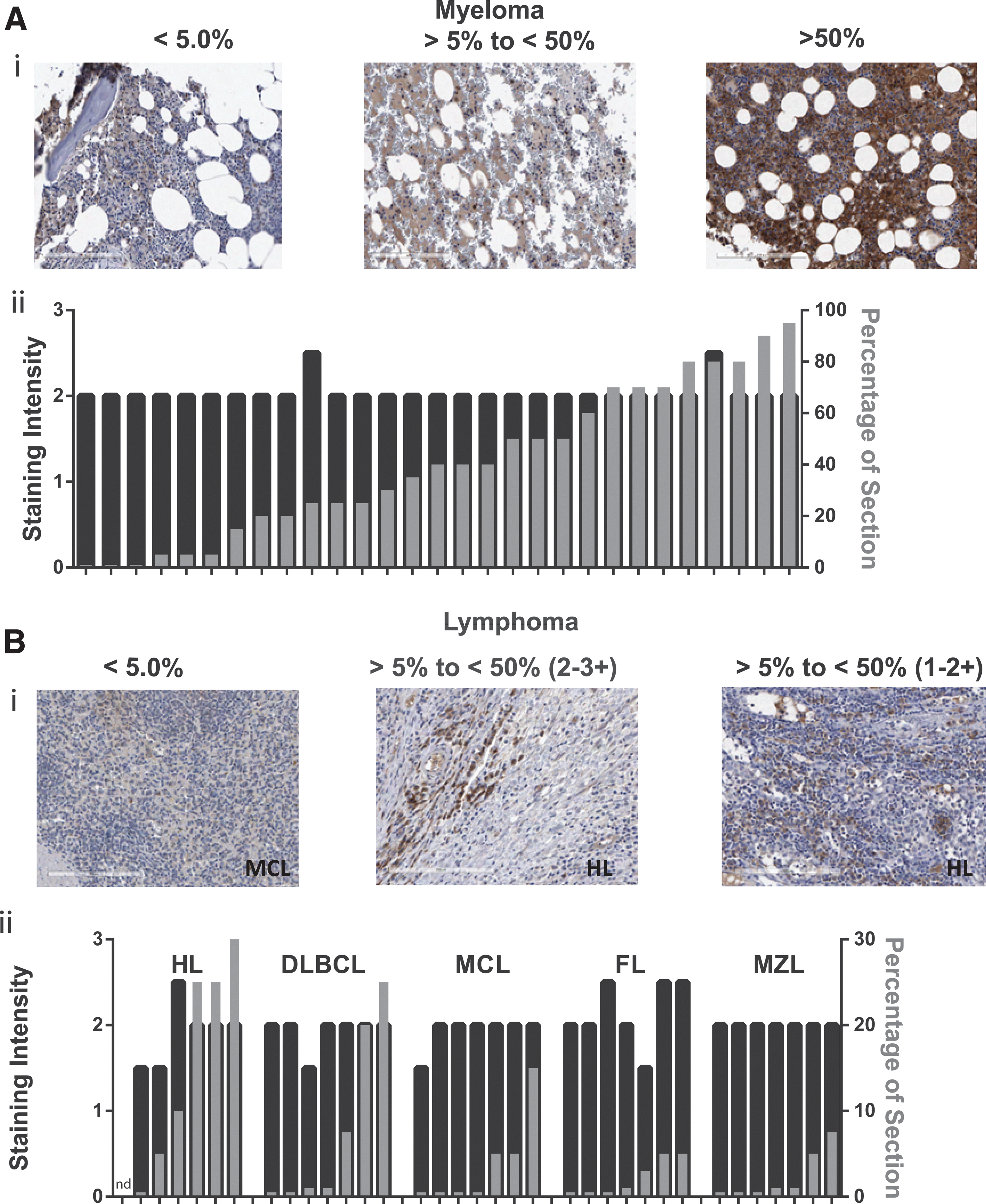
BCMA was detected in biopsies of patients diagnosed with MM and five subtypes of lymphoma.
The prevalence of BCMA expression in various primary lymphoma types has not been previously reported. To investigate this, tumor biopsies were obtained from 35 lymphoma patients representing five distinct disease subsets and assessed for BCMA expression by IHC (Fig. 2B, representative examples shown). Samples consisted of HL or non-HL (NHL), including MCL, MZL, DLBCL, and FL (seven samples for each subtype). BCMA expression was demonstrated in 34/35 lymphoma samples. BCMA staining intensity on individual cells in lymphoma biopsies was absent in one HL sample, weak-to-moderate in five samples, moderate in most samples, and moderate-to-strong in four samples by standard pathology scoring (0–4 scale; Fig. 2B). Compared to MM, the frequency of BCMA expression was low in these lymphoma samples. BCMA+ cells were observed in >5% of the tumor cells in 57% of HL and 18% of NHL samples. No lymphoma biopsy displayed >30% BCMA+ cells. These results suggested that BCMA protein expression (determined by IHC staining) may be more prevalent in HL than NHL biopsies.
Anti-BCMA CAR expression and activity
Given the widespread expression of BCMA in MM and in a subset of largely incurable lymphomas, (e.g., DLBCL, as shown in Fig. 2), this study sought to produce anti-BCMA CAR encoding LVVs to generate anti-BCMA CAR T cells. Four anti-BCMA CARs were constructed using distinct anti-human BCMA monoclonal antibodies (mAbs) shown to have high specificity to human plasma cells. 34 Sequences from the mAbs were used to construct scFvs (in orientation VL-linker-VH), which were assembled into a CAR architecture using a CD8α extracellular hinge and transmembrane domain followed by 4-1BB costimulatory domain and CD3ζ T-cell signaling elements (Fig. 3A). LVVs encoding anti-BCMA CAR constructs BB2120, BB2121, BB2122, and BB2123 and, as controls, an anti-CD19 CAR and an anti-CD19Δ CAR (signaling deficient) were produced as replication-incompetent vector supernatants and used to transduce T cells from normal donor PBMCs. CAR T cells for each anti-BCMA CAR construct and each control were cultured in parallel from the same donor. All LVVs effectively transduced T cells with an average number of integrated vector copies per diploid cell (VCN) ranging from one to three (Table 2 and Fig. 3B). Despite showing equivalent transduction efficiencies, as determined by VCN, transduction with BB2122 resulted in significantly fewer CAR+ cells (range 12–20%) than the other anti-BCMA CAR LVVs (range 38–74%). T-cell expansion and cell phenotype of CAR T cells were similar to control transduced and untransduced T-cell cultures.
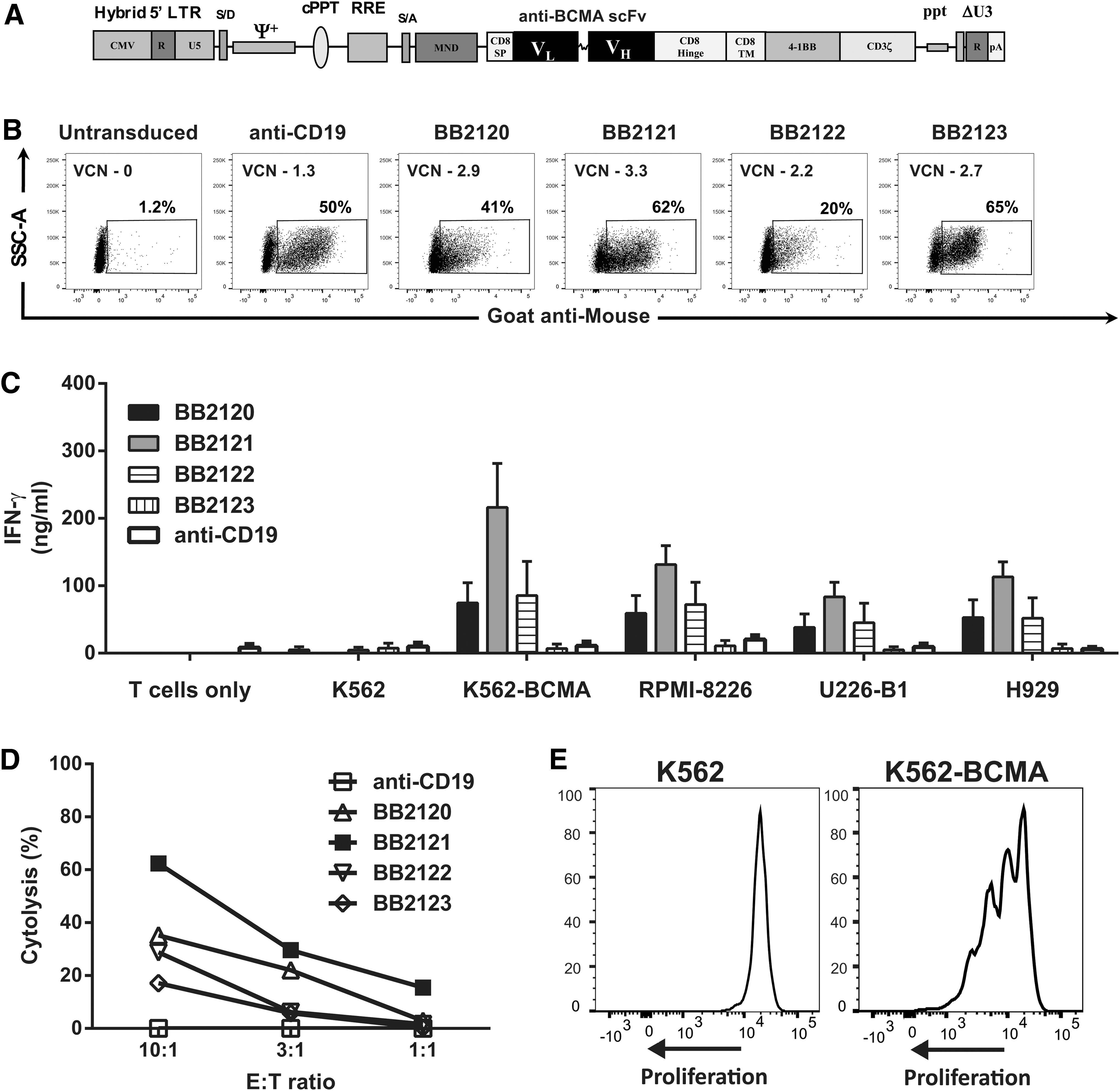
Anti-BCMA chimeric antigen receptor (CAR) T cells elicit antitumor reactivity against BCMA-expressing targets.
Frequency of transduced T cells and average number of vector integrations
% = percentage of CD3+ T cells positive for the CAR determined by flow cytometry.
VCN, vector copy number; ND, not done.
Next, the biological activity of the anti-BCMA CAR T cells were assessed when co-cultured with BCMA expressing cell lines using standard tests for effector T-cell function. Transduced T cells generated by three of four anti-BCMA CAR LVVs (BB2120, BB2121, and BB2122) released IFN-γ in response to BCMA-expressing K562 cells and MM cell lines (Fig. 3C), while CAR T cells transduced with BB2123 did not exhibit reactivity. T cells transduced with anti-BCMA CAR vectors demonstrated antigen-specific BCMA+ tumor cell line recognition, and there was no evidence of antigen-independent activity (tonic signaling). Notably, BB2121-transduced T cells showed higher cytokine levels compared to the other anti-BCMA CAR constructs (p < 0.0001). The ability of anti-BCMA CAR T cells to kill BCMA-expressing target cells was determined using K562 cell engineered to express BCMA. Similar to the IFN-γ co-culture assays, BB2121-transduced T cells demonstrated the highest level of cell killing (Fig. 3D). No activity was observed against cell lines that did not express BCMA. In both studies, anti-CD19 CAR T cells showed only background activity against cell lines lacking CD19 expression. As a final test of antigen-dependent activity, BB2121-transduced CAR T cells were tested for proliferation against a BCMA-expressing tumor. As shown in Fig. 3E, robust proliferation of BB2121-transduced CAR T cells was observed upon co-culture with K562-BCMA+ cells, but not unmodified K562 cells. Based on gene transfer efficiency, antigen-specific cytokine release, and cytolytic activity of anti-BCMA CAR T cells, BB2121 was selected for additional studies.
Reactivity of anti-BCMA CAR T cells against MM/lymphoma cell lines and primary CLL
T cells transduced with the anti-BCMA CAR LVV BB2121 (bb2121) were tested for cytotoxic activity against three different MM and two lymphoma cell lines. In a 4 h cytotoxicity assay, bb2121 specifically killed RPMI-8226, NCI-H929, and U266-B1 MM cells (Fig. 4A). Cytotoxic activity was not observed from control vector transduced T cells (anti-CD19 or anti-CD19Δ CAR T cells). By contrast, both anti-BCMA and anti-CD19 CAR T cells showed specific and equivalent activity against the BCMA and CD19 double-positive BL cells (Daudi) and MCL cells (JeKo; Fig. 4A). Activity against Daudi cells confirmed BCMA expression seen by flow cytometry, despite a lack of BCMA detection by IHC. These results suggest that flow cytometry may be a more sensitive method for the detection of low levels of BCMA expression.
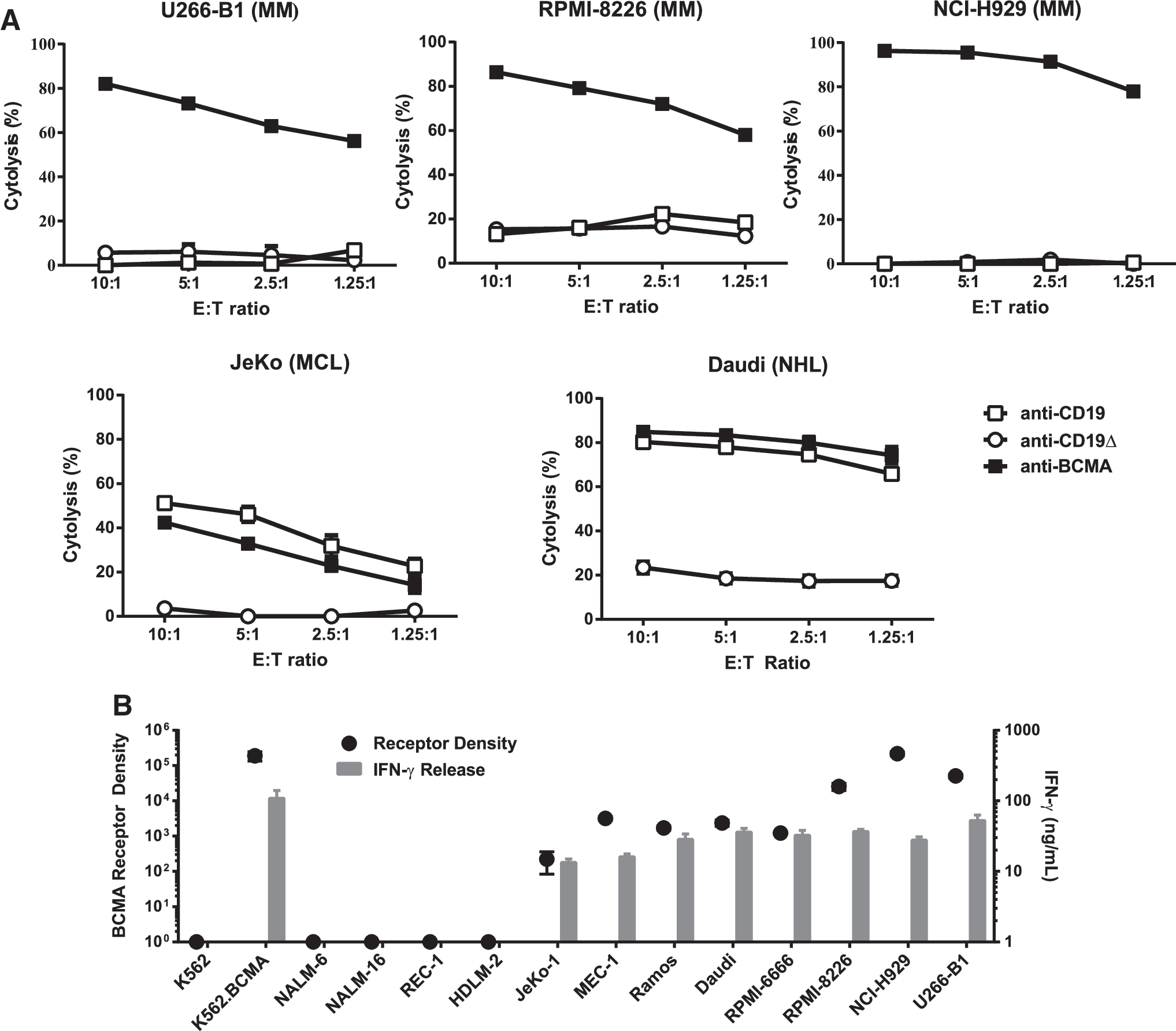
Anti-BCMA CAR T cells elicit potent reactivity against BCMA+ MM and lymphoma cell lines.
Given the high level of activity of anti-BCMA CAR T cells and the expression of BCMA on a variety of B-cell neoplasms, antitumor activity was evaluated against a panel of cell lines with quantified BCMA expression (Fig. 4B). The cell lines included: BCMA- controls representing the NHL subtypes ML (K562), ALL (NALM-6 and NALM-16), and MCL (REC-1), as well as HL (HDLM-2). BCMA+ controls including K562 clones engineered to express BCMA (K562-BCMA) and BCMA+ MM (RPMI-8226, NCI-H929, and U266B1), and NHL subtypes B-CLL (MEC-1), BL (Daudi and Ramos), and MCL (JeKo-1), as well as HL (RPMI-6666). As demonstrated by IFN-γ production following co-culture, anti-BCMA CAR T-cell activity was observed against all cell lines expressing BCMA, as assayed by flow cytometry (Fig. 4A and B). Of interest, IFN-γ release to the JeKo-1 MCL cell line demonstrated that as few as 222 BCMA molecules/cell were sufficient to elicit anti-BCMA CAR T-cell activity.
To investigate the correlation further between BCMA expression and recognition by CAR T cells quantitative reverse transcription PCR (qRT-PCR) assay was developed to quantitate BCMA RNA levels. Multiple lymphoma cell lines were subject to simultaneous co-culture and BCMA RNA quantitation. As shown in Fig. 5A, two ALL cell lines (NALM-6 and NALM-16) had low but detectable levels of BCMA RNA (ΔCt 5.0 and 6.5, respectively), but were not recognized by anti-BCMA CAR T cells. Cell lines with a BCMA ΔCt value of at least 2.0 (including JeKo-1, with a receptor density value of 222 molecules/cell) were recognized in this assay. A commercially available lymphoma RNA panel was obtained and tested simultaneously with the cell lines using the same BCMA qRT-PCR assay (Fig. 5B). As shown, 21/48 samples tested had a BCMA ΔCt value of at least 2.0, suggesting that up to 44% of lymphoma tumors could potentially be recognized by anti-BCMA CAR T cells. The sensitivity of BB2121-transduced CAR T cells to minimal BCMA levels led to the study determining the reactivity anti-BCMA CAR T cells to B-CLL cells. PBMCs from two primary B-CLL patients were tested for BCMA expression by flow cytometry, and one was shown to be weakly BCMA+ (9.8% BCMA+; Fig. 5C). These cells were then co-cultured with anti-BCMA CAR transduced T cells, and specific IFN-γ release was observed to the BCMA+ primary B-CLL samples, which was comparable to the B-CLL cell line, MEC-1 (Fig. 5D). Consistent with prior reports showing absence of BCMA expression in healthy PBMCs, no activity against normal donor PBMCs was observed.
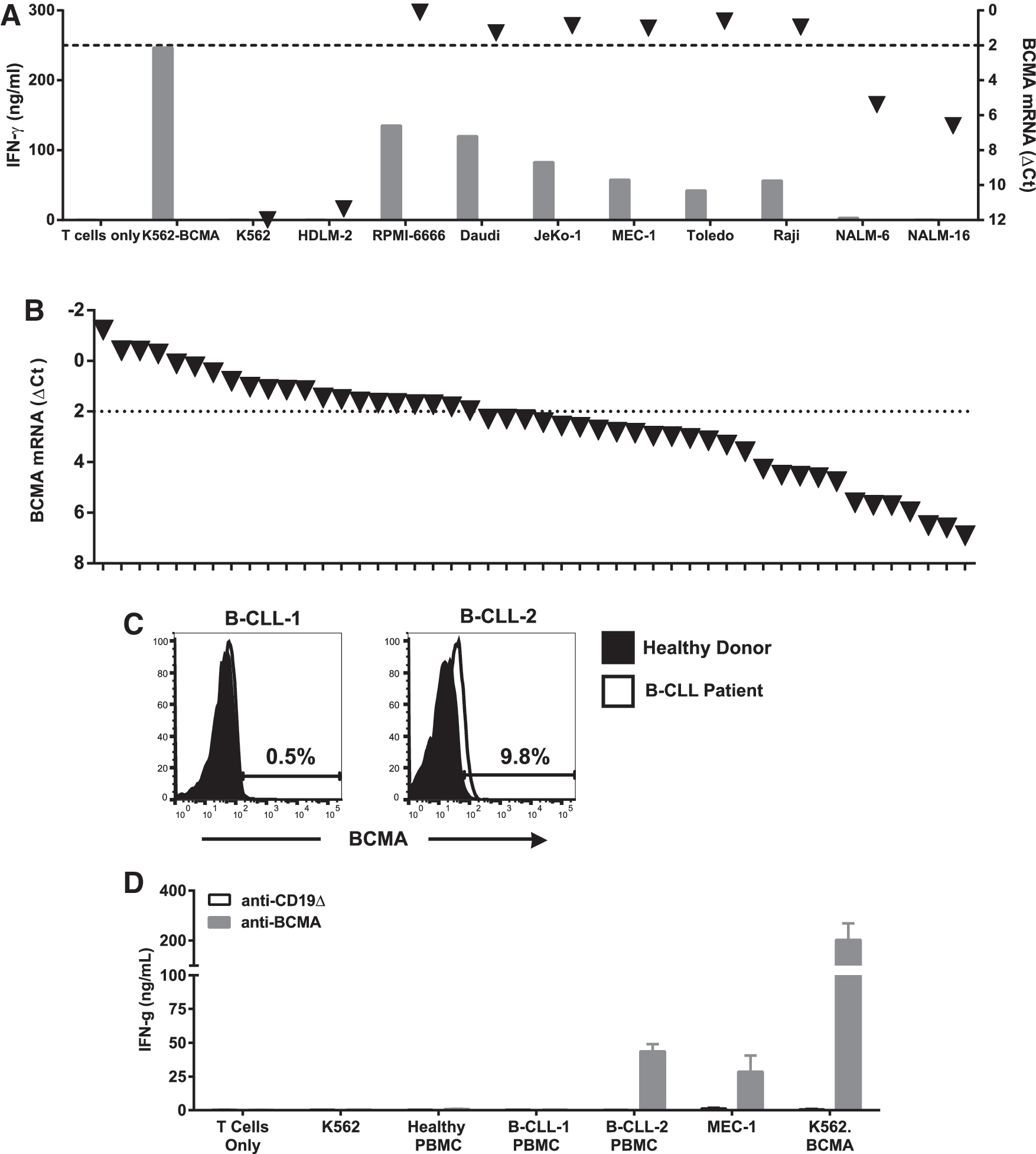
Anti-BCMA CAR T cells respond to low expression levels of BCMA in lymphoma cell lines and primary tumors.
Anti-BCMA CAR T cells rapidly expand and eliminate MM xenografts despite soluble BCMA protein
Next, the study sought to determine the in vivo activity of the anti-BCMA CAR T cells. The antitumor efficacy of bb2121 was evaluated in NSG mice bearing well-established (∼100 mm3) tumors using RPMI-8226 human MM s.c. xenografts (Fig. 6A). Mice received a single i.v. administration (5 × 106 CAR + T cells/mouse) of either anti-BCMA CAR T cells, control anti-CD19Δ CAR T cells, or repeated i.v. administration of protease inhibitor bortezomib (Velcade®; 1 mg/kg twice weekly for 4 weeks). Mice treated with bb2121 had complete tumor elimination and long-term survival (up to day 85 post CAR T treatment, at which point the experiment was terminated; Fig. 6B). In contrast, mice treated with anti-CD19Δ control CAR T cells had mean tumor volumes of 3,200 mm3 on day 33 post adoptive transfer and did not survive beyond day 40, similar to vehicle-treated mice. Mice treated with bortezomib had low tumor volumes (mean 20–40 mm3) during time of treatment (days 1–28), but once treatment was discontinued, tumors began to grow and were 1,800 mm3 by day 57, with only 50% survival at day 64 and no mice surviving at the end of the study (day 85).
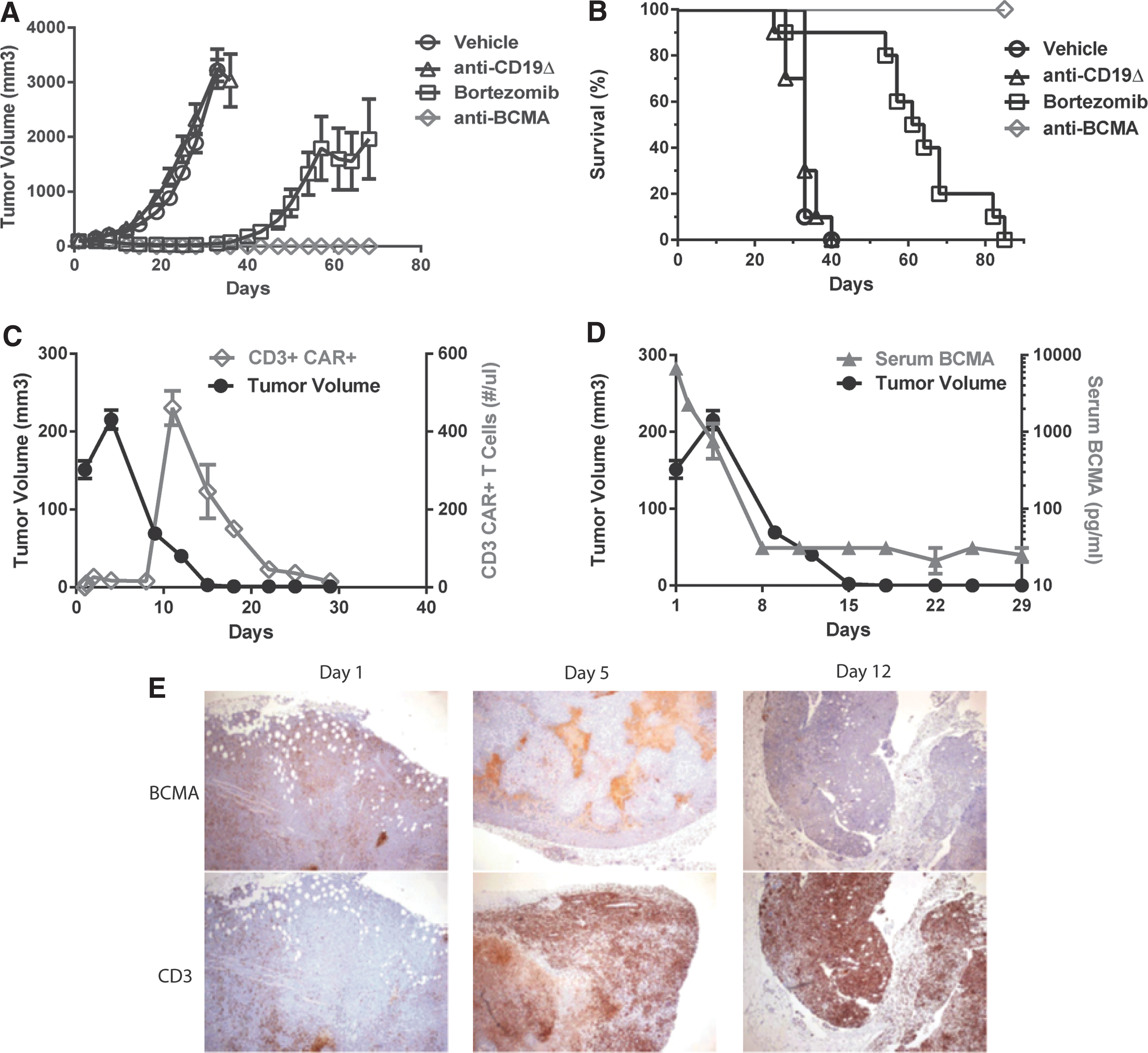
Anti-BCMA CAR T cells rapidly expand and eliminate MM xenografts, despite soluble BCMA protein in vivo.
To investigate further the kinetics of anti-BCMA CAR T-cell treatment of MM xenografts in NSG mice, the presence of CAR T cells in blood and tumor was determined. Mice bearing RPMI-8226 MM tumors that received a single i.v. administration of bb2121 (5 × 106 CAR + T cells/mouse) showed an initial increase in tumor volume followed by rapid and complete tumor regression by 15 days post infusion (Fig. 6C). CD3+/CAR+ T cells were observed in peripheral blood (PB) starting at day 2 and markedly increased at 11 days after adoptive transfer and then declined over the next 3 weeks. In comparison of anti-CD19Δ and anti-BCMA CAR T cells, the CD19Δ CAR T cells peaked on day 2 at a level of 36 CD3+ T cells/μL. In contrast, the anti-BCMA CAR T cells peaked at day 11 at a level of 461 CD3+ T cells/μL. The presence of soluble BCMA (sBCMA) in serum has been suggested as a potential biomarker in MM but could also represent a hurdle to full anti-BCMA CAR T-cell activity. The concentration of sBCMA was measured in this same in vivo study, and at the time of T-cell infusion, serum levels of sBCMA were 6,700 pg/mL. Post CAR T-cell infusion, sBCMA levels precipitously declined in parallel with tumor regression (Fig. 6D). The levels of sBCMA post day 8 were at or near the background detection level of this assay. These data are consistent with prior reports that anti-BCMA CAR T cells were not inhibited by soluble BCMA protein. 23 In a separate cohort of animals, tumors were excised on days 1, 5, and 12 post treatment with bb2121, and IHC was performed for BCMA and human T cell marker CD3. As shown in Fig. 6E, CD3+ human T cells (representative of anti-BCMA CAR T cells) actively trafficked to tumor by day 5 and were widespread by day 12. In contrast, BCMA+ tumor staining revealed patchy areas of BCMA+ tumor at day 5 and near complete elimination of all BCMA+ tumor by day 12.
Anti-BCMA CAR T cells control lymphoma xenografts comparably to anti-CD19 CAR T cells
Lastly, given the findings that some lymphoma cell lines also express BCMA and that anti-BCMA CAR T cells can recognize these same cell lines in vitro (Figs. 4 and 5), the ability of anti-BCMA CAR T cells to support in vivo anti-lymphoma activity was determined. Anti-lymphoma activity of bb2121 was evaluated in NSG mice bearing systemic BCMA+ Daudi BL xenografts (Fig. 7A). Mice were initially injected i.v. with 2 × 106 Daudi cells labeled with FFluc. After waiting for a large systemic lymphoma tumor burden to be established (day 18 post tumor cell injection), mice received a single i.v. injection of anti-BCMA CAR T cells, anti-CD19, or anti-CD19Δ CAR T cells (6 × 106 CAR + T cells per mouse). Interestingly, while both anti-BCMA and anti-CD19 CAR T cells were able to control tumor growth, different kinetics of tumor regression were observed. Anti-CD19 CAR T cells resulted in the rapid decrease of Daudi tumor levels, whereas anti-BCMA CAR T cells stabilized lymphoma growth for about 10 days before ultimately mediating tumor regression. The more rapid decline in lymphoma mediated by anti-CD19 CAR T cells was not sustained, and eventually tumor regrowth was observed. While no difference in survival was noted during the study period (Fig. 7B), only treatment with bb2121 resulted in complete tumor clearance by study termination (day 51).
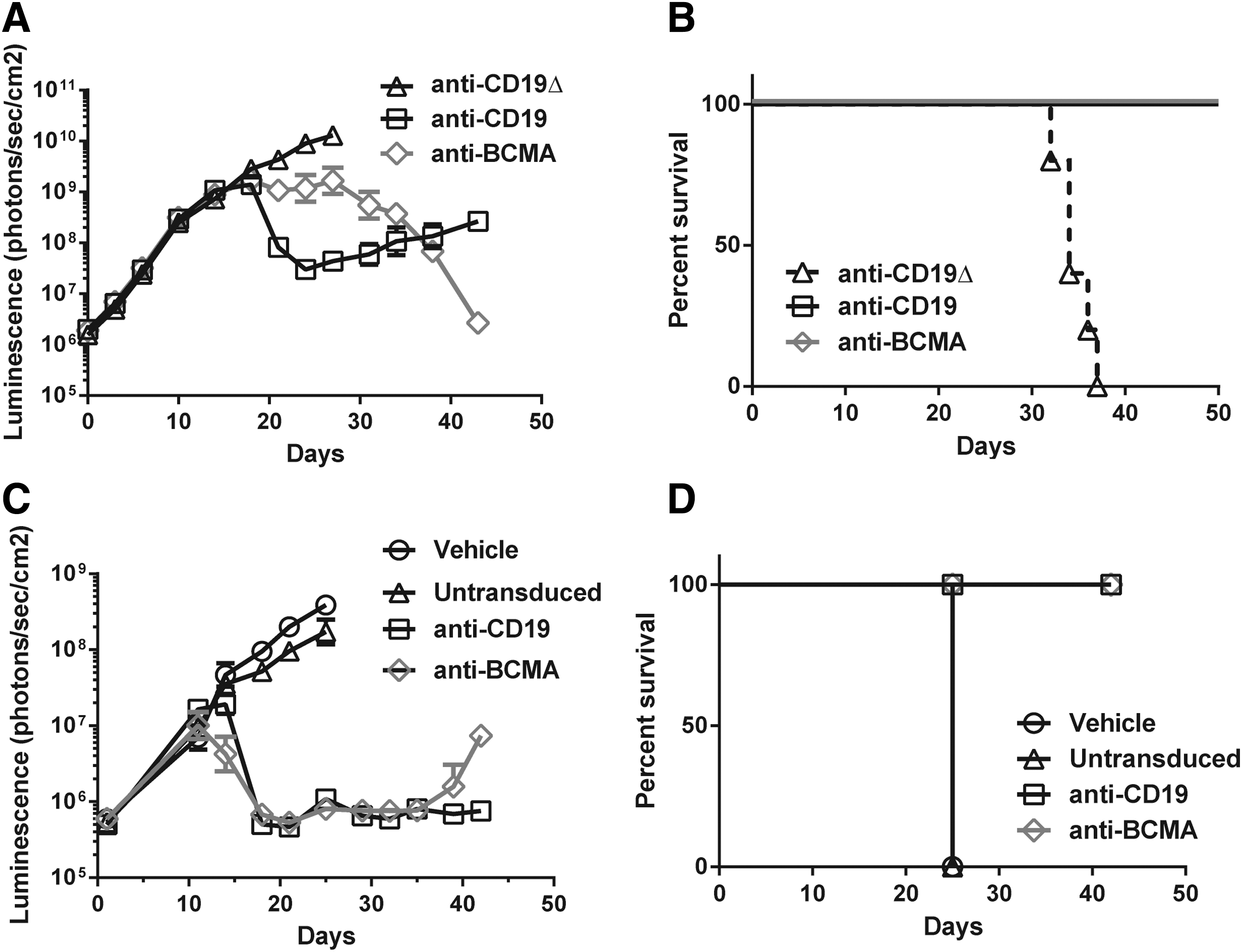
Anti-BCMA CAR T cells show robust antitumor activity against lymphoma xenografts.
In a second lymphoma model, well-established JeKo-1 MCL xenografts were treated with anti-CD19 or anti-BCMA CAR T cells (Fig. 7C). Mice were injected i.v. with 1 × 106 JeKo-1 cells labeled with FFluc and allowed to establish a measurable tumor burden (as determined by imaging). On day 11 post tumor cell injection, mice received a single i.v. injection of anti-BCMA or anti-CD19 CAR T cells (1 × 107 CAR + T cells per mouse). Vehicle and control-treated animals rapidly progressed and were sacrificed by day 25. Treatment with either CAR T cell resulted in a rapid decrease in tumor burden. At later time points, tumor regrowth was observed in some animals treated with anti-BCMA CAR T cells. A 100% survival rate was observed for both anti-CD19 and anti-BCMA CAR T-cell treatment groups during the study period (study terminated on day 43).
Discussion
The therapeutic potential of CAR T cells has been demonstrated using CAR constructs targeting CD19-expressing tumors. Dramatic and prolonged tumor regression in leukemia and lymphoma patients was observed in clinical trials, with nearly 90% of ALL patients achieving a complete response after treatment with anti-CD19 CAR T cells. 35,36 In multiple studies, robust clinical responses were observed after treatment with a variety of anti-CD19 CAR vectors, using different scFvs combined with either CD28 or 4-1BB co-signaling domains. Recently, some patients responding to these anti-CD19 CAR T cells have had recurrence of CD19 antigen-negative tumors, suggesting the need to develop additional CAR-based therapies for these hematological malignancies. 37
BCMA has drawn increasing interest as a therapeutic target in MM. A number of biological factors suggest that, particularly in MM, BCMA may be an ideal target for immunological-based therapies. 19 BCMA signaling promotes MM survival via AKT1, mitogen-activated protein kinase (MAPK), and the nuclear factor (NF)-κB pathways, and recent reports further suggest that in the bone-marrow microenvironment of MM, BCMA signaling upregulates immune inhibitory molecules such as programmed death ligand (PD-L)1, IL-10, and transforming growth factor (TGF)-β. 38,39 BCMA-targeting antibody drug conjugates (ADC), bispecific BCMA/CD3 antibodies, and BCMA-targeted CAR T cells have all been reported to have anti-MM cell reactivity. 23,26,40,41 Recently, in a study of anti-BCMA bi-specific antibodies, Lee et al. used ABC-bead quantitation methods to determine BCMA receptor density on 39 MM patient samples. These investigators reported that all patient samples tested were BCMA+ and found a mean of 1,085 BCMA molecules per MM cell (range 349–4,268). 41 These values are well in the range of the reactivity of the anti-BCMA CAR T cells in this study, which it was demonstrated recognized BCMA+ cell lines that expressed as little as 222 BCMA molecules per cell (Table 1 and Fig. 4).
Many parameters are likely involved in the successful development of CAR T-cell therapy, with vector design elements being an important factor. Several lines of evidence suggest that 4-1BB co-stimulation may be superior to CD28 in the context of CAR T cells. CAR constructs using CD28 co-stimulatory domains were associated with increased levels of Th2-like cytokines (including inhibitory cytokines IL-4 and IL-10), and their kinetics of activation are more rapid than 4-1BB-containing CAR vectors. 30,42,43 The rapid activation of CAR T cells using the CD28 co-stimulation pathway post infusion may make these cells more prone to initiate an early-onset cytokine release syndrome in treated patients. At a more fundamental level, it is becoming increasingly clear that T-cell metabolism can have a significant influence on the function of T cells in both viral and tumor treatment models. Recently, Kawalekar et al. reported that when comparing CAR constructs with identical scFvs in tumor treatment models, compared to CD28 co-stimulatory containing vectors, 4-1BB containing vector-transduced T cells developed into more central memory-like T cells with enhanced respiratory capacity, increased fatty acid oxidation, and enhanced mitochondrial activity. 44 Initial clinical outcomes with CAR architectures exploiting 4-1BB co-stimulatory domains showed increased T-cell persistence, reported as a significant survival increase for 4-1BB-containing CARs versus those using CD28. 45 It should be noted, however, that such clinical comparisons must be viewed cautiously, as they are based on trials studying multiple independent variables that may also contribute to the persistence of the T cells. No head-to-head comparison of CD28 and 4-1BB-containing, but otherwise identical, CARs have yet been performed in humans.
The specific scFv used to target the CAR T cells also affects T-cell effector function. For example, analysis of the influence of scFv affinity on CAR T-cell avidity has demonstrated that scFvs with lower binding constants (Kd) were associated with enhanced T-cell activity. 46,47 In the analysis of the four scFvs used in this report, it was observed that despite similar transduction efficiency, as shown by VCN, CAR expression mediated by one of the vectors, BB2122, was less than the other vectors. Most interestingly, in comparison of the biological activity of these four anti-BCMA CAR constructs, and despite good CAR expression, T cells transduced with the BB2123 vector did not display significant cytokine release in co-culture assays (Fig. 3C) and had minimal lytic activity (Fig. 3D). As the sequences selected to construct these CARs were from mAbs shown to bind to BCMA+ cells in flow cytometry assays, an explanation for this observation is lacking, since it was not possible to access the mAbs in order to confirm BCMA binding specificity. Taking the properties of CAR expression, cytokine release upon co-culture, and lytic activity into consideration, BB2121 vector–transduced T cells were uniformly superior to T cells transduced by other vectors.
As a final consideration in CAR design elements, it was recently reported that the biophysical properties of the CAR molecule were also essential in determining CAR T-cell function. Long et al. demonstrated that an anti-GD2 CAR can cluster on the surface of CAR engineered T cells without antigen exposure and that this surface clustering was associated with antigen-independent tonic signaling, shown by the spontaneous release of effector cytokines and the expression of surface markers associated with T-cell exhaustion. 48 This tonic signaling property resulted in poor in vivo activity, and interestingly, this lack of in vivo function was reversed when the co-stimulatory domain was changed from CD28 to 41BB. The lack of antigen-independent tonic signaling was an additional variable investigated in the choice of CAR architecture used to select the final anti-BCMA CAR vector (BB2121) utilized in this report.
In consideration of clinical applications, Ali et al. recently reported the first-in-man results of anti-BCMA CAR T cells to treat patients with relapse refractory MM. 49 In this Phase I dose-escalation trial, 12 patients were treated, and three of six patients in the two highest dose levels responded to therapy (two very good partial responses and one complete response) with clearance of BCMA+ MM cells from bone marrow and >95% decrease in serum monoclonal proteins. Responding patients at the highest dose level had concomitant cytokine release syndrome (from which they all recovered) consistent with a robust T cell–mediated response to MM. The anti-BCMA CAR T-cell approach reported here was designed to extend these initial clinical observations in the context of a CAR architecture using the 4-1BB co-stimulatory domain and lentiviral transduction rather than the CD28 co-stimulation and γ-retroviral vector transduction. In contrast to γ-retroviral vectors, LVVs have a number of distinct properties that lend themselves to human T-cell gene therapy applications. For example, LVVs can transduce minimally activated T cells, and in the present procedures, an overnight stimulation of T cells was sufficient to lead to robust transduction efficiency (>50% CAR + T cells) facilitated by the simple addition of viral vector supernatant to the culture media (i.e., neither polycations nor co-localizing enhancements such as fibronectin fragments were needed for efficient T-cell engineering). The ability to transduce early/non-replicating T cells may contribute to the establishment of CAR T cells with desirable memory-like phenotypes, which may be associated with better T-cell persistence. Furthermore, long-term patient safety data have been demonstrated in studies using LVV gene therapy for multiple diseases, including adrenoleukodystrophy, β-thalassemia, and severe combined immunodeficiency. 50 –52
Based on the predominant role of BCMA in MM, the strong preclinical data presented here, and initial anti-BCMA CAR T-cell trial results, a multi-center clinical trial has been initiated with centralized manufacturing to evaluate the safety and efficacy of bb2121 as a potential therapy for relapsed refractory MM (
Footnotes
Acknowledgments
We thank the staff of the preclinical development and cellular process development departments for their assistance in this project, Dr. Philip Gregory for helpful comments and suggestions on this manuscript, and Kate Lewis for diligent editing.
Author Disclosure
No competing financial interests exist.