
Abstract
The clinical success of gene-engineered T cells expressing a chimeric antigen receptor (CAR), as manifested in several clinical trials for the treatment of B cell malignancies, warrants the development of a simple and robust manufacturing procedure capable of reducing to a minimum the challenges associated with its complexity. Conventional protocols comprise many open handling steps, are labor intensive, and are difficult to upscale for large numbers of patients. Furthermore, extensive training of personnel is required to avoid operator variations. An automated current Good Manufacturing Practice–compliant process has therefore been developed for the generation of gene-engineered T cells. Upon installation of the closed, single-use tubing set on the CliniMACS Prodigy™, sterile welding of the starting cell product, and sterile connection of the required reagents, T cells are magnetically enriched, stimulated, transduced using lentiviral vectors, expanded, and formulated. Starting from healthy donor (HD) or lymphoma or melanoma patient material (PM), the robustness and reproducibility of the manufacturing of anti-CD20 specific CAR T cells were verified. Independent of the starting material, operator, or device, the process consistently yielded a therapeutic dose of highly viable CAR T cells. Interestingly, the formulated product obtained with PM was comparable to that of HD with respect to cell composition, phenotype, and function, even though the starting material differed significantly. Potent antitumor reactivity of the produced anti-CD20 CAR T cells was shown in vitro as well as in vivo. In summary, the automated T cell transduction process meets the requirements for clinical manufacturing that the authors intend to use in two separate clinical trials for the treatment of melanoma and B cell lymphoma.
Introduction
M
CD20 is stably expressed at a high level on >90% of patients with B cell lymphomas,
13
as well as on a minor subset of cancer-sustaining melanoma cells.
14,15
In line with this observation, rituximab, an anti-CD20 specific monoclonal antibody, has a major clinical impact on lymphomas, including diffuse large B cell lymphoma (DLBCL), follicular lymphoma, and chronic lymphocytic leukemia (CLL).
16
–18
Interestingly, in a case study, rituximab also induced established metastatic melanoma regression in patients,
19,20
leading to a clinical Phase II trial (
The authors therefore set out to develop anti-CD20 CAR T cells for such indications and generated a second-generation CAR construct comprising an extracellular single-chain variable fragment (scFv) directed against CD20 and linked to intracellular signaling domains (4-1BB_CD3ζ).
Manufacturing CAR T cells is complex. It requires skilled operators, as well as a dedicated infrastructure. Current processes face challenges toward meeting potentially large demand upon growing success. 21 This study sought to develop, optimize, and verify the fully closed, automated T cell transduction (TCT) process on the CliniMACS Prodigy™ platform to prepare genetically engineered T cells in <2 weeks under closed conditions, starting from leukapheresis (LP), buffy coat (BC), or whole blood (WB). This system has already been demonstrated as a current Good Manufacturing Practice (cGMP)-compliant protocol for the generation of engineered T cells on a clinical scale, starting from LP from healthy donors (HD). 22,23 However, this study demonstrates the eligibility of the TCT process for the manufacture of anti-CD20 CAR T cells, whereby starting material is also obtained from heavily pretreated patient-derived material (PM). Furthermore, the robustness and reproducibility were assessed by using diverse devices on different days controlled by varying operators. This article reports on the cell composition, phenotype, and function of CAR T cells generated from HD and PM, and shows the adequacy of the developed process.
Material and Methods
Starting blood products
Fresh BC (DRK Dortmund) or non-mobilized LP (Universitätsklinikum Köln or DRK Ulm) was obtained from HD and typically used after 24–48 h after blood donation. Transportation was performed at room temperature (RT). For T cell preparation from PM, either non-mobilized LP product derived from DLBCL patient or WB sample derived from a melanoma patient was provided from Universitätsklinikum Köln. DLBCL patients were heavily pretreated and were undergoing relapse at time of harvest. No exclusion criteria were set for LP or WB samples regarding prior treatments (e.g., lymphodepletion, chemotherapy, or therapeutic antibodies) or the presence or numbers of leukemia blast in the peripheral blood.
Cryopreservation
Peripheral blood mononuclear cells were prepared from fresh non-mobilized LP by density gradient centrifugation and formulated in TexMACS Medium (Miltenyi Biotec) supplemented with 20% human AB serum (Gemcell) and 10% dimethyl sulfoxide (Sigma–Aldrich) at a density of 2 × 107 white blood cells (WBC)/mL. For freezing, cells were stored at −70°C using a freezing container (Mr. Frosty; Nalgene) and transferred afterwards for long-term storage in liquid nitrogen. After thawing (water bath at 37°C), cells were immediately washed twice in pre-warmed (37°C) TexMACS medium without supplements, plated with 5 × 106 WBC/mL in a T175 flask (Corning), and allowed to recover for 24 h at 37°C at 5% CO2. After recovery, cells were harvested, washed, and reformulated in 50 mL 1 × phosphate-buffered saline (PBS) supplemented with 1 mM of EDTA. Cell suspension was transferred into a 150 mL transfer bag (Miltenyi Biotec) and connected to the installed tubing set for following the automated enrichment process.
Cell line
JeKo-1 cell line was obtained from Leibniz Institute DSMZ (DSMZ No. ACC 553) and cultured in RPMI 1640 supplemented with 10% fetal bovine serum (Biochrome).
Automated generation of genetically modified T cells
The TCT process on the CliniMACS Prodigy™ in combination with the TS520 was used for the entire process. Upon selection of the TCT application program, all parameters of stimulation, washing, and feedings used in the current study are available to the user as preloaded conditions. A description of the process is given below. The single-use disposable tubing set TS520 consists of a pump tubing, a separation column for magnetic cell selection, the chamber for cell processing and cultivation, several input and output lines for buffer, media, reagents, product bags, and waste. Technical features have previously been described in detail by Apel et al. 24
Blood products were analyzed for total WBC concentration and target cell frequency (CD4+ plus CD8+ among CD45+) prior to process, and a maximum of 3 × 109 target cells were processed on the CliniMACS Prodigy™. Process buffer CliniMACS PBS/EDTA (Miltenyi Biotec) supplemented with 0.5% bovine serum albumin (Miltenyi Biotec) or 0.5% human serum albumin (HSA; Grifols), as well as TexMACS GMP medium (Miltenyi Biotec) supplemented with 3% heat-inactivated human AB serum (Gemini Bio-Products; pooled from a maximum of 25 donors), 12.5 ng/mL of recombinant human interleukin (IL)-7, and 12.5 ng/mL of recombinant human IL-15 (Miltenyi Biotec) were prepared before starting the automated process. Process parameters were entered into the “activity matrix” in order to define the conditions of the automated run. Initial priming, sample preparation, and cell labeling was performed by the instrument at 4–8°C. Cell labeling with magnetic beads was performed using CliniMACS CD4 Reagent and CliniMACS CD8 Reagent (Miltenyi Biotec) for 30 min at 4–8°C. After magnetic separation, target cells were eluted in TexMACS GMP medium, and cell concentration of the enriched fraction was determined by the operator and input to the application program. For the automated cultivation, 2 × 107 to a maximum of 1 × 108 cells were automatically transferred into the chamber of the CliniMACS Prodigy™ from the bag containing the enriched T cells, washed, and the T cells were then activated with the contents of one vial (4 mL) of the MACS GMP T Cell TransAct (Miltenyi Biotec) in final volume of 70 mL of media containing cells (corresponding to a 17.5-fold dilution of the reagent), according to the manufacturer's instructions. T cells were transduced 24 h after T cell stimulation using lentiviral vectors (LV). To remove excess stimulation reagent and LV, culture wash was automatically performed 3 days after stimulation, and culture was switched from static culture to agitated culture. Cultivation volume was increased subsequently through automated feeding to 250 mL. Automated media exchange via centrifugation was executed every day via replacement of a maximum of 180 mL of culture medium. After 6 days of cultivation, the media bag was exchanged (TexMACS GMP supplemented with 12.5 ng/mL of recombinant human IL-7 and 12.5 ng/mL of recombinant human IL-15 (Miltenyi Biotec); no heat-inactivated human AB serum was used for the second media bag). Formulation and harvest of cells was performed using either TexMACS medium or Composol (Fresenius) supplemented with 2.84% HSA (Grifols). Cultivation samples were taken frequently. Cell count as well as viability were analyzed via hemocytometer and erythrosine B staining. The pH value of the culture samples was measured using pH test strips (MColorpHast, pH 6.5–10; Merck). Glucose concentration was measured using a handheld blood sugar meter (ACCU-CHECK Aviva). For the enriched fraction (day 0), the in-process control (day 5/6), and for the final cellular product (day 12), samples were taken for flow cytometric analysis to determine cellular composition, T cell phenotype, and transduction efficiency.
Antibodies and flow cytometry
Cellular composition, T cell phenotype, and transduction efficiency were determined using the following antibodies (all fluorescent antibodies were obtained from Miltenyi Biotec) and protocols. Cellular composition of the starting material and the enriched fraction was determined after red blood cell lysis (10 min at RT with red blood cell lysis solution; Miltenyi Biotec). Subsequently, cells were labeled with fluorescent antibodies against CD45, CD56, CD16, CD3, CD4, CD8, and CD14 (10 min at 4–8°C). T cell phenotype and composition were analyzed for the enriched fraction, the in-process control, and the final product using fluorescent antibodies against CD62L, CD45RO, CD95, CD3, CD4, and CD8. Transduction efficiency of the in-process control and the final product was determined using fluorescent antibodies against CD45, CD3, CD4, CD8, and CD14. CD20 CAR staining was performed in the same sample using anti-CD20 CAR peptide linked to PE. 7-AAD was added to each staining panel to discriminate death cells. Flow cytometric analysis was performed with the MACSQuant Analyzer 10 and analyzed with the MACSQuantify 2.8 software.
Anti-CD20 CAR LV
T cells were automatically transduced (for each run 200 × 106 transducing units were formulated in 10 mL of culture medium, transferred to a 150 mL transfer bag [Miltenyi Biotec] and sterile connected to the tubing set) to express the PGK promoter-driven CAR targeting CD20+ cells. Transducing units were defined by titrating concentrated LV preparations on SupT1 cells. Therefore, a lentiviral anti-CD20 CAR encoding construct comprising a Leu-16-derived scFv linked to a CD8 spacer domain, a CD8 transmembrane region, and the signaling modules of 4-1BB, and CD3ζ was designed on the basis of a retroviral anti-CD20 CAR vector kindly provided by H. Abken (ZMMK, Cologne) and originally described by Schmidt et al. 15 VSV-G pseudotyped LV was produced using HEK293T cells, and the supernatant was concentrated and stored at −70°C until transduction. A Tagman duplex real-time quantitative polymerase chain reaction was performed on a CFX96 Touch™ Real-Time PCR Detection System (Biorad) to determine the vector copy number (VCN). For the quantification of the integrated provirus, a primer and probe were designed, specifically amplifying a 89 bp fragment between 5′ LTR and the internal promoter. A cellular reference gene was analogously quantified, as described in Maetzig et al. 25 A plasmid containing both the viral and cellular target sequences was constructed and served as standard. The ratio of viral to cellular copies was calculated, and the transduction efficiency was also taken into account to determine the VCN for the transduced cells only.
Cytotoxicity assay
To assess the cytolytic activity of the engineered T cells, 1 × 104 CD20+ JeKo-1 cells were labeled with 1 μM CellTrace™ Violet (Life Technologies) and co-cultured for 24 h with anti-CD20 CAR T cells at indicated effector-to-target ratios in round-bottom 96-well plates. Specific lysis was detected by quantification of viable violet-labeled target cells via flow cytometry. Mock-transduced T cells were used as control in the same effector-to-target ratios.
Cytokine secretion
A total of 1 × 105 JeKo-1 cells and 1 × 105 anti-CD20 CAR T cells were co-cultured for 24 h in 96-well round bottom plates. The cytokine secretion of anti-CD20 CAR T cells was determined in the supernatant using the MACSPlex Cytokine 12 Kit, human (Miltenyi Biotec), according to the manufacturer's instructions.
In vivo experiments
All experiments performed on animals follow institutional guidelines and regulations.
To analyze the in vivo functionality of the manufactured T cells, 5 × 105 RajiffLuc cells were injected via the tail vain into NOD SCID gamma (NSG; NOD.Cg-PrkdcscidIl2rgtm1Wjl /SzJ) mice. After 7 days, either 1 × 106 anti-CD20 CAR T cells or mock T cells were infused intravenously (i.v.). Antitumor response was measured frequently using an in vivo imaging system (PerkinElmer) after injecting 100 μL (30 mg/mL) XenoLight D-Luciferin (K+ Salt) Bioluminescent Substrate (PerkinElmer). Bone-marrow samples were collected from the femur and tibia of both legs. Spleens were manually dissociated. Red blood cells were lysed, and samples were stained using CD45, CD3, CD4, CD8, and CD19 (Miltenyi Biotec) prior to flow cytometric analysis.
Ethical concerns
The Institutional Review Board and ethics committee of the University of Cologne approved sample collection for this study. Each patient provided written informed consent before sample collection in accordance with the Declaration of Helsinki.
Results
Automated TCT process
LP, WB, and BC derived from HD or PM were utilized as starting material to assess the feasibility and the robustness of the fully automated TCT application on the CliniMACS Prodigy™ (Fig. 1). On day 0, cryopreserved or fresh material was sterile welded onto the closed tubing set (TS520) to isolate CD4+ and CD8+ T cells magnetically, which were subsequently activated using MACS GMP T Cell TransAct, a nanomatrix-based polyclonal T cell stimulation reagent that significantly improves the transduction process by inducing CD3-CD28-mediated cell activation. For viral transduction, 24 h after T cell stimulation, VSV-G pseudotyped anti-CD20 CAR lentiviral particles were aseptically connected to the tubing set. During the subsequent expansion phase, feeding with T cell medium TexMACS supplemented with recombinant human cytokines IL-7 and IL-1526 (and until day 6 with human 3% [v/v] AB serum) takes place daily from days 6–11. On day 12, the final cell product was washed, formulated in Composol (Fresenius), and harvested. Over the entire period of the TCT process, QC pouches enabled in-process assessment of cell count, viability, pH value, and glucose consumption.
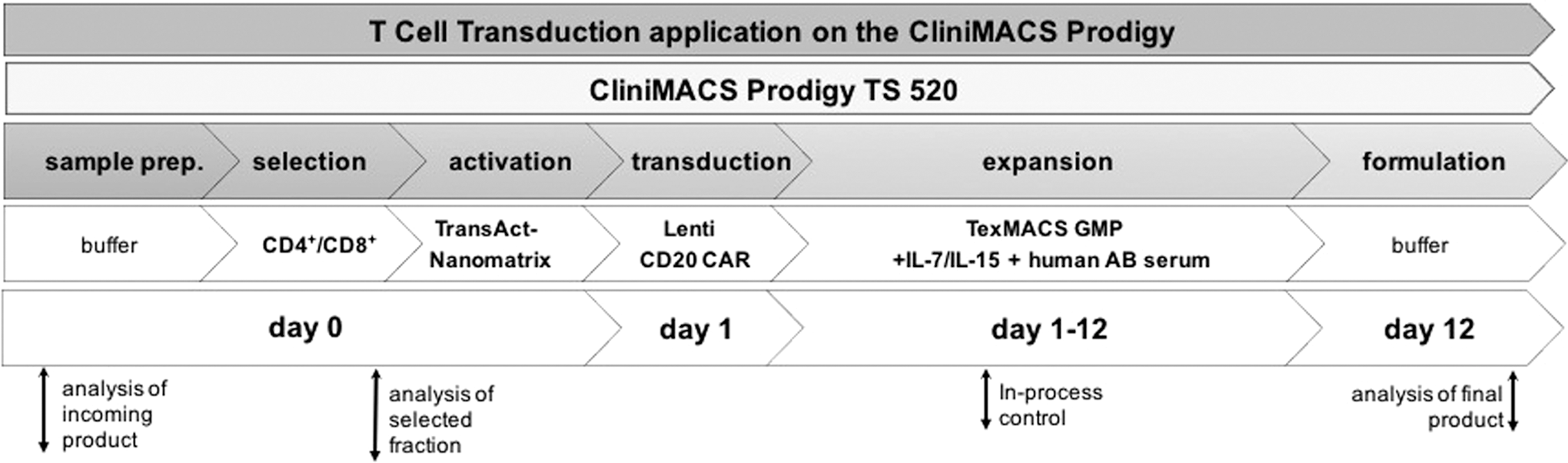
Workflow of automated T cell transduction (TCT) process. The manufacturing of genetically modified T cells is performed on the CliniMACS Prodigy™ platform, allowing the entire 12-day process in one single closed tubing set (TS520). Day 0: Starting from leukapheresis, whole blood, or buffy coat, samples were prepared, and T cells were enriched by positive magnetic selection combining CD4 and CD8. Subsequently, T cells were activated using MACS GMP T cell TransAct for polyclonal T cell stimulation. Day 1: Lentiviral transduction using vectors encoding for an anti-CD20 CAR. Days 1–12: T cell expansion phase, including feeding steps via adding (feed) or replacing (medium exchange) T cell medium TexMACS supplemented with recombinant human cytokines interleukin (IL)-7 and IL-15 and 3% human AB serum (until day 6). Day 12: Cells were washed and harvested during formulation. A static culture was performed to enable optimal stimulation and transduction (until day 3), followed by culture agitation, allowing sufficient gas support at higher culture volumes. In-process control and quality controls were performed at the indicated time points.
CliniMACS Prodigy™ enables reproducible cGMP-compliant manufacturing of gene-engineered T cells starting from different cell samples and conditions
The TCT process was evaluated in 15 runs using four LP, as well as one BC from HD, two LP from DLBCL patients, and one melanoma-patient-derived WB-sample (Table 1), in order to test the reproducibility of the process when challenged with high variance in starting material, not only with regard to their status (fresh or frozen), their abnormal cell phenotypes, or varying cellular compositions that might hamper the TCT process, but also in terms of limited amount of material being available to start the process. Flow cytometric analysis of the incoming product revealed strong differences in the cellular composition. Starting viable CD3+ cell frequencies varied from 4% (PM; WB-3) to 83% (HD; LP-11 and LP-12; Table 1). Upon combined CD4 and CD8 enrichment, the majority of T cells were obtained. However, natural killer (NK) cells and natural killer T (NKT) cells were co-enriched due to their low CD8 expression. Monocytes were greatly reduced, even though they are CD4dim. Granulocytes and B cells were generally significantly reduced (Supplementary Fig. S1; Supplementary Data are available online at
Overview of material used for automated manufacturing of gene-modified T cells on the CliniMACS Prodigy
Split pack run: enriched T cell fraction was used for manufacturing runs on different CliniMACS Prodigy™ devices.
LP, leukapheresis; BC, buffy coat; WB, whole blood; HD, healthy donor; TNC, total nucleated cells; DLBCL, diffuse large B cell lymphoma.
After enrichment of CD4+ and CD8+ T cells, typically, 1 × 108 cells were redirected to the chamber for stimulation unless a reduced amount of cells was available (Table 1), while the rest of the enriched cells remained in the target cell fraction bag to be cryopreserved for later use. T cell activation status was detected by assessing the typical clustering of activated T cells using the integrated microscope camera (Fig. 2A). This qualitative measurement during the early static culture phase enabled a first in-process control without perturbing the initial T cell activation phase or requiring sampling (and thus avoiding early cell loss). Measurements of cell numbers during the dynamic expansion phase (past day 3) showed a robust cell expansion, with exponential growth starting on day 4/5 (Fig. 2B). All HD and DLBCL runs yielded comparable expansion curves. Only the run with melanoma-patient blood showed a delay, which is associated with the fivefold lower starting cell counts (2 × 107 cells). The final T cell numbers after 12 days ranged from 2.4 × 109 for the melanoma patient to 3.2 × 109–4.9 × 109 for DLBCL patients and 2.9 × 109–6.3 × 109 for HD (Fig. 2C). No impact on the expansion was observed for run LP-9 and LP-10 that utilized cryopreserved LP from a DLBCL patient with only 6 × 107 cells as the starting number instead of the usual 1 × 108 cells. In addition, ranging from 14.0 × 106 ± 3.7 × 106 cells/mL for PM compared to 17.2 × 106 ± 5.5 × 106 cells/mL for HD, no significant differences could be observed for the final cell densities (Fig. 2D). Viability remained high (Fig. 2E), and no statistically significant differences in viable leukocytes were observed in the final product with 90.4 ± 4.3% for HD and 88.4 ± 7.4% for PM (Fig. 2F). The cryopreserved product, however, showed a frequency of viable cells that started with 84.2% and which peaked at 93.1 ± 4.2% on day 6 followed by a continuous decrease to 80.7 ± 2.4% on day 12 (data given for LP-9 and LP-10). Fresh cells from DLBCL patients behaved in an opposite fashion, starting with a high viability of 97.8% that meanwhile dropped to 81.3 ± 2.5% and went back to 91.8 ± 0.7% (data given for LP-4 and LP-5). Overall, comparable expansion rates for PM (65 ± 36-fold) and HD (43 ± 14-fold) were achieved (Fig. 2G).
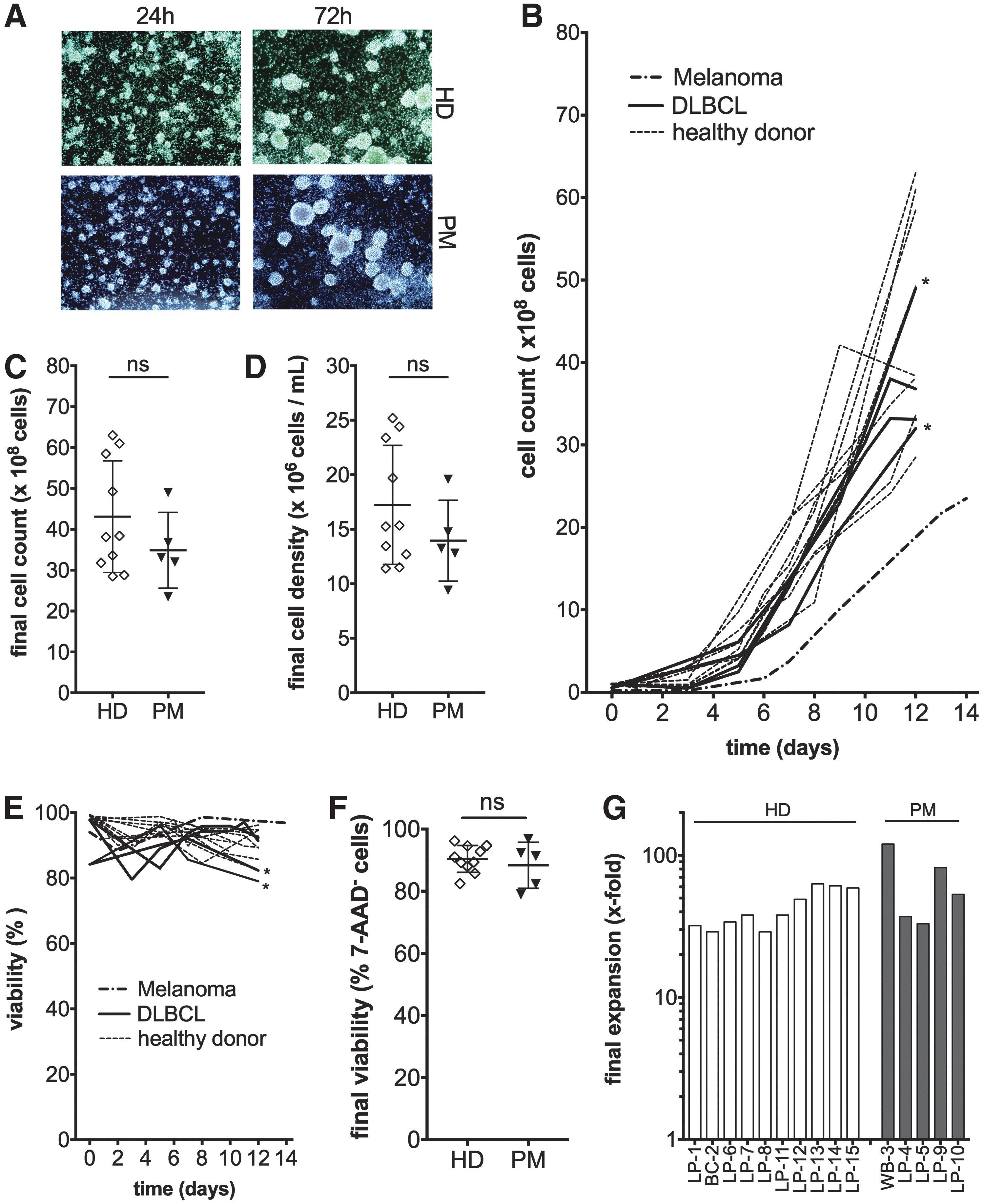
Fully automated TCT process on the CliniMACS Prodigy™ enables generation of large numbers of viable T cells independently of the starting material. T cells derived from healthy donor (HD) or patient-derived material (PM) were reproducibly generated in a clinical scale setting. Qualitative analysis of the activation status of T cells via cluster formation. Pictures were taken with the CliniMACS Prodigy™ integrated camera 24 and 72 h after polyclonal stimulation under static culture conditions
TCT process yields predominately T cells with a favorable TSCM and TCM phenotype
The cellular composition of the CD4/CD8 enriched cells was analyzed via flow cytometry, as exemplarily shown for HD-derived LP (Supplementary Fig. S2). The enriched CD4/CD8 fractions derived from HD were mainly composed of CD3+ T cells (83.4 ± 9.6%), whereas only 54.8 ± 11.5% T cells could be detected for PM (Fig. 3A), which directly reflects the increased frequency of NK and NKT present in the patient starting material (both populations are CD8dim and partially co-enriched). In addition, the frequency of CD3+ T cells in the starting material of PM was 23.0 ± 21.3% compared to 72.2 ± 10.7% for HD. Furthermore, significantly more B cells (CD19+), monocytes (CD14+), NK cells (CD56+), NKT cells (CD3+/CD56+), and granulocytes (CD16±/CD56±/SSChi) were measured for PM compared to HD. Differences in the enriched cells were abrogated during the 12 days of automated expansion in favor of the T cell outgrowth (Fig. 3B). Accordingly, a comparable T cell purity of 91.3 ± 5.0% for HD and 88.3 ± 7.1% for PM was detected in the final product. As already detected in the enriched fraction, NKT cells remained the predominant contaminating cells at a frequency of 5.1 ± 2.2% for HD and 8.0 ± 4.3% for PM. T cell phenotypes were analyzed based on the expression of CD62L, CD45RO, and CD95 among viable CD3+ cells using flow cytometry, as exemplified for run WB-3 (Supplementary Fig. S3). While naïve T cells (TN: CD45RO–CD62L+CD95–; 26.1 ± 16.5%), central memory T cells (TCM: CD45RO+CD62L+CD95+; 22.0 ± 13.0%), and effector memory T cells (TEM: CD45RO+CD62L–CD95+; 28.5 ± 10.6%) were represented at comparable levels for the enriched fraction of HD, analysis of the PM-enriched cells revealed a phenotypical shift toward the more differentiated effector T cells (TEFF: CD45RO–CD62L–CD95+; 24.7 ± 11.3%), whereas TN cells (7.3 ± 0.5%) were significantly underrepresented in the enriched fraction (Fig. 3C). Interestingly, despite these phenotypic differences in the enriched cells, a predominant appearance of stem-cell memory T cells (TSCM: CD45RO–CD62L+CD95+; 45.0 ± 11.2%) and TCM (33.99 ± 15.31%) for HD and TCM (64.7 ± 5.8%) for PM was detected in the final product (Fig. 3D). Nevertheless, the combined frequencies of memory T cells for HD with 79.0 ± 13.5% and PM with 87.1 ± 7.7% were comparable. Analyzing the frequency of CD4+ and CD8+ T cells (Fig. 3E) as well as the CD4/CD8 ratio (Fig. 3F) revealed significant differences between HD and PM in the enriched fraction. The ratios, however, changed from 3.8 ± 0.8 (HD) and 2.3 ± 0.8 (PM) in the enriched fraction to 2.0 ± 1.6 for HD and 1.9 ± 0.9 for PM in the final product where the differences disappeared (Fig. 3G and H).
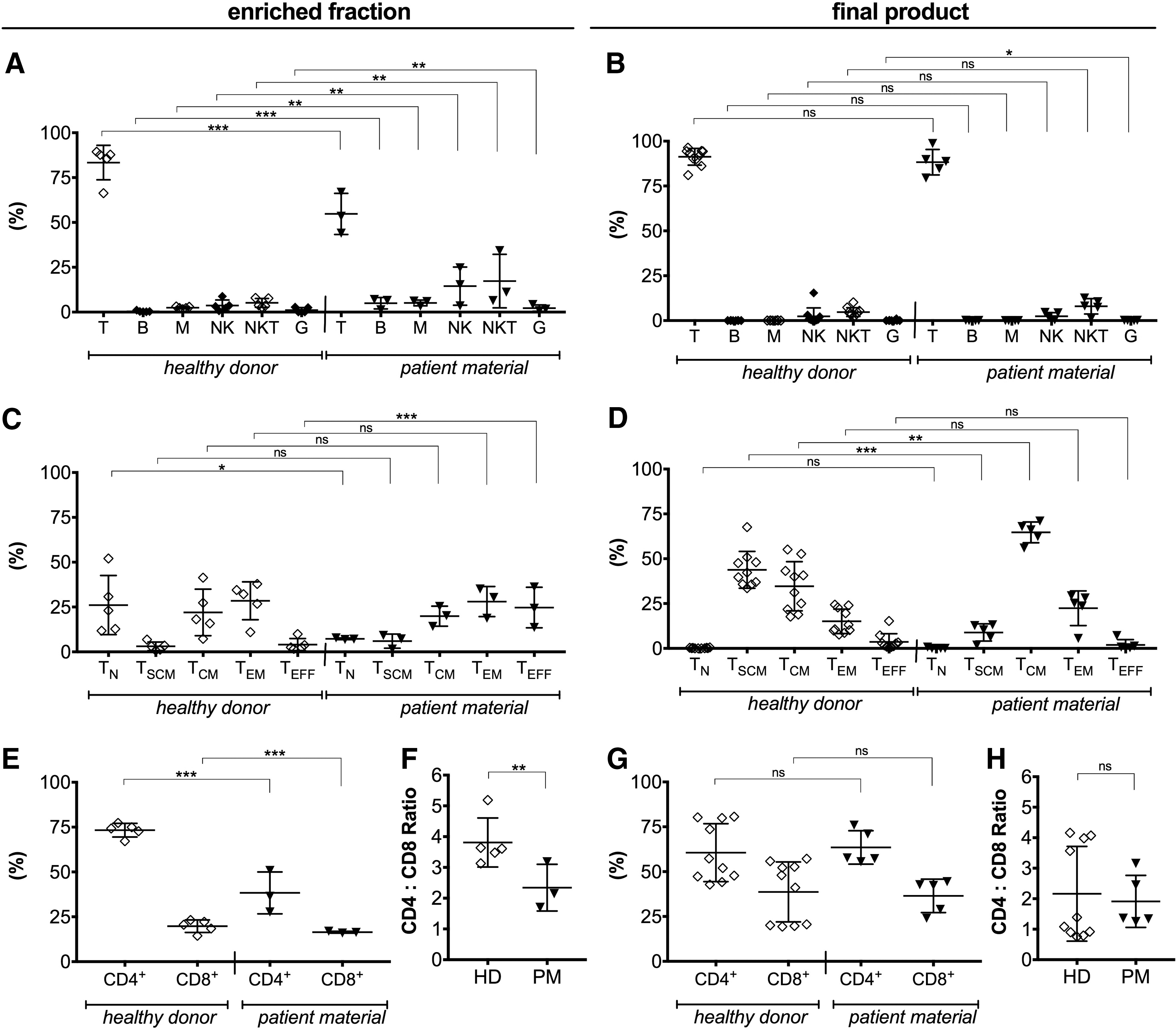
Analyzing the phenotypic characteristics of T cells cultured in the presence of IL-7 and IL-15 revealed a predominant appearance of long-term persisting stem-cell memory T cells (TSCM) and central memory T cells (TCM) in the final product. Cellular composition of the enriched fraction
TCT process generates polyfunctional CAR T cells with potent in vitro antitumor functionality
Polyclonal activated T cells were transduced with an anti-CD20 CAR encoding LV. Transduction efficiencies were determined during the process on day 5, as well as in the final product by flow cytometry using a fluorophore (PE)-linked CD20 CAR peptide, and an automated analysis algorithm (express mode), as representatively demonstrated for the runs LP-4 and LP-5 (Supplementary Fig. S4). Overall, the frequencies of anti-CD20 CAR-expressing T cells on day 5 ranged from 14% to 28% (M = 25.2 ± 7.8%) for HD (Fig. 4A) and 21.4% to 24.0% (M = 22.2 ± 1.2%) for PM (Fig. 4B). These frequencies considerably increased to an average of 34.5 ± 11.7% for HD and 36.4 ± 17.7% for PM on day 12 (Supplementary Table S1). VCN was determined in five runs (LP-8 to LP-12, with respective VCN values of 0.7, 2.1, 1.9, 1.3, and 1.3) and showed on average 1.5 ± 0.6 copies per transduced T cells. Accordingly, starting from HD, 1.4 ± 0.7 × 109 anti-CD20 CAR-expressing T cells were manufactured during the 12 days of the TCT process. Comparable numbers with 1.0 ± 0.4 × 109 engineered T cells were achieved for the PM runs. Remarkably, even though the transduction efficiencies widely differed, the final T cell count for both HD (4.4 × 109 ± 1.4 × 109) and PM (3.5 × 109 ± 1.1 × 109) confirmed the robustness of the automated manufacturing process using several types of starting material.
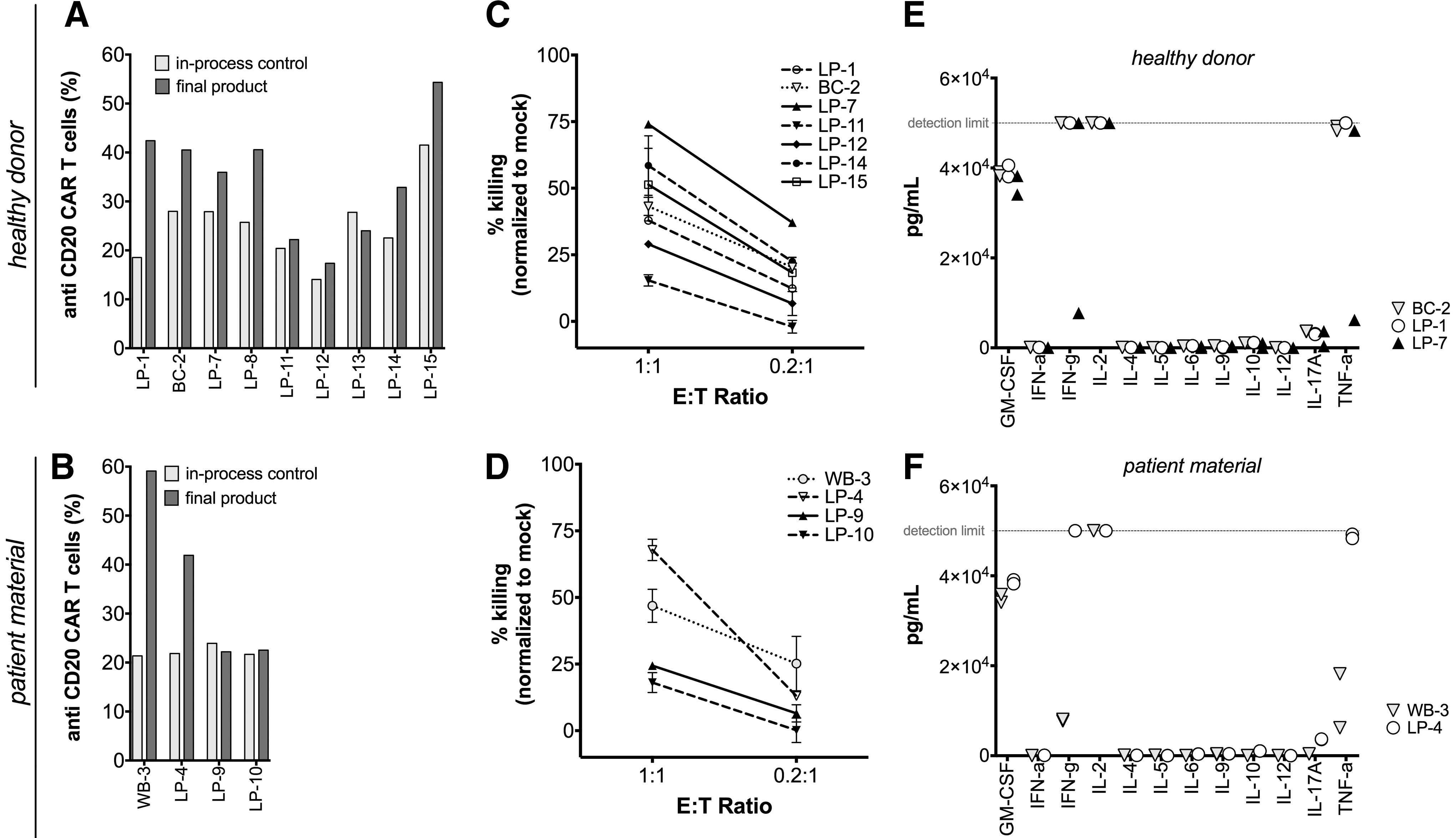
CliniMACS Prodigy™ gene-engineered T cells showed potent in vitro functionality. Comparison of anti-CD20 CAR transduction efficiency during manufacturing (in-process control) and at the end of each process for HD
The cytolytic potential of CliniMACS Prodigy™ manufactured anti-CD20 CAR T cells against tumor cells was evaluated using in vitro functionality assays. For this purpose, CD20+ JeKo-1 cells were co-cultured with anti-CD20 CAR-expressing T cells in different effector-to-target ratios for 24 h. Using flow cytometry, a specific killing of labeled target cells was detected for HD- (Fig. 4C) as well as for PM-derived CAR T cells (Fig. 4D), with an effector-to-target ratio of 1:1 ranging from 13.9% to 75.0% (M = 44.2 ± 19.3%) for HD and from 15.4% to 70.7% (M = 39.3 ± 21.3%) for PM, respectively. In addition, pro-inflammatory and inhibitory cytokines were measured in the supernatant. Both HD- and PM-derived CAR T cells released comparable levels of granulocyte macrophage colony-stimulating factor, interferon gamma, IL-2, and tumor necrosis factor alpha upon encountering JeKo-1 cell (Fig. 4E and F), while no type 2 cytokines such as IL-4, IL-5, or IL-10 were detected. In summary, no significant difference between the runs were ascertainable regarding the potency of the genetically engineered T cells, independent of the starting material or starting condition.
The produced gene-engineered T cells are effective in vivo
The functionality of anti-CD20 CAR-engineered T cells was analyzed in a lymphoma xenograft model. For this purpose, initially 5 × 105 RajiffLuc cells were injected i.v. into NSG mice followed by i.v. inoculation of 1 × 106 anti-CD20 CAR-expressing T cells or mock T cells after 1 week (Fig. 5A). While no antitumor effect could be detected in mice receiving mock T cells, treatment of mice with manufactured anti-CD20 CAR T cells resulted in an efficient eradication of tumor cells, as assessed using an in vivo imaging system (Fig. 5B and C) and flow cytometry (Fig. 5D). While tumorigenic target cells were completely eliminated, CAR T cells persisted in the blood, spleen, and bone marrow (Fig. 5E–G).
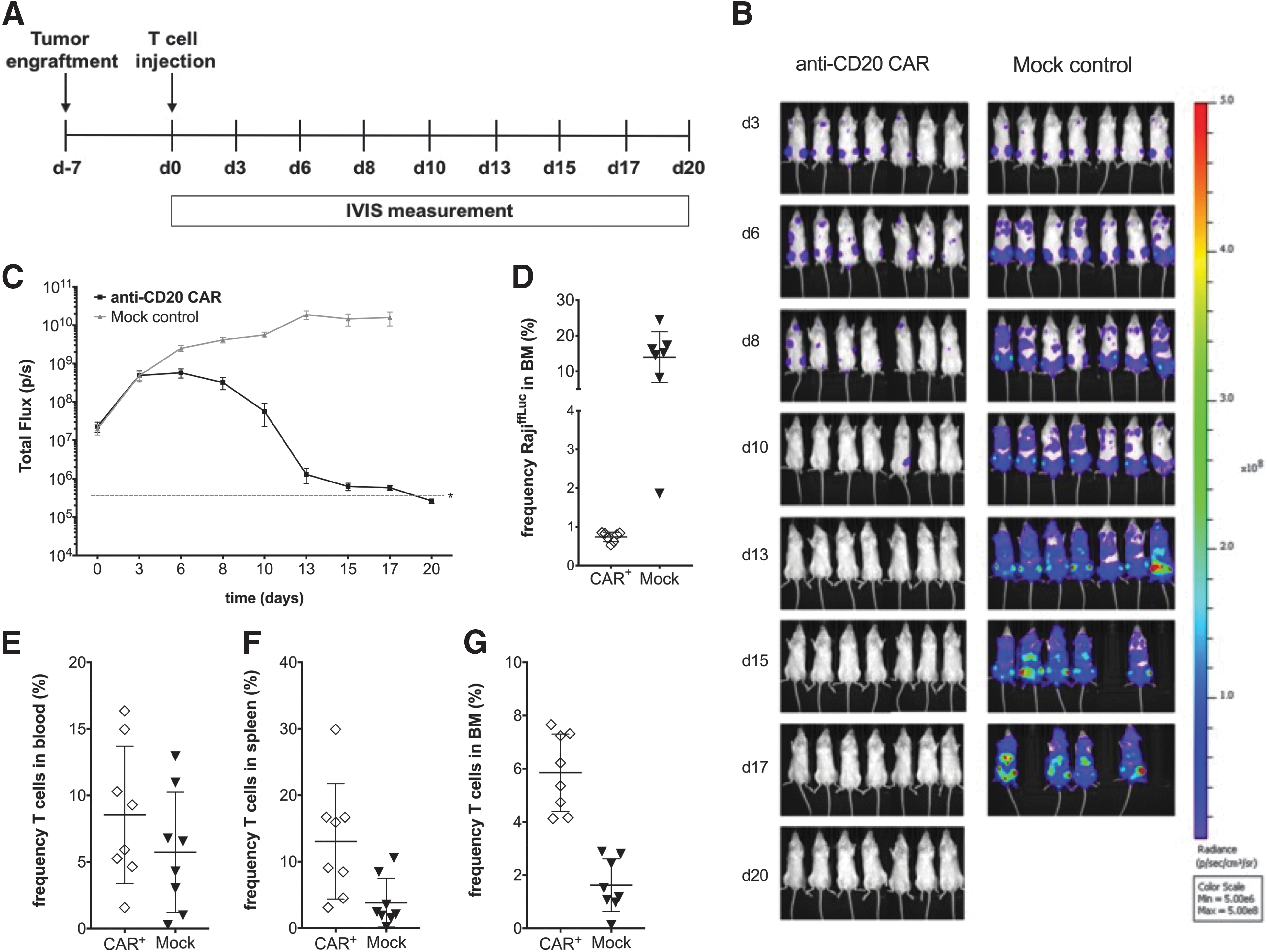
Antitumor reactivity of the manufactured anti-CD20 CAR T cells. In vivo experimental work flow
Discussion
The potential of engineered T cells expressing a CAR has been shown in several clinical trials. In particular, anti-CD19 CAR T cells have resulted in high remission rates in patients with relapsed and refractory B cell malignancies such as ALL, 5 –7 CLL, 8,9 and non-Hodgkin lymphoma. 10 However, despite this clinical success, Ruella et al. 11 recently reported that a significant number of patients still fail the treatment with this CD19-specific CAR due to the loss of CD19 on the tumor cells that is associated with a poor prognosis for the affected patients. The current study developed a CD20-directed therapy enabling the treatment of B cell lymphomas 13,27 or melanomas. 15,19,20 Furthermore, a manufacturing process was optimized using the CliniMACS Prodigy™. The possibility of generating functional gene-modified T cells using the cGMP-compliant, automated TCT process was recently demonstrated by Mock et al. 22 and Priesner et al. 23 However, this study used LP-derived from DLBCL patients, WB from a melanoma patient, or HD-derived LP as the starting material for the manufacturing of anti-CD20 CAR T cells. The run performed with melanoma-patient cells suggested the possibility of starting with cell counts as low as 2 × 107. However, an increased time was required for outgrowth of the cells. Further experiments are required to define the lowest number of starting cells. Here, it is shown that neither differences regarding the cellular composition nor disease-specific phenotypic characteristics negatively influenced the quality or quantity of the manufactured clinical product. Thus, the final cells were not only highly comparable, but also high and analogous expansion rates for PM (65 ± 36-fold) and HD (43 ± 14-fold) were achieved.
Overall. this demonstrated that LP, BC, or WB, cryopreserved as well as fresh samples, can be used as starting material in the executed process, which provided ideal conditions, including pH and glucose concentration (Supplementary Fig. S5) that supported T cell growth. Noteworthy, these rates of growth actually confirmed that even with only 2 × 107 enriched cells, compared to the standard of 1 × 108, the TCT process works successfully, and enough T cells could be manufactured covering the clinical need to treat patients.
With the aim of generating a highly functional and persisting T cell product for the treatment of patients, the expansion protocol used IL-7/IL-15 instead of IL-2. 26 So, either IL-15 or to a lesser extent IL-2 are capable of inducing T cell expansion. However, IL-7 is compulsory for generating and maintaining memory T cells, the most potent cells for ACT. Interestingly, despite the identical culture conditions for HD and PM runs using IL-7 and IL-15, significantly more TSCM were detected in the final product, starting from HD than PM, which is likely to be a direct consequence of the higher frequency of TN present in the starting material of HD. However, this needs to be investigated further. Cieri et al. 26 made the hypothesis that those TSCM, due to their stemness-associated potential of self-renewal, persist longer and are more tumor reactive than other T cell phenotypes, which might improve the efficacy of engineered T cells in ACT. Nevertheless, Wang et al. 28 in accordance with Berger et al. 29 also reported for mice as well as for macaques a comparable potential of TCM to preserve the memory T cell pool and the herewith associated ability of anti-tumor reactivity after T cell inoculation. Generally, the differences between HD and PM, although present with regard to the cellular composition, the phenotype, and the CD4/CD8 ratio in the enriched fraction, were limited to the characteristics of the generated memory phenotype of the T cells in the final product. The manufactured cells thus contained either a major population of TSCM (HD) or TCM (PM), both T cell subsets that share inherent functional advantages for ACT. 26,28,29 The overall frequency of memory T cells in the final product was similar for HD (45.0% TSCM + 34.0% TCM) and PM (8.9% TSCM + 64.7% TCM). Consequently, neither starting material derived from LP, BC, or WB nor cryopreserving influenced the phenotypic characteristics of the final product. Accordingly, we not only demonstrated the robustness of the TCT process, but also verified with a limited number of patients that cellular products can efficiently be generated from patient-derived T cells.
In contrast to Priesner et al. 23 a direct CD4/CD8 selection was applied instead of a CD62L enrichment to prevent an unintended co-enrichment of additional hematopoietic cells, including granulocytes, B cells, or monocytes also expressing CD62L. Thus, we did not enrich contaminating monocytes that impair the transduction process (Fig. S6) as well as T cell expansion, as shown here in accordance with Stroncek et al. 30 Furthermore, a CD4/CD8 selection avoided the risk of enriching and consequently genetically engineering for instance malignant B cells, which could lead to an indisputable safety risk for ACT. In addition, a freezing-induced downregulation of CD62L on T cells 23 was circumvented and the herewith associated issues for the enrichment step.
In addition, the study confirmed a cytolytic in vitro potential for both HD- and PM-derived CAR T cells and demonstrated the in vivo functionality of the HD-derived anti-CD20 CAR T cells. These findings were consistent with the data reported by Mock et al. 22 for their anti-CD19 CAR T cells, which they also manufactured using the TCT process. Accordingly, the study proved that independently of the operator or device, the TCT process yields a therapeutic dose of potent gene-modified T cells. In addition, this study demonstrated that the manufacturing protocol offers solutions to the current manufacturing problems. Thus, a fully closed, automated, and cGMP-compliant procedure was established, which works independently of the starting material. In addition, by utilizing MACS GMP T Cell TransAct for the polyclonal T cell activation instead of anti-CD3/anti-CD28 Dynabeads®, no magnetic bead removal is required for the final harvest. Although only low serum volumes were used (60 mL/run), further refinements will focus on the feasibility of manufacturing entirely serum free in the future.
In conclusion, the TCT process on the CliniMACS Prodigy™ can be used to manufacture potent engineered T cells in a clinical relevant scale under state-of-the art conditions. Thus, this clinic-ready manufacturing process will hopefully contribute to the dissemination of individualized therapy approaches to a broad range of patients.
Footnotes
Acknowledgments
The authors thank Hinrich Abken for providing the retroviral anti-CD20 CAR construct, Ian Johnston for his scientific advice as well as Anne Richter, Julia Milleck and Michaela Niemöller for generating the PE-linked CD20 peptide. Furthermore, we acknowledge the technical support of Juliane Stuth, Monika Winkels, Jörg Mittelstät, Britta Drees, Sandra Dapa and Katrin Lange. This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No 667980 (project „CARAT“) and by the German Federal Ministry of Education and Research (BMBF) under the project “CD20 CAR-TIME” in the program „innovations for individualized medicine“, grant identifier 01EK1507A.
Author Disclosure
D.L., N.M-T., K.D., C.B., D.M., T.S., C.K., W.A.R., J.B., O.H., M.A., and A.K. are employees of Miltenyi Biotec. No competing financial interests exist for the remaining authors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.