
Abstract
Transposons have emerged as promising vectors for gene therapy that can potentially overcome some of the limitations of commonly used viral vectors. Transposons stably integrate into the target cell genome, enabling persistent expression of therapeutic genes. Transposons have evolved from being used as basic tools in biomedical research to bona fide therapeutics. Currently, the most promising transposons for gene therapy applications are derived from Sleeping Beauty (SB) or piggyBac (PB). Stable transposition requires co-delivery of the transposon DNA with the corresponding transposase gene, mRNA, or protein. Stable transposition efficiency can be substantially increased by using “next-generation” transposon systems that combine codon-usage optimization with hyper-activating mutations in the SB or PB transposases. By virtue of their relatively large capacity, gene therapy applications with relatively large therapeutic transgenes, such as full-length dystrophin, can now be envisaged. The authors and others have shown that efficient and stable gene transfer can be achieved with these next-generation transposons in several clinically relevant primary cells, such as CD34+ hematopoietic stem/progenitor cells, T cells, and mesenchymal and myogenic stem/progenitor cells that are amenable for ex vivo transfection. Alternatively, in vivo transposon gene delivery has been explored using non-viral vectors or nanoparticles or in combination with viral vectors. The therapeutic potential of these SB- and PB-based transposons has been demonstrated in preclinical models that mimic the cognate human diseases. However, there are still challenges impeding clinical translation of transposons pertaining mainly to the typical limiting efficiencies of most non-viral transfection methods and the intrinsic DNA toxicity. Nevertheless, it is particularly encouraging that transposons have now been used in gene therapy clinical trials. In particular, transposon-engineered T cells expressing chimeric antigen receptors are starting to yield promising results in patients with hematological malignancies.
Introduction
G
To overcome some of the limitations of these viral vector systems, non-viral vectors have been used instead. 14 Typically, these non-viral vectors systems are fully synthetic and do not rely on any viral component or mammalian cell-dependent manufacturing. This dramatically reduced the manufacturing costs while alleviating at least some of the immune concerns associated with the use of viral vectors. Though transfection of naked DNA into cells is notoriously inefficient, a plethora of non-viral transfection methods have been developed to improve gene transfer efficacy. Their purpose is to prevent DNA from degradation and to facilitate the uptake of the therapeutic DNA in the target cells. Typically, the therapeutic genes are transferred in forms of plasmids specifically tailored to maximize gene expression in the intended somatic target cells. However, even in optimal conditions of high transfection efficiencies, expression of the gene of interest by non-viral vectors is only short-term due to its degradation and gradual dilution of the episomal DNA in dividing target cells. This justifies the development of an efficient, integrating non-viral vector system based on mobile DNA element or transposons that stably integrate the therapeutic gene in the target cell chromosomes. 15 –19 Consequently, non-viral transfection of transposons combines some of the advantages of viral vectors (i.e., sustained expression of the therapeutic gene) with those of non-viral vectors (i.e., reduced immunogenicity and cost-effective manufacturing). Nevertheless, the incoming and transfected DNA by itself can activate the innate immune system. 20,21
Transposable elements are divided into two categories: retrotransposons and DNA transposons. Transposable elements can alter the genome of the host cells through insertions, duplications, deletions, and translocations. 22,23 Retrotransposons are described as mobile elements that employ an RNA intermediate that is first reverse transcribed into a complementary single-stranded (c) DNA strand by a reverse transcriptase encoded by the retrotransposon. Subsequently, the single-stranded DNA is converted into a double-stranded DNA that then integrates into the host genome. This so-called “replicative mechanism” yields several new copies of retrotransposons expanding throughout the target genome over evolutionary time. Retrotransposons are categorized into many subtypes according to the DNA sequences of the long terminal repeats and its open reading frames. 24 Retrotransposons were employed to enable transgene integration into the target cell DNA, in some cases relying on adenoviral delivery. 25 Nevertheless, retrotransposons can actively spread through the human genome and create genomic instability in different ways, even potentially contributing to disease. 26 Alternatively, DNA transposons translocate via a “non-replicative mechanism,” whereby two Terminal Inverted Repeats (TIRs) are recognized and cleaved by a transposase enzyme, releasing the cognate DNA transposons with free DNA ends. The excised DNA transposons then integrate into a new genomic region where target sites are recognized and cut by the same transposase. This cut-and-paste mechanism usually duplicates DNA target sites upon insertion, leaving target site duplications (TSDs). 19 The most recent transposition of DNA transposons present in the human genome was estimated to have occurred 80 million years ago during evolution. 27 Hence, endogenous DNA transposons are therefore considered to be silent in mammalian genomes, obviating concerns associated with transposon spreading and genome instability as in the case of retrotransposons.
Dna Transposons
The typical architecture of DNA transposons includes a gene encoding a transposase for transposition, flanked by two TIRs. DNA transposons can be classified into diverse families, which differ in terms of target site recognition, TSDs, TIRs, and transposon DNA sequences. Some of these DNA transposon subgroups are employed for human gene therapy to achieve sustained gene expression of the therapeutic transgene. 15,19
Sleeping Beauty (SB)
The Sleeping Beauty (SB) transposon belongs to the Tc1/mariner superfamily, which constitutes a large group of transposable elements that are widespread in several eukaryotic organisms. Salmonid transposons exhibit high DNA sequence homology to the Caenorhabditis elegans Tc1 transposon and were first identified in fish genomes in 1994. 28 However, these transposons were initially silent, primarily because of the accumulation of inactivating mutations in transposase genes. Thus, the functional transposase gene was originally reconstructed by “reverse evolution” based on consensus DNA sequence of different transposons derived from eight fish species by Ivics et al. 29 They showed, for the first time that this resurrected transposase exerted transposition activity specifically to TIRs of salmonid transposons. 29
Originally, the SB transposon was about 1.6 kb in total size and was mainly composed of two inverted repeat/direct repeats (IR/DRs) flanking the gene encoding a 360-amino acid SB transposase (SBase). Two transposase-binding regions, referred as direct repeats (DRs), are present in each IR/DR, and both of them are important for transposase recognition. 19 In contrast to the right IR/DR, the left IR/DR contains a 11 bp DNA sequence similar to 3′-half of the DR, namely the HDR motif, which significantly enhances transposition rate. 30
The SBase contain several distinctive structural motifs, including two DNA-binding helix-turn-helix (HTH) domains, consisting of PAI and RED subdomains, and catalytic domains harboring Asp-Asp-Glu (DDE) residues for DNA hydrolysis. 31 A GRPR-like sequence is located between two HTH domains and is highly conserved among Tc1/mariner membered transposases. 32 This GRPR motif is responsible for Hin invertase–DNA interaction. 33 The cut-and-paste transposition mechanism begins with multiple interactions between the transposase subunits and specific DNA motifs, that is, binding of PAI together with the RED subdomains to the DRs, and binding of the individual PAI subdomain to HDR. This ensures the specificity of the SB transposase activity toward the cognate SB transposon. This is followed by formation of tetrameric structure between the SB transposase and the SB transposon, allowing DNA and enzymes to be in close proximity for further DNA cleavage. 30 The synaptic transposome complex is also stabilized by the high mobility group box 1 protein (HMGB1). 34 The two strands of the transposon are cleaved by hydrolysis generating 5′-trinucleotide overhangs at both ends of transposon. The SBase also cleaves the genomic DNA at a TA dinucleotide consensus site to create the integration site for transposition. The cleaved transposon is then ligated to the integration site ends, resulting in the generation of a TA dinucleotide duplication. 31 Double-stranded breaks with non-identical 3′-trinucleotide overhangs at the original integration site can be repaired through multiple pathways, such as DNA-PK-dependent end-joining, Ku-independent end-joining, and homology-dependent gap repair, which may result in DNA deletion or transposon footprints depending on the actual DNA repair mechanism. 35
To generate a tool for gene delivery, the original SB system was converted into a two-component system by separating the two IR/DRs flanking the gene of interest from the SBase gene onto two different plasmids (Fig. 1). Typically, these two plasmids are co-delivered into target cells, and transposition occurs through a cut-and-paste mechanism, just as with the native transposon. 19 Expressing the SBase from an expression plasmid carries the risk that this plasmid may potentially integrate. Moreover, even non-integrated expression plasmids can give rise to low-level sustained SBase expression. Sustained SBase expression may result in the continuous transposon mobilization and integration. Consequently, this may result in steadily increasing integrated transposon copies per cell that may in turn augment the risk of insertional oncogenesis. As a safer alternative, it is also possible to deliver the SBase as an mRNA. 36 –38 The SBase mRNA is typically transfected into the cells by electroporation. An improved approach of retrovirus particle-mediated mRNA transfer (designated as RMT) was recently developed that allowed for transient and dose-controlled expression of SBase. 39,40 Interestingly, RMT provides a mean to support efficient transposition while preventing overt cell damage. In this case, the short-term presence of the SBase would create a window of opportunity, enabling transposition while substantially reducing the risk of insertional oncogenesis.
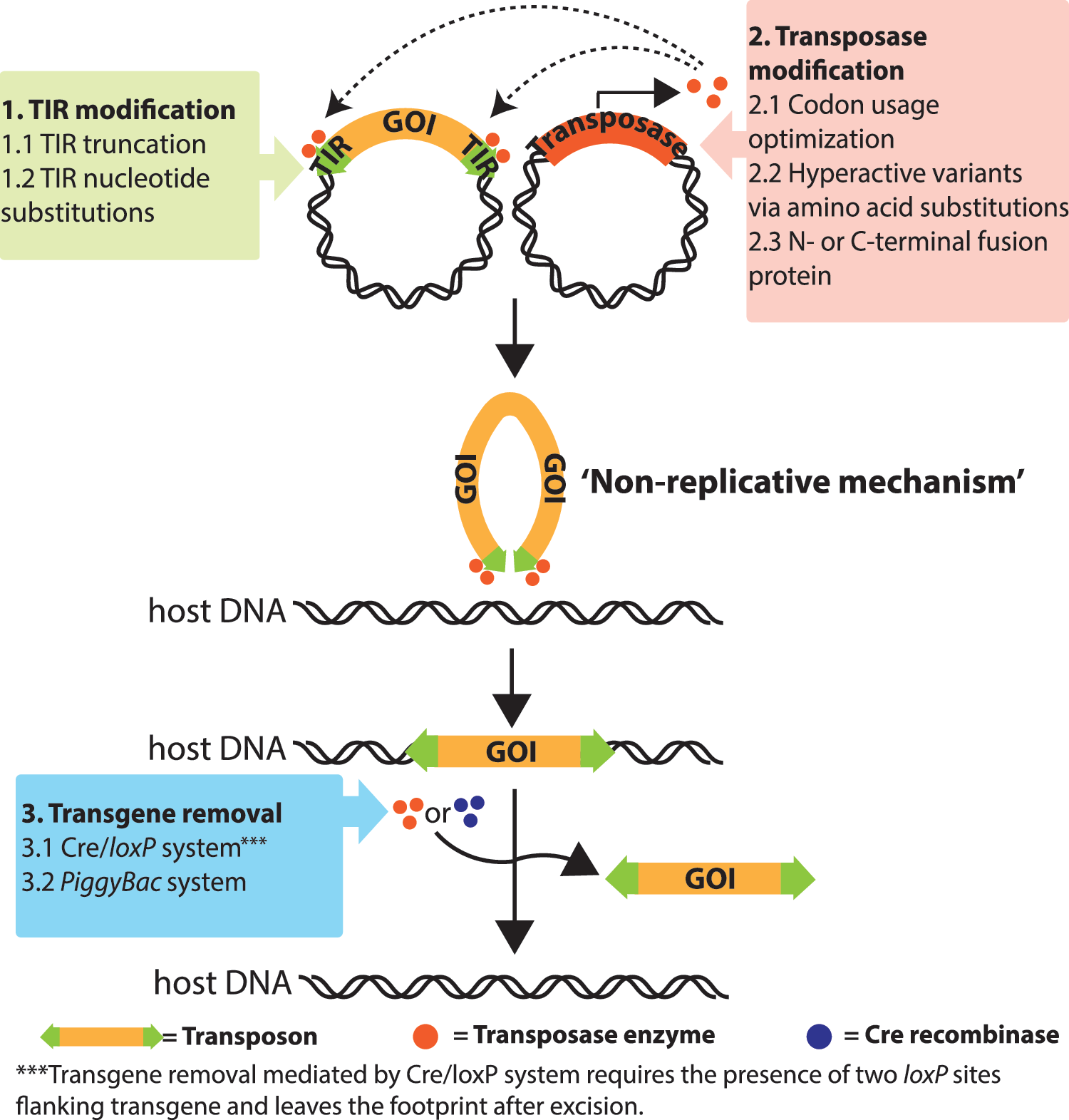
Schematic representation of two-component transposon system for gene delivery and improvements. Terminal inverted repeats (TIRs) and transposase gene of Sleeping Beauty (SB), piggyBac (PB), and Tol2 transposons are located in two different plasmids. The gene of interest (GOI) is flanked by two TIRs. Upon co-delivery of transposon-GOI and transposase vectors into host cells, transposases bind to TIRs for cleavage and form the transpososome complex with the transposon containing the GOI. The transposase subsequently introduces double-strand breaks and inserts the transposon with the GOI into the target site in the host DNA through a non-replicative (cut-and-paste) transposition mechanism, resulting in transposon integration. Several strategies have been developed to improve transposition activity, including TIR and transposase modification. The use of transposon/transposases also allows for specific excision of GOIs by supplying the transposase transiently. In the case of PB, the excision is seamless, leaving no genetic trace.
Several improvements have been reported to enhance SB transposition efficiency, including the generation of hyperactive transposase mutants. First-generation hyperactive SBase, called SB11, was generated by introduction of various amino acid mutations into the original transposase (SB10), leading to a relatively modest approximately threefold higher transposition efficiency. 41 Similarly, consensus amino acids of transposases from several species were incorporated into the SBase, yielding more hyperactive versions such as HSB5 42 and SB100X. 43 What distinguishes SB100X in particular from the other SB transposases is that it was generated by in vitro molecular evolution and selection. SB100X was 100-fold more efficient in transposon mobilization using a marker rescue assay than the original SB10. The authors were the first to demonstrate the superiority of this SB100X system in clinically relevant primary cells, such as CD34+ HSC, muscle stem/progenitor cells, mesenchymal and mesoangioblasts (MABs), hepatocytes, and induced pluripotent stem cell (iPS)-derived myogenic cells. SB100X 43 –45 significantly outperformed HSB5 46,47 and is considered the most hyperactive SBase to date. The authors have recently combined several other hyper-activating mutations within SB100X, but they did not further increase the overall transposition efficacy (VandenDriessche and Chuah, unpublished observations).
Generally, there is no strict limitation with respect to the transgene size, at least up to 8 kb. In contrast to transposons, viral vectors have a more limited packaging capacity. Nevertheless, transposition efficiency does decrease with increased transgene size beyond 8 kb. 41 However, it is possible to expand the transgene capacity of transposons by using a so-called sandwich configuration whereby the expression cassette is flanked with two complete pairs of IR/DRs in inverted orientation. 48 Consequently, this transposon payload can be further increased to at least 10 kb DNA while maintaining a relatively efficient transposition. Additionally, the payload capacity of SB transposon can be increased up to 12 kb when SB transposon is delivered in the context of a herpes simplex virus (HSV) amplicon vector 49 or a helper-dependent adenoviral vector. 47
PiggyBac (PB)
The piggyBac (PB) transposon was first identified and isolated from the cabbage looper moth Trichoplusia ni genome. 50,51 The original PB transposon is 2,475 bp in length and encodes a 594-amino acid PB transposase (PBase) gene. The PB transposon is flanked by a 311 bp 5′ end and 235 bp 3′ end, each containing TIR sequences. 52 Similar to SBase, PBase contains a catalytic unit harboring a characteristic DDE/D motif, 53 which interacts with Mg2+ ions during transposition. 54 The C-terminal region of the PBase possesses a Zn2+-binding “plant homeodomain” (PHD) finger with conserved cysteine residues. 55 The PB system also undergoes a typical cut-and-paste transposition reaction, as in the case of SB, whereby the PBase recognizes the two TIRs of the PB transposon. Unlike SB transposons, the PB transposon preferential genomic integration site contains a TTAA consensus motif. 19 The binding of the PBase to the transposon is followed by the 3′-strand excision of DNA, leaving the free 3′-OH group, which further forms the hairpin structure with 5′-TTAA overhangs of the transposon. The hairpin transposome complex is then released from the genome and binds to the site of integration in which the TTAA consensus sequence is present. The hairpin chemical bonds are subsequently cleaved by the transposase enzyme, and free 3′-OH attack the target site to generate TTAA overhangs in the target genome. The TTAA overhangs of the PB transposon and the target genome eventually anneal to complete the transposition reaction. Through this mechanism, the PB transposon system also leads to TGDs upon completion of the transposition process. However, in contrast to SB, PB transposon does not leave 3 bp footprints at the original transposition site, since the two 5′ overhangs generated by PBase excision are perfectly complementary. 52
A two-component PB platform has been developed and demonstrated, analogous to SB, yielding relatively robust stable gene transfer efficiencies in several independent studies 19 (Fig. 1). Additionally, the PBase has been modified to boost transposition activity in mammalian systems. The first strategy consisted of engineering the PBase DNA sequence by codon optimization for use in mammalian cells. For instance, mouse (m) and human (h) codon-optimized PBases were derived from the original PBase 56,57 and are expressed at higher levels in mammalian cells, significantly improving the overall transposition efficacy. Alternatively, novel amino acid mutations that were screened for increased transposition efficacy were introduced into the PBase. These mutants were generated by using error-prone polymerase chain reaction (PCR), which were subsequently validated in yeast cells. Ultimately, this resulted in a hyperactive PBase (hyPBase) that contained seven amino acid substitutions that synergistically enhanced transposition efficiency. 58 Comparative in vitro studies indicated that the hyPBase outperformed the mPBase by 10-fold, at least in mouse ES cells. 58 Similarly, a 20-fold increase in liver-directed expression has been shown with the hyPBase compared to when the mPBase was employed. 59 In an attempt to increase transposition further, the TIRs of the PB transposons were engineered. The 5′ and 3′ TIRs were trimmed to retain only those functional regions that were sufficient for transposition, thus reducing the total size of the PB vectors. 60 The smallest TIRs were defined as having only 40 and 57 bp of 3′ and 5′ IRs (referred as “IRmicro”), respectively, and they exhibited relatively robust transposition activity compared to the native IRs. 61 Alternatively, IR variants bearing nucleotide substitutions were generated by random PCR amplification to investigate the effects of substituted nucleotides on PB transposition. The 5′ IR harboring T53C and C146T mutations (referred as “IRmut”) substantially improved transposition when used in combination with the hPBase. 56 Though side-by-side comparison in vivo demonstrated an increase of transgene integration mediated by IRmicro relative to original IRs, this was not the case with IRmut. 59,62 In terms of foreign DNA payload, in vitro studies of human and murine models showed that the PB system is capable of carrying up to 100 kb transgenes. 59,63,64 The PB platform was adapted to accommodate even 200 kb of bacterial artificial chromosomes for animal transgenesis. 64 Delivery of such large transgenes is not possible with viral vectors. Nevertheless, transfection efficiencies decrease with increased transgene size, warranting selective enrichment of those cells that had undergone stable transposition. 44 Taken together, the attributes of PB transposon make them particularly attractive for non-viral vector–based gene transfer. 19,65 –67
Tol2
The Tol2 transposable element belongs to the hAT (hobo/Ac/Tam3) superfamily and was discovered in 1996 in the medaka fish (Orizyas latipes) genome as the first autonomous transposon in vertebrate hosts. 68 Tol2 is approximately 4.7 kb in size and is composed of a gene encoding the transposase flanked by 12 bp TIRs. Tol2 mRNA contains four exons, which give rise to several transposase isoforms of which the 649-amino acid isoform is the most active variant. 69 Tol2 transposition generates a 8 bp TGD at the integration site. 70 Tol2 transposon is able to deliver a genetic cargo up to 10–11 kb in several mammalian cells without compromising transposition efficiency. 71 –74 Moreover, the Tol2- two-component system has been used successfully to generate transgenic animals such as mouse, chicken, frog, 75 –79 and particularly zebrafish models. 72,77,80,81 Like SB and PB, the Tol2 system enables sustained transgene expression after gene delivery, thereby providing the advantage of being suitable vector for gene therapy 72,73 (Fig. 1). To improve the efficacy of Tol2 system for gene transfer, Tol2 transposase was codon-optimized for enhancement of transposase expression in mammalian systems, including mouse and human. 69,82 Furthermore, the original Tol2 transposon was truncated to carry a 200 bp 5′ end and a 150 bp 3′ end to minimize the overall size of the Tol2 mobile element while retaining the transposition capacity (referred as “minimal Tol2”). This minimal Tol2 resulted in an approximately threefold increased transposition activity compared to the full-length Tol2 system. 61
Taken together, SB, PB, and Tol2 transposons are considered relatively efficient non-viral vectors for stable integration of the gene of interest into the target cell genomes, enabling long-term gene expression, even of relatively large transgenes. However, different transposon platforms do not function at comparable efficiencies, especially in the context of primary human cells or ultimately in clinical trials. The relative transposition efficiency of these different transposon systems varies depending on several confounding variables, including the cell type, transfection efficiency, DNA concentration, transposon/transposase ratio, and read-out (i.e., mobilization or rescue, gene expression, and antibiotic resistance). Consequently, head-to-head comparisons under the same conditions with rigorous controls, including inactive transposases, are necessary to determine the relative efficacy of different transposon systems. For instance, head-to-head comparative analysis showed that the SB100X and mPBase transposase-based systems were more robust than Tol2, at least in HeLa cells. 83 In addition, transposition in human CD34+ HSC showed that SB100X outperformed mPBase. 83
Ex vivo transposon delivery
Several approaches have been used to deliver transposons into the desired target cells to achieve sustained transgene expression. The introduction of transposons into host cells has to overcome the challenges of the intrinsic physiological and cellular barriers, including cell membranes and the instability of naked DNA due to DNA degradation. The delivery methods that have been employed to transfect transposons in vitro can be classified into DNA–chemical complexes (i.e., calcium phosphate, liposome, polymers, and nanoparticles), electroporation, and microinjection. Calcium phosphate transfection allows the formation of DNA–CaCl2 complexes that bind to the cell surface and are engulfed by host cells through endocytosis. This method is relatively simple compared to other techniques, and is therefore used in easy-to-transfect cell lines, yielding high transposition efficiency. 84,85 Similarly, chemical-based transfection can also be achieved by cationic liposomes and polyethyleneimine (PEI), 86 –89 which effectively binds to the transposon/transposase DNA and facilitates their interaction with the target cell membranes. However, most clinically relevant primary target cells such as CD34+ HSC, immune cells, and muscle cell progenitors are relatively refractory to these types of transfection methods. Electroporation has been used as an alternative transfection method for transposons in primary cells. It results in reversible membrane pore formation that facilitates transposon/transposase DNA uptake into the primary target cells, including CD34+ HSC, 43 T cells, 73 mesenchymal stem/progenitor cells, 45 iPS, 45 myoblasts, 45,66 MABs, 44 and iPS-derived progenitors (VandenDriessche, unpublished observations). Nevertheless, one limitation of electroporation is that it results in significant cell mortality, particularly when DNA is used. Magnetofection, which relies on magnetic nanoparticles delivered into cells using magnetic fields, is sometimes used for gene transfer into difficult-to-transfect cells and offers similar transfection efficiency to electroporation, depending on the target cells.
HSC
because of their self-renewal potential and ability to differentiate into different lympho-hematopoietic lineages, HSC are promising target cells for gene therapy. Initially, an early generation SB transposon (i.e., SB10) had been used to deliver genes stably into HSC, though the overall efficiencies were relatively modest, and no evidence was provided to demonstrate efficient hematopoietic reconstitution and multi-lineage marking in vivo. 90 Later, it was demonstrated, for the first time, that transposon-modified CD34+ HSC contributed to efficient and sustained multi-lineage gene marking in vivo using the SB100X system. 43 This demonstrated the potential of the hyperactive SB100X system for stable gene transfer in bona fide HSC that outperformed the early generation SB systems, creating new perspectives for HSC-based gene therapy. This was later independently confirmed in other studies. 91,92 It was then shown that the ability to use transposons to delivery genes stably into CD34+ HSC can be extended to the PB system, though SB100X was more efficient. 83 However, comparative studies with the latest generation hyPB would be required to determine which of the two systems is ultimately the most efficient. As a clinically relevant application, transposons were used to drive β-globin expression in patient-derived hematopoietic progenitors, resulting in a reduced sickling phenotype in red blood cells in vitro. 93
Mesenchymal and myogenic stem/progenitor cells
Myoblasts are self-renewing adult muscle progenitor cells that can undergo terminal myogenic differentiation into skeletal muscle fibers 94,95 that have been explored for cell therapy of muscle disorders. SB100X-mediated gene transfer was optimized to improve transposition efficiency up to 10-fold in myoblasts compared to SB11. 45 Correspondingly, the SB platform was used to deliver therapeutic genes stably, including human dysferlin 96 and micro-dystrophin, 97 a truncated dystrophin variant, in myoblasts that stably expressed the corresponding transgenes and retained their myogenic characteristics upon engraftment in mice. 96,97 Similarly, the PB system was also validated in myoblasts 44,98 at least in vitro.
SB- or PB-based transposon-based gene transfer enabled stable gene integration in mesenchymal stem cells (MSCs). 45,99 –103 In particular, successful transgene transposition in MSCs was demonstrated using SB100X technology, which resulted in up to 10-fold higher transposition efficiency compared to the SB11 system. The genetically engineered MSCs maintained the ability to undergo osteogenic, myogenic, and adipogenic differentiation after electroporation with the SB100X or PB transposon system. 45,99 The engineered MSC efficiently expressed coagulation factor IX (FIX), indicating that it is a suitable platform for production of secreted proteins. 45 Recently, adipose-derived MSCs secreting interferon gamma were generated using the hyPBase system as a basis for cancer immunotherapy in murine models. 103
MABs are capable of penetrating blood vessels during intra-arterial delivery, in contrast to myoblasts, which can only be delivered directly in the muscle tissue itself. MABs can undergo myogenic differentiation to promote muscle regeneration in dystrophic animal models. 104 –106 They can readily be engineered using transposon, resulting in persistent transgene expression in vivo upon transplantation in mouse models. 89 By virtue of their capacity to accommodate large inserts, it has been shown that PB transposons were able to deliver and express the full-length codon-optimized human dystrophin cDNA (∼11.1 kb) in dystrophic MABs when used in combination with the hyPBase. 44 The main advantage is that all of the functional domains were retained in contrast to when truncated dystrophins were employed, but the overall efficiency was reduced due to the intrinsic DNA toxicity after electroporation.
iPS cells
iPS cells can be generated by de novo expression of the quintessential reprogramming factors Oct4, Sox2, Klf4, and c-Myc. 107 Both SB- and PB-based transposons have been employed to express these reprogramming factors and successfully reprogram mouse, 108 –110 bovine, 111,112 porcine, 113 and human 110,114,115 fibroblasts into iPS cells. Direct comparative analysis between hyPBase- and SB100X-mediated gene transfer showed that both systems displayed comparable reprogramming efficiency. 109 One particular advantage of using transposons for iPS generation is that the reprogramming cassette can be excised using Cre/loxP, clustered regularly interspaced short palindromic repeats/CRISPR-associated protein-9 nuclease (CRISPR/Cas9) systems, or reintroduction of the transposase 108,116 (Fig. 2). Consequently, so-called transgene-free or genetically unmodified iPS cells can be generated that cannot re-express any potentially oncogenic reprogramming genes. In addition, this also minimizing potential concerns associated with insertional oncogenesis. The PB system generally restores the site of integration without duplicating or adding footprint in host genome upon removal of the reprogramming cassette in contrast to when SB or Cre/loxP technology was employed. This constitutes a “seamless” excision strategy for the generation of genetically unmodified iPS cells. 115
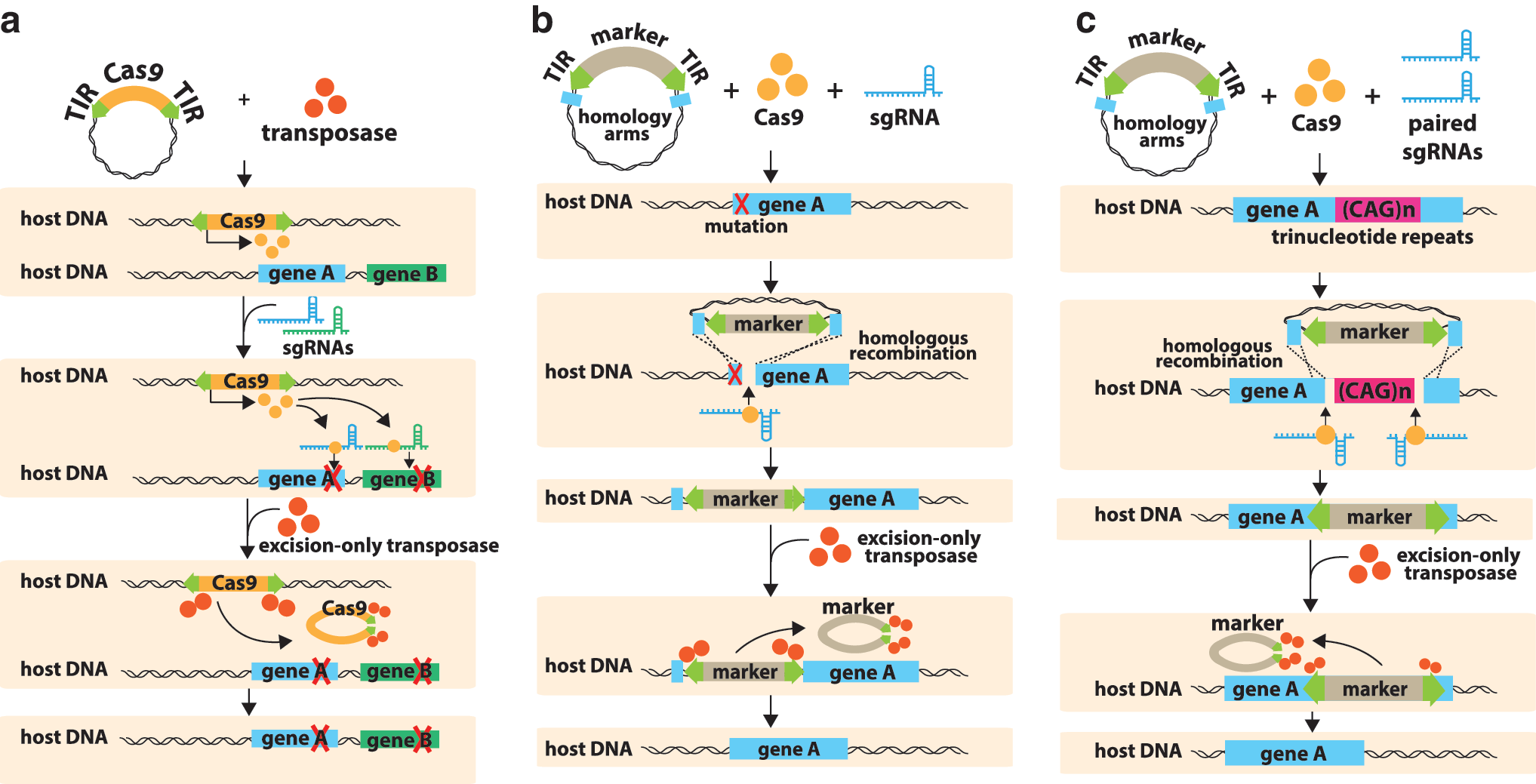
Schematic representation of possible strategies of combined PB-CRISPR/Cas9 for genome editing with seamless excision.
Transposons have also been used to introduce functional gene copies in patient-derived iPS cells containing defective genes. For instance, SB-mediated ectopic expression of micro-utrophin in dystrophic iPS-derived skeletal muscle progenitors restored the underlying muscle pathology by contributing to dystrophin–glycoprotein complex formation. 117 This can be achieved following transplantation of transposon-modified iPS-derived muscle progenitors that resulted in improved muscle contraction strength. Additionally, the authors and others have shown that transposons can be used to coax the differentiation of iPS cells into their relevant differentiated cells. For instance, transposon-based ectopic expression of myogenic transcription factors such as PAX3 and MYOD1 induced the differentiation of iPS cells into muscle cell progenitors. 45,118
Gene editing of an aberrant allele in iPS cells can be achieved following co-delivery of a PB transposon carrying a correct donor sequence together with a site-specific designer nuclease, such as a transcription activator-like effector nuclease (TALEN), zinc finger nuclease (ZFN), or CRISPR/Cas9 (Fig. 2). The sequence-specific double-strand DNA break facilitates homology-directed integration of the donor DNA at this target locus. Subsequent transient excision-only PBase activity allows for the seamless excision of selectable marker genes that are part of the targeting cassette. 119 This hybrid platform was previously used for gene editing in iPS cells derived from patients with sickle cell disease caused by a β-globin (HBB) gene mutation or Huntington's disease caused by trinucleotide repeat expansions in the huntingtin gene. 120 –122
Primary T-lymphocytes
Immunotherapy based on redirected T cells expressing chimeric antigen receptors (CARs) holds great promise for cancer therapy. 123 The first use of transposons for T cell engineering was based on the use of SB10 to target the CD19 antigen. Transposon-modified redirected T cells efficiently mediated specific target cell lysis and cytokine production upon the exposure to CD19-positive leukemia cells and contributed to potent tumor remission in a murine model xenografted with tumor cells expressing CD19. 124,125 Subsequently, SB100X for CAR T cell generation exerted superior transposition activity (fourfold higher) compared to SB11 and retained efficient target cell lysis. 126 SB-based CAR T cells were also successfully generated to treat acute lymphocytic leukemia 127 and sarcomas. 128 –130 Similarly, PB transposons were explored to generate CAR T cells redirected against different antigens and cancer types, 131 –133 including myelomonocytic leukemia, 134 breast cancer, 135 and cholangiocarcinoma. 136 Tol2-mediated CD19-specific CAR expression in T-lymphocytes was recently demonstrated to suppress B cell lymphoma progression both in vitro and in vivo. 73 Comparative analysis indicated that PB was more robust than SB11 and Tol2 systems. 137
In vivo transposon delivery
Transposon-mediated gene transfer has also been validated for in vivo transfection. Typically, for liver-directed gene delivery, transposon and transposase constructs were administered by hydrodynamic (HD) injection that involves rapid delivery of large volumes of DNA solutions. 138 HD transfection of transposons resulted in sustained gene expression in liver and kidney. 59,62,139 –142 In the absence of an active transposase, expression declined, perhaps due, at least in part, to epigenetic silencing, indicating that genomic integration was required to circumvent this and achieve stable expression. Moreover, in vivo stable inducible gene expression system can be established by transposon technology, allowing temporal control of transgene expression. 143
HD-based transposon gene transfer has been utilized to direct long-term therapeutic gene expression in various inherited and acquired genetic disease models, including hemophilia A 144,145 and B, 141,146 sickle cell disease, 147 mucopolysaccharidosis (MPS), 148 –150 type I tyrosinemia, 151 type I diabetes, 152 familial hypercholesterolemia (FH), 153 and type I Crigler–Najjar syndrome 154 (Table 1). SB transposon in conjunction with wild-type SBase or first-generation hyperactive SBase such as SB11 and HSB5/17 were initially used and were sufficient to enhance therapeutic gene expression and correct deficient phenotypes. For example, insulin expression mediated by native SB system significantly restored blood glucose to normal levels following glucose infusion in diabetic mice. 152 In particular, hemophilia B treatment in mouse and canine models can be achieved by standard SB 141 or HSB5 146 platforms to alleviate bleeding diathesis and sustain physiological FIX levels. 146 SB technology for in vivo gene therapy in MPS and hemophilia A was also validated and led to phenotypic correction following gene delivery. 145,148, 149,155 However, transgene expression dramatically decreased over time because of the subsequent development of immune response against therapeutic gene products. 148 To overcome this limitation, immunomodulatory agents such as gadolinium chloride, and cyclophosphamide were used. 148,156 Alternatively, the immunosuppressive enzyme indoleamine 2,3-dioxygenase (IDO) 144 was employed to boost immune tolerance and prolong transgene expression in mouse models. In a recent study in MPS mice, transposition efficacy of transgene gene mediated by SB100X was 15-fold more efficient than with the SB11 system and contributed to persistent therapeutic gene expression levels for 1 year. 156 In addition, the SB100X technology encoding low-density and very-low-density lipoprotein receptors contributed to long-term therapeutic effect in FH mice. 153 Therefore, the SB100X system holds great promise for further development of in vivo gene therapy to enhance transposition efficacy and stable expression of therapeutic genes.
Sleeping Beauty (SB) transposon application in gene therapy research
If not specifically indicated, two-component SB transposon system was used in conjunction with SB10.
FPL, fusogenic galactose-terminated F-glycoprotein of the Sendai virus; HD, hydrodynamic injection; HSC, hematopoietic stem cell; IV, intravenous injection; MSC, mesenchymal stem cell; RPE, retinal pigment epithelium; PEI, polyethylenimine.
Comparative studies indicated that hyPBase contributed to a threefold increase of transposition efficiency compared to the SB100X system, at least in one experimental model system. 157 PB transposons were successfully used to correct von Willebrand's disease (vWD), 158 unilateral ureteral obstruction (UUO), 159,160 and hemophilia 161 (Table 2). It has been demonstrated that PB transposons efficiently directed expression of insulin-like growth factor-1 receptor (IGF-1R) and glutathione transferase isozyme A4 (GSTA4) and subsequently attenuate renal fibrosis formation, one of the major deficient symptoms in mice with UUO. 159,160 In addition to wild-type PBase, 162 hyPBase system mediated long-term expression of blood clotting factor VIII (FVIII) and FIX and correct coagulation defects in hemophilia A and B mice, respectively. 59,62,163 Comparative analysis in the hemophilia B murine model indicated that the hyPBase was more efficient than mPBase, resulting in higher stable FIX expression levels. 59,62 PB-mediated expression of FIX and FVIII was long lasting (>300 days) and did not induce an immunological reaction. 59,62,162
PiggyBac (PB) transposon application in gene therapy research
If not specifically indicated, two-component PB transposon system was used in conjunction with wild-type PBase.
CRISPR/Cas9, clustered regularly interspaced short palindromic repeats/CRISPR-associated protein-9 nuclease; iPS, induced pluripotent stem cell; MAB, mesoangioblast; TALEN, transcription activator-like effector nucleases; ZFN, zinc finger nuclease.
As a clinically feasible approach for transposon-based gene therapy, HD injection can be assisted by catheterization for local delivery of DNA in larger animal models such as pig, 164 dog, 165,166 and monkey. 60 It remains to be seen whether this method could eventually be applied safely and efficaciously in a clinical setting, given that hepatotoxicity ensued following HD injection. In some cases, immune suppression (e.g., with gadolinium chloride [GdCl3]) was warranted to prevent immune responses against the transgene product, as in the case of α-L-iduronidase (IDUA) and β-glucuronidase (GUSB 166 ). Long-term FIX expression was also seen in the canine model (in the presence of GdCl3). Sustained therapeutic IDUA, GUSB, and FIX expression levels were able to be attained after SB transposition in dogs, which is an important step toward future clinical translation. As an alternative, PEI transfection has been explored as an alternative to HD in order to mediate in vivo transposon delivery through intravenous infusion into mice but was at least 100-fold less efficient compared to HD delivery. 167 Recently, in vivo administration of nanoparticles carrying transposons has emerged as an efficient method to deliver transgenes into circulating T cells in mice, and in situ engineered T cells exhibited similar activity to conventional engineered T cells generated by ex vivo gene transfer. 168 Nanoparticles represents a potentially attractive technology to deliver transposons into liver sinusoidal endothelial cells. 169 Intramuscular electroporation of transposons in murine models resulted in transient and localized transgene expression. 89 To overcome the intrinsic challenges of non-viral transfection, transposons have also been accommodated in conventional viral vectors, including adenoviral, 47,146,170,171 herpes-simplex viral, 172 AAV, 170 and integration-defective lentiviral vectors. 173 –175 Some of these viral vector-based transposon delivery approaches were used for in vivo delivery and resulted in high transduction efficiency and persistent expression in murine 146,170,172 and canine models. 146 Nevertheless, these hybrid systems inevitably result in some disadvantages due to the viral vector components, such as potential immunogenicity.
Transposon-Based Gene Therapy in Clinical Trials
There are multiple ongoing clinical trials using SB transposons for ex vivo gene therapy. The first approved transposon-based clinical application was based on early-generation SB technology to generate CAR T cells for adoptive immunotherapy in patients with CD19+ B-lymphoid malignancies (i.e., non-Hodgkin lymphoma and acute lymphoblastic leukemia), spearheaded by Cooper et al. 176 In an attempt to decrease the relapse rate in these patients, they were injected with autologous transposon-modified CAR T cells along with autologous HSC. Alternatively, patients were treated with allogeneic transposon-modified CAR T cells and allogeneic HSC. 173,174 The CAR T cells were selectively expanded in vitro >1,000-fold, resulting in 80–90% CAR expressing T cells. This required a protracted expansion of the gene-modified CAR T cells on an “artificial” antigen-presenting cell line (derived from K562) that expressed CD19, CD86, CD137L, and membrane-bound IL-15. Thirty months after autologous HSC transplantation, progression-free survival (PFS) 173 was 83%, and overall survival (OS) was 100%. PFS and OS corresponded to 53% and 63%, respectively, following allogeneic HSC transplantation. This is lower than in the autologous setting, possibly due to the need for immune-suppression to prevent graft-versus-host disease (GvHD). Regardless, it would appear than the adoptive transfer of the transposon-modified CAR T cells significantly increased PFS and OS compared to historical controls based on autologous or allogeneic HSC transplantation, without CAR T cell therapy. There was no evidence of cytokine release syndrome, exacerbated GvHD, or other overt toxicities. Nevertheless, mild GvHD was reported in a few allogeneic recipients. The transposon-modified CAR T cells persisted for an average of 201 days in the autologous and 51 days in the allogeneic transplantation setting, with no evidence of integration hotspots of the transposon in the CAR T cell genome or malignant transformation of the CAR T cells. Control trials are needed to corroborate these encouraging results. Several clinical protocols have been initiated based on the same principle use of CD19-specific CAR T cells that varied based on the origin of the T cells (e.g., autologous, allogeneic, cord blood), cell dose, or lympho-depletion therapy (NCT00968760, NCT01497184, NCT01653717, NCT02529813, and NCT01362452).
One potential caveat of the original CAR T cell in vitro expansion protocol is that it takes several weeks. Consequently, this protracted cell culture period may potentially compromise the effectiveness of the CAR T cells, at least in part due to cellular exhaustion and/or impaired engraftment of fully functional CAR T cells. Increasing the overall transposition efficiency by using hyperactive SB100X, mini-circle plasmids or combinations thereof may also require a less prolonged in vitro enrichment period of the transposon-modified CAR T cells. 126,177,178 Even mini-circle plasmids entirely devoid of antibiotic resistance genes can potentially be used. 177
Alternatively, co-transfection of the SB transposase construct with two distinct transposons, one encoding CAR and the other mIL15, can potentially shorten the in vitro expansion period. 178 This is based on the premise that the proliferation of the CAR/mIL15 transposon-modified T cells continues in vivo after transplantation, offering a selective survival advantage. Adoptive transfer of transposon-modified T cells that express both CAR and mIL15 in CD19+ tumor-bearing mouse models are more effective and persistent longer than the first-generation CAR T cells. Interestingly, they also display a transcriptional profile and marker gene expression closely resembling that of bona fide memory T cell,s which would be advantageous with respect to enhancing in vivo persistence of the modified CAR T cells. It is likely that this CAR T cell concept based on transposon-modified T cells will benefit from the advances made in lentiviral vector modified CAR T cells that are now poised to target other lymphohematopoietic malignancies and solid tumors. In addition to using CAR-modified T cells, SB transposons have also been explored to express T cell receptors specific for MHC class I-restricted neoplastic neoantigens that are frequently observed in epithelial malignancies. 179 Though cancer has so far been the main target disease for transposon-based therapies, this sets the stage for targeting other diseases with transposon-modified cells. In particular, a clinical trial is imminent based on the use of SB100-modified pigment epithelial cells expressing anti-angiogenic factors to treat age-related macular degeneration (AMD). 174,180
Insertional Oncogenesis
Insertional mutagenesis is one of the main concerns using integrative-based gene therapy. Genomic integration may potentially activate oncogenes or interrupt tumor-suppressor genes and hence contribute to malignant transformation. Several studies have been performed to identify the preference of DNA integration sites mediated by transposons. SB transposon appears to integrate at central TA dinucleotide surrounded by palindromic AT repeats, which contribute to bendable DNA conformation. 181 Target site of PB transposon is known to be TTAA-rich region, regardless of the surrounding AT content, 182 whereas Tol2 transposon tends to integrate at weak consensus palindromic-like TNA(C/G)TTATAA(G/C)TNA octanucleotides. 69,183 In terms of genomic integration pattern, PB transposon demonstrated highest integration preference in transcriptional units (∼50% into RefSeq genes) relative to SB and Tol2 (ranging from 40% to 45% into RefSeq genes). Whereas SB seemed integrate quite randomly in the target genome, PB and Tol2 transposons exhibited a stronger biased integration toward DNaseI hypersensitivity site, CpG islands, and transcriptional start sites. 137 Therefore, SB seems to have a more favorable integration profile compared to PB and Tol2 transposons, though this may also be influenced by the cell type. Ultimately, the safety and estimated risk of insertional oncogenesis of a given transposon construct were tested in tumor-prone mouse models. 59 Minimizing the risk of insertional mutagenesis mediated by transposition is an important priority to move this technology forward. The first strategy is to design the expression cassette itself carefully and avoid the use of elements such as strong viral enhancers, which potentially increase cis-activation of neighboring oncogenes at the integration locus. Alternatively, it is possible to redirect transposon integration toward validated “safe-harbor” sites. This can be achieved by transposase modification at N- or C-terminal regions to carry site-directed DNA binding domain, thereby enabling site-specific transposition at these designated target regions in the genome. 184 –186 From a safety perspective, it is encouraging that the transposon TIRs have only weak enhancer activity, in contrast to the γ-retroviral vector long terminal repeats that carry a greater risk of insertional oncogenesis. 187 Nevertheless, to augment the overall safety of transposon further and to minimize the risk of insertional oncogenesis, insulators have been considered. 188 However, these insulators may also have adverse epigenetic effects or may contribute to vector instability, which would need to be weighed against their potential benefits.
Conclusions and Perspectives
DNA transposon systems, particularly those based on SB, PB, and Tol2, have emerged as promising alternatives to overcome some of the manufacturing constraints associated with the use of viral vectors. Preclinical and clinical studies rely on the transient expression of the transposase enabling stable transposition of the corresponding transposon in the target cell genome. Though initial studies relied on the co-transfection of the transposase-containing expression construct with the transposon containing the gene of interest, subsequent studies showed that transient delivery of the transposase by mRNA would be preferred, since this would minimize the risk of continued transposon remobilization. The original SB and PB systems have been engineered to augment their overall transposition efficacy further by introducing specific mutations giving rise to the SB100X and hyPB systems that are among the most efficient ones for preclinical studies and clinical applications. The DNA transposons are very versatile and have been validated for ex vivo and in vivo gene therapy for many disease targets. Despite their promise, delivery remains a challenge requiring the use of non-viral transfection methods that often result in significant cytotoxicity. Though direct in vivo applications using transposons have often relied on the use of viral vectors, it would be preferred to use a safe and efficient non-viral transfection system instead, given the known immune ramifications associated with the use of viral components. Unfortunately, the overall efficacy of non-viral in vivo transfection is often limited and/or the transfection methods used are not readily amenable for clinical applications (e.g., hydrodynamic transfection). Nevertheless, the development of novel non-viral nanoparticle technologies may open new perspectives for in vivo delivery of transposons. This warrants further investigation in preclinical disease models and requires robust optimized methods for large-scale nanoparticle manufacturing. The use of transposons should also be considered in comparison with gene editing technologies based on designer nucleases (i.e., ZFN, TALEN, CRISPR/Cas9, homing endonucleases). The relative efficacy and safety of transposons versus gene editing should be carefully assessed on a case-by-case basis and depends on several confounding variables, including the disease target, cell type, transfection method, and so on. Nevertheless, for some applications, transposons can be used in combination with these gene editing technologies (e.g., “seamless” gene editing of iPS cells). The use of transposon technology to generate CAR T cells for cancer immunotherapy constitutes one of the most promising clinical applications to date. Moreover, these advances set the stage for future clinical trials to treat patients suffering from other genetic or acquired diseases.
Footnotes
Acknowledgments
We thank the members of the Department of Gene Therapy and Regenerative Medicine and our collaborators for their various contributions to some of the work presented in this review. We also wish to thank FWO, AFM, VUB-IOF-GEAR (GENEFIX), SRP-Groeier, and Willy Gepts for providing financial support. We thank Dr. Chai for critical reading of the manuscript.
Author Disclosure
No competing financial interests exist.