
Abstract
The non-viral, integrating Sleeping Beauty (SB) transposon system is efficient in treating systemic monogenic disease in mice, including hemophilia A and B caused by deficiency of blood clotting factors and mucopolysaccharidosis types I and VII caused by α-L-iduronidase (IDUA) and β-glucuronidase (GUSB) deficiency, respectively. Modified approaches of the hydrodynamics-based procedure to deliver transposons to the liver in dogs were recently reported. Using the transgenic canine reporter secreted alkaline phosphatase (cSEAP), transgenic protein in the plasma was demonstrated for up to 6 weeks post infusion. This study reports that immunosuppression of dogs with gadolinium chloride (GdCl3) prolonged the presence of cSEAP in the circulation up to 5.5 months after a single vector infusion. Transgene expression declined gradually but appeared to stabilize after about 2 months at approximately fourfold baseline level. Durability of transgenic protein expression in the plasma was inversely associated with transient increase of liver enzymes alanine transaminase and aspartate transaminase in response to the plasmid delivery procedure, which suggests a deleterious effect of hepatocellular toxicity on transgene expression. GdCl3 treatment was ineffective for repeat vector infusions. In parallel studies, dogs were infused with potentially therapeutic transposons. Activities of transgenic IDUA and GUSB in plasma peaked at 50–350% of wildtype, but in the absence of immunosuppression lasted only a few days. Transposition was detectable by excision assay only when the most efficient transposase, SB100X, was used. Dogs infused with transposons encoding canine clotting factor IX (cFIX) were treated with GdCl3 and showed expression profiles similar to those in cSEAP-infused dogs, with expression peaking at 40% wt (2 μg/mL). It is concluded that GdCl3 can support extended transgene expression after hydrodynamic introduction of SB transposons in dogs, but that alternative regimens will be required to achieve therapeutic levels of transgene products.
Introduction
N
The Sleeping Beauty (SB) transposon system is a non-viral vector that can mediate stable integration of therapeutic transgenes into the genomes of treated animal cells 7 –10 and provides sustained expression up to an entire lifetime. 11 –13 Importantly, a nearly random integration profile makes this vector safer than its viral counterparts. 1,2,4,10,14 The SB transposon system has proved effective in mouse models of several monogenic diseases, including hemophilia A 15 and B 11,16,17 and mucopolysaccharidoses (MPS) types I and VII. 18,19 Clotting factor IX (FIX), in particular, has been a transgene of choice in many gene therapy studies due to its low immunogenicity. In addition, a low concentration of FIX in the blood is needed to attain therapeutic efficacy, and the relatively small size of its coding sequence permits easy subcloning into a variety of therapeutic expression cassettes. 20 The current standard of care for hemophilia B is lifelong FIX replacement therapy to maintain the circulating levels at above 1% of normal. This treatment prevents hemorrhages but is not curative. 21 MPS I and MPS VII are progressive lysosomal storage disorders caused by deficiency of lysosomal enzymes α-L-iduronidase (IDUA; EC 3.2.1.76) and β-glucuronidase (GUSB; EC 3.2.1.31), respectively. The current standard of care for MPS I is bone-marrow transplantation and/or enzyme replacement therapy, 22 and while MPS VII is extremely rare in humans (1:250,000), its animal models are useful for testing novel therapies due to the availability of assays for quantifying and localizing GUSB activity. 23 Preclinical studies using SB transposons in MPS I mice have demonstrated lifelong IDUA expression in the liver at about 100-fold wild type (WT), which results in therapeutic effects comparable to those attained with retroviral liver-directed gene therapy. 13,19 However, mouse data are insufficient for translation to human clinical trials due to many differences, including organ size, metabolism, and life-span. 24
Dog models exist for both MPS I 25,26 and MPS VII. 27,28 These animals have been used as translational models for preclinical studies with gamma-retroviral and adeno-associated viral vectors. 29 –36 Amelioration of the disease has been achieved with liver-directed gamma retroviral gene therapy of neonatal dogs. 36 However, intravenous (i.v.) injection of gamma-retroviral vectors into adult animals was ineffective, even in mice, unless immune responses were blocked. 37 Immune responses can override achievements of gene therapy by eliminating either the transgenic protein activity or the transduced cells. 36 –38 Construction of vectors with species-specific therapeutic transgenes and liver-specific promoters that restrict expression to hepatocytes have been shown to reduce, although not eliminate, deleterious immunological responses. 38 –40
In mice, success of hydrodynamics-based DNA delivery to the liver depends primarily on two parameters: volume and speed of infusion. The tail-vein injection is routinely performed with a syringe. In the dog, delivery of plasmids is accomplished by isolation of the liver using balloon catheters with infusion directly into the hepatic veins, which is a surgical procedure performed by an interventional team under radiologic guidance. The injection route, properties of the vasculature and of the target organ (the liver), as well as the direction of flow after hydrodynamics-based injection are important parameters that differ substantially between mice and large animals. 4,41,42
Until recently, the efficacy of non-viral DNA delivery to treat large animals has been discouraging. 4,42 –44 The authors developed and examined the potential of catheter-mediated infusion via the hepatic veins wherein targeted liver tissues are isolated from the blood flow by balloon occlusion of blood vessels. In dogs aged ≥3 months, 41 detectable expression of the reporter enzyme canine serum alkaline phosphatase (cSEAP) was achieved for up to 6 weeks, a duration that is too short for clinical application, which requires extended gene expression. Consequent delivery of therapeutic transposons carrying IDUA and GUSB reported here yielded a low albeit therapeutically relevant level of initial expression, but was measurable for only a few days.
It was hypothesized that with immune suppression, transgene expression attained by the delivery method would be prolonged and might be maintained at therapeutic levels for systemic diseases. Here, in cSEAP- and cFIX-treated dogs, prolonged transgene expression after balloon catheter–mediated delivery is demonstrated for the first time, which lays the groundwork for further improvement of immunosuppression and transposition in large animal models.
Materials and Methods
Animals and infusions
All animal procedures were reviewed and approved by the University of Minnesota Institutional Animal Care and Use Committee. C57BL/6 mice were obtained from Jackson Laboratories, maintained under specific-pathogen-free conditions, and provided food and water ad libitum. Male beagles were procured from a USDA class-A vendor for the described procedures. The MPS VII mixed breed dog was produced at the Department of Animal Science of Iowa State University. The dogs were fasted for 12 h prior to anesthesia, then sedated with acepromazine 0.4 mg/kg SQ and atropine 0.05 mg/kg SQ or torbugesic at 0.05–0.5 mg/kg SQ. Anesthesia was induced with propofol at 4 mg/kg i.v. followed by intubation and general anesthesia using isoflurane 1–2% with 3–4 L/min of oxygen at approximately 15 cc/kg tidal volume. Animals were continuously monitored throughout the procedure by visual assessment, electrocardiogram, and intravascular venous pressure recording.
Two different strategies were used for balloon-mediated occlusion of the liver with subsequent infusion of DNA into the hepatic venous circulation. The double-balloon system of Hyland et al. 41(Fig. 1A) was applied in Dog9-1 and Dog13. A custom double-balloon catheter with infusion ports was introduced through the right femoral vein into the inferior vena cava, and a second pressure-sensing catheter was introduced through the left femoral vein, with the end of the catheter positioned between the two balloons. This procedure delivered therapeutic plasmids to both right and left lobes of the liver. 41 The single-balloon system (Fig. 1) was used in all dog experiments except Dogs 9–1 and 13. This catheter was introduced through the right jugular vein to the inferior vena cava and positioned just inside the left hepatic vein for delivery to the left side of the liver. A second pressure-sensing catheter was introduced alongside the balloon catheter from the right jugular vein, with the end of the catheter positioned distal to the balloon. This procedure was designed to provide focused plasmid delivery to the left side of the liver.
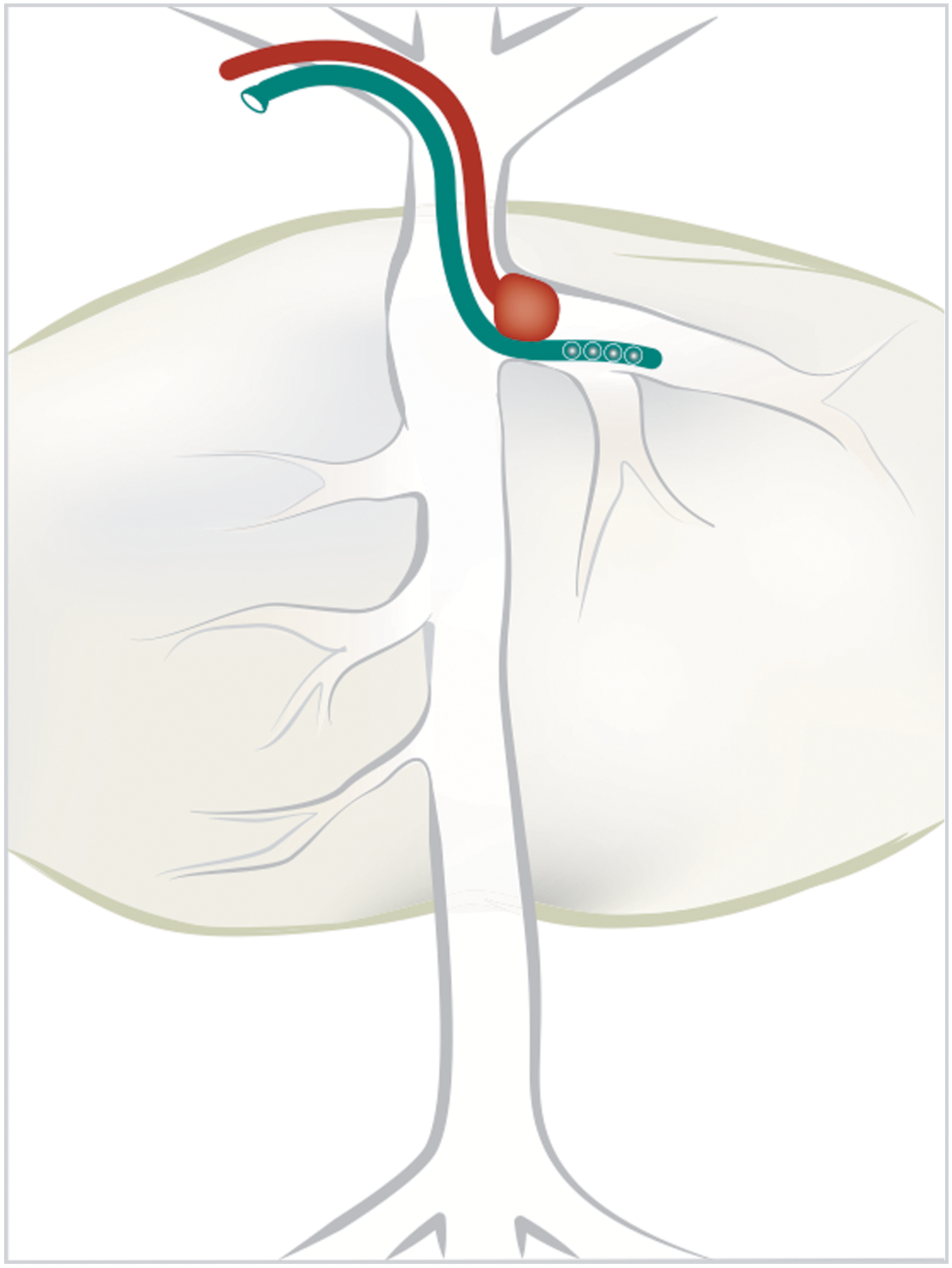
Single-balloon strategy used for infusion of DNA into the left side of the liver through the left hepatic vein in all dogs listed in Table 1 except Dogs 9–1 and 13. The single-balloon catheter is indicated in red while the catheter for DNA delivery is shown in teal. An additional catheter introduced to monitor intravascular pressure is not shown.
The infusion parameters developed by Hyland et al. 41 served as guidelines: DNA concentration of 2 mg/kg in a total infusion solution volume of 200 mL was delivered in 10–20 s to achieve intravascular pressure peaks of 100–200 mmHg (Table 1). The DNA dose in all but one dog was within a 1.5-fold range of 2 mg/kg, which Hyland et al. found to be nearly saturating. The MPS VII Dog28 received a triple dose of plasmid, as explained below.
Summary of balloon catheter-mediated DNA delivery to liver in dogs
SB transposon delivered reporter is indicated. All dogs were normal, with the exception of Dog28 affected by MPS VII, with GUSB activity <1% WT due to a missense R166H mutation.
IVC, DNA delivery to the entire liver via inferior vena; LHV, delivery to the left liver side via left hepatic vein.
Peak of reporter protein or activity on days 2–4 post treatment.
For cSEAP-treated dogs, fold increase in activity relative to pre-injection activity level. For FIXmyc-treated dogs, fold increase in FIX protein relative to a WT dog (5000 ng/mL 72 ). For GUSB-treated dogs, fold increase in GUSB activity compared to the WT untreated control dog (about 100 nmoles 4MU/mL/h) determined in the same study, as the treated dog.
FIX, factor IX; GUSB, β-glucuronidase; IDUA, α-L-iduronidase; IVC, inferior vena cava; LHV, left hepatic vein; N/A, not available; SB, Sleeping Beauty; WT, wild type.
Immunomodulation
Cyclophosphamide (CP; Sigma–Aldrich, St. Louis, MO) was administered i.v. at 10 mg/kg 1 day before and then again immediately prior to plasmid injection and then once weekly for 30 days. GdCl3x6H2O (10 mg/kg; Sigma–Aldrich) was administered i.v. 1 day before and then immediately prior to plasmid injection and then twice weekly for 30 days. Dexamethasone (Dex) was injected at 1 mg/kg for 10 days, starting 1 day prior to plasmid infusion. Dogs receiving GdCl3 were implanted with a port to prevent damage of small veins and spare animals discomfort and pain due to multiple injections and blood draws.
SB vectors
Vectors used in this study are shown in Fig. 2. SB transposons for cSEAP expression and human (h)GUSB and hIDUA have been described previously, 13,18,19,41,45 as have plasmids for co-delivery of expression cassettes for SB transposases, pCMV-SB11, mini(m)Ub-SB11, 19,46 and pCMV-SB100X. 47 Liver-specific promoter (LSP) ApoEHCRhAAT 40 was a gift from Dr. Mark Kay (Stanford University, CA). The SB transposon pKT2/ApoEHCRhAAT-cGUSB was engineered by inserting the full-length canine (c)GUSB into a unique EcoRI site of the pKT2-ApoE-hAAT-BGintron plasmid. 13 The plasmid containing cGUSB was a gift from Dr. Kathy Ponder (Washington University, St. Louis, MO). Prior to injection into dogs, plasmids were hydrodynamically infused into C57BL/6 mice to ensure transgene expression and transposition.
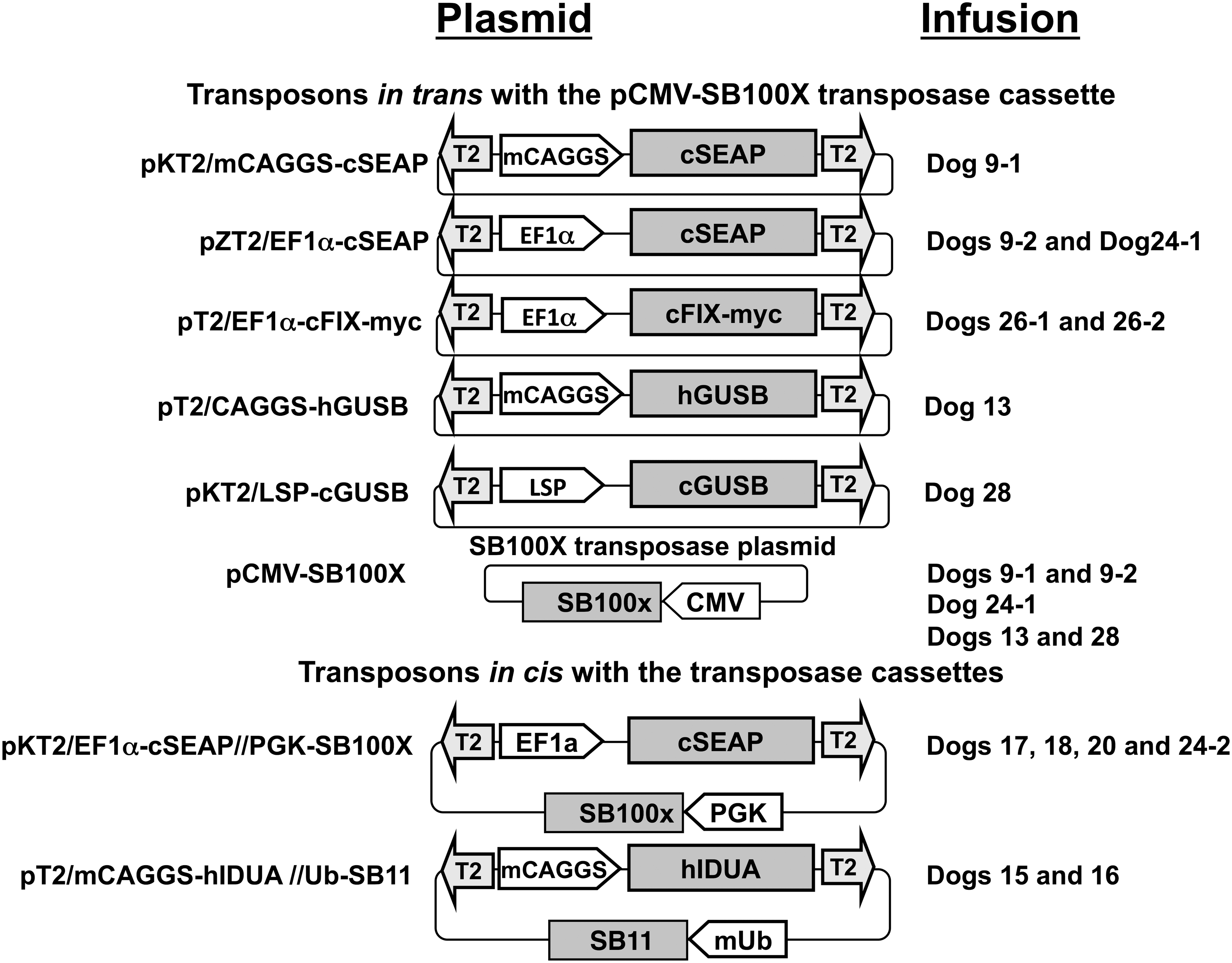
Transposon and transposase-encoding plasmids. Transposons contain T2 inverted terminal repeats (lightly shaded arrows) and a reporter gene sequence (darker shaded boxes), encoding canine secreted alkaline phosphatase (cSEAP), canine factor IX (cFIX), human or canine β-glucuronidase (h or cGUSB), or human α-L-iduronidase (hIDUA). Reporter gene expression was regulated by the ubiquitously expressed CAGGS or mini(m)CAGGS promoters, 19,46 liver-specific promoter (LSP) ApoEHCRhAAT, 40 or by a CpG-less variant of EF1α promoter (unshaded block arrows); pKT2/EF1α-cSEAP//PGK-SB100X 41 contains a PGK-regulated SB100X gene, exterior to the cSEAP-encoding transposon, while pT2/mCAGGS-hIDUA//mUb-SB1119 contains a mini-ubiquitin-regulated SB11 gene, 45 downstream of the transgene-encoding transposon (i.e., in cis). For the other transposons, Sleeping Beauty (SB) transposase was provided in trans by co-infusion of pCMV-SB100X plasmid. 47 Plasmids with pKT2- or pZT2-backbones were kanamycin- or zeocin-resistant, respectively; other plasmids were ampicillin-resistant.
Blood and tissue collection
Heparinized venous blood samples were collected before, during, and after the procedure for analysis, as described below. Plasma was prepared for enzyme assay, and whole-blood aliquots were shipped to the Marshfield Labs (Marshfield, WI) for complete blood chemistries including alanine transaminase (ALT) and aspartate transaminase (AST) level determination. At the end of each experiment, dogs were given heparin at 150 units/kg i.v. and anesthetized with propofol at 2–6 mg/kg i.v. The animals were euthanized using euthasol at 1 mL (390 mg pentobarbital +50 mg phenytoin)/4.5 kg. The liver was perfused with 1,000 mL of saline to attain virtually uncolored outflow, and then 20–100 liver samples were collected for individual analysis. Plasma and tissue samples were stored at −80°C.
cSEAP enzyme assay
Plasma samples were heated for 10 min at 65°C and then assayed for alkaline phosphatase enzymatic activity using Applied Biosystems Tropix® Phospha phospholuminescent substrate (Thermo Fisher Scientific, Waltham, MA) with human SEAP as a standard for determination of results in ng/mL.
cFIX-myc enzyme-linked immunosorbent assay
Canine plasma samples were assayed in duplicate by sandwich enzyme-linked immunosorbent assay (ELISA) for canine FIX-myc protein. High binding polystyrene 96-well plates (Costar 3369) were coated with goat polyclonal anti-myc tag capture antibody (Ab9132; Abcam, Cambridge, United Kingdom), diluted to 2.5 μg/mL in 50 mM of carbonate buffer, pH 9.6, and incubated overnight at 4°C. Plasma samples were diluted 20-fold with sample diluent (100 mM of HEPES, 100 mM of NaCl, 10 mM of disodium EDTA, 150 mM of bovine serum albumin, and 0.1% Tween-20, final pH 7.2). A total of 100 μL was added to each well, and the plate was incubated for 1 h at room temperature. After three washes with wash buffer (Bio-Techne, Minneapolis, MN; #895126), detection antibody was added (100 μL at 1/400 dilution of horseradish peroxidase-conjugated anti-canine FIX antibody, CFIX-EIA-D; Enzyme Research Laboratories, South Bend, IN), and the plate was incubated for 1 h at room temperature. After three washes with Bio-Techne wash buffer, 100 μL of tetramethylbenzidine/hydrogen peroxide substrate (Bio-Techne) was added to each well and stopped with 100μL of 2N sulfuric acid after 20 min at room temperature. Plates were read at 450–490 nm (Spectromax), and cFIX levels were reported in ng/mL. To generate a standard for canine FIX-myc, plasma samples were pooled after collection from three mice 1 day after hydrodynamic injection with pKT2/EF1α-cFIX-myc plasmid. The concentration of total cFIX in the pooled plasma sample was determined by ELISA (CFIX-EIA; Enzyme Research Labs, South Bend, IN). Freshly diluted mouse plasma was used as a standard in each ELISA assay.
IDUA and GUSB enzyme assays
IDUA and GUSB activities were measured in tissue homogenates and plasma using 4-methylumbelliferyl-β-D-glucuronide (Sigma–Aldrich) and 4-methylumbelliferyl-α-iduronide (Glycosynth, Warrington, United Kingdom), respectively, in fluorometric assays using a 96-well plate reader, as previously described. 13,48
Quantification of liver glycosoaminoglycans
Liver lysates prepared from collected pieces were incubated overnight with Proteinase K, DNase I, and RNase, as previously described. 48 Glycosoaminoglycan (GAG) concentrations were determined using the Blyscan Sulfated Glycosaminoglycan Assay (Accurate Chemical & Scientific Corp., Westbury, NY) according to the manufacturer's instructions.
Polymerase chain reaction analysis
DNA copy numbers of GUSB and IDUA were determined by real-time quantitative polymerase chain reaction (qPCR) using 2X IQ"SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA), as previously described. 13,19 DNA was isolated by phenol-chloroform extraction from approximately 50 mg of liver tissue samples pulverized in liquid nitrogen. Canine glyceraldehyde-3-phosphate dehydrogenase (cGAPDH) sequence served as an internal control for genomic DNA content. Canine GAPDH standard curve consisted of serial dilutions of dog genomic DNA. The canine cSEAP qPCR was performed using Taqman technology, as previously described. 41 The limit of detection was 2 × 10–4 copies per diploid genome equivalent in dog liver.
Quantitative image analysis of hGUSB transgene expression and lysosomal storage in the liver
Histochemical localization of GUSB was performed using AS-BI-naphthol-β-D-glucuronic acid (Sigma–Aldrich), as described. 49 Stained tissue samples were photographed at 20 × magnification using a digital camera (Nikon, Melville, NY). Images were contrast enhanced so as to delineate image details clearly using Photoshop 6.0 image processing software (Adobe Systems, San Jose, CA), as previously described. 18
Transmission electron microscopy and toluidine blue staining
For detection of storage vacuoles, liver tissue samples of approximately 2 mm × 2 mm × 2 mm collected at necropsy were immediately fixed with 2.5% glutaraldehyde overnight at 4°C. After washing three times with 0.1 M of sodium cacodylate, the samples were post fixed with 1% OsO4 for 1 h, rinsed three times, and dehydrated in a graded series of ethanol solutions. The samples were embedded in Epon812 resin. Ultrathin sections (65 nm) were stained with uranyl acetate and lead citrate, and then examined by JEOL 1200EX II electron microscopy. Thin sections were cut at 1 mm from the same blocks, stained with toluidine blue, and evaluated for lysosomal distension. 18
Statistical analysis
Data were analyzed using GraphPad Prism v6.0 (GraphPad Software, Inc., San Diego, CA). The significance of differences between groups was determined based on two-tailed p-values obtained with Welch's t-test. A p-value of <0.05 was considered statistically significant.
Results
Transient GdCl3 regimen prolonged persistence of the cSEAP reporter in plasma
To evaluate the impact of immunosuppression on duration of cSEAP expression, dogs were treated with immunosuppressive drugs that had been efficient in the mouse gene therapy studies. 13,18,50 Dog17 was administered cyclophosphamide (CP) i.v. at a dose of 10 mg/kg 1 day before DNA infusion and then once weekly for 30 days. In addition, dexamethasone (Dex), a commonly used postoperative anti-inflammatory agent, was injected at a dose of 1 mg/kg for 10 days, starting 1 day prior to plasmid infusion. This combination of drugs did not prolong cSEAP expression (Table 2). Figure 3B shows that the profile of cSEAP activity in Dog17 is virtually indistinguishable from that in Dog9, which was not immune suppressed. Therefore, the immunosuppression regimen was modified to add gadolinium chloride (GdCl3), an agent that blocks liver and spleen macrophages. GdCl3 was previously shown to extend IDUA expression in IDUA-deficient mice. 18 GdCl3x6H2O at a dose of 10 mg/kg was administered i.v. 1 day prior and immediately prior to plasmid infusion and then twice weekly for 30 days. In Dog18, it was observed that the addition of GdCl3 treatment prolonged cSEAP expression up to 5.5 months. cSEAP expression in this animal was associated with a gradual decline, but appeared to stabilize after about 2 months at approximately fourfold higher than the baseline level (Fig. 3A). The half-life (T1/2) of cSEAP activity profiles was calculated to quantify the decline of expression in plasma independently of peak height. 13 GdCl3 regimen extended the cSEAP half-life in plasma by about 2.5-fold, from 6 to 15 days (Table 2).
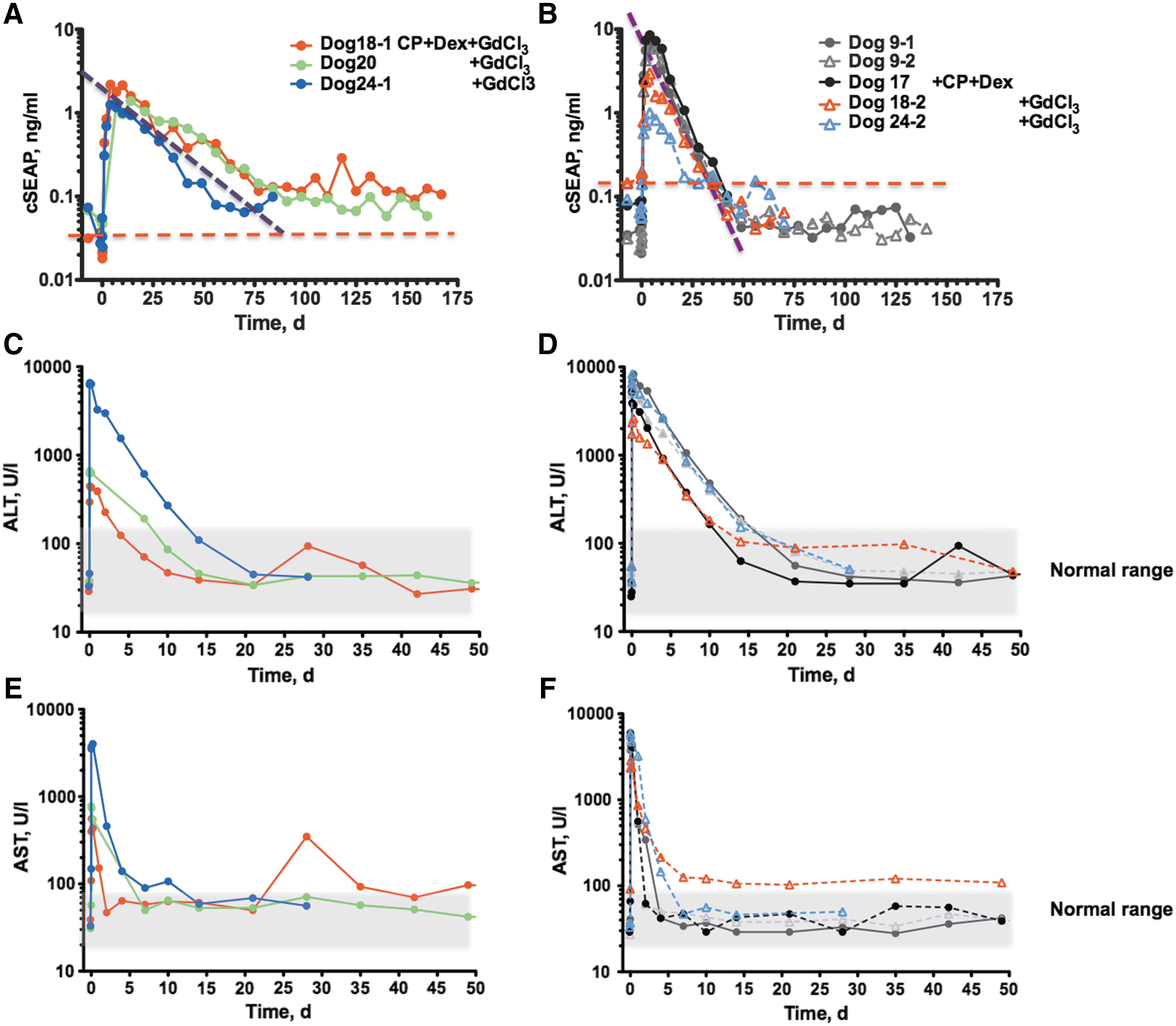
Transgene expression and liver toxicity in dogs infused with cSEAP transposons with and without GdCl3 immunomodulation.
Maintenance of transgene expression in dogs with respect to immunomodulation regimen and elevation of ALT and AST
Fold increase over pre-first infusion ALT and AST level, respectively, in the same dog; normal levels in dogs: ALT 14–151 IU/L; AST 18–86 IU/L.
Half-life of protein/enzyme activity in plasma.
ALT, alanine aminotransferase; AST, aspartate transaminase.
Next, the study tested if GdCl3 alone would extend the profile of cSEAP activity after hydrodynamic injection of cSEAP transposon DNA to dogs. Indeed, in the next two treated dogs (Dog20 and Dog24), the cSEAP expression half-life values were higher than those observed in Dog9 and Dog17, but re-infusion in Dog24 (Dog24-2) did not result in prolonged transgene expression. The same effect was observed in Dog18 (Dog18-2) upon transposon re-infusion on day 167. Despite repeated GdCl3 regimen, cSEAP declined as rapidly (T1/2 ≈ 6 days) as in Dog9 and Dog17 that did not receive GdCl3. Rapid decay of cSEAP expression upon DNA re-infusion cannot be explained by lower transposon delivery. Plasma cSEAP maximal levels after the first and second infusions were comparable, suggesting that the increased liver volume at the time of the second infusion had little effect on gene delivery (Dog9, Dog17, Dog18, and Dog24; Table 1).
Noteworthy, at the peak of activity (2–4 days post infusion), nine separate infusions of cSEAP transposon plasmids resulted in cSEAP plasma levels 14- to 200-fold greater than the pre-infusion level of heat-stable alkaline phosphatase (Table 1 and Fig. 3A and B), supporting the data of Hyland et al. 41 There was no relationship between recorded peak i.v. pressure and maximum plasma cSEAP, which suggests that high pressure alone is not a critical parameter for extent of DNA delivery to cells in the liver.
DNA qPCR showed that transposon plasmid encoding cSEAP peaked in plasma within 1 day post infusion and was rapidly cleared from the blood by 5 days (Supplementary Fig. S1A; Supplementary Data are available online at
Impact of transient hepatocellular toxicity on transgene expression
Achieving prolonged expression of cSEAP after a single infusion of vector permitted factors to be evaluated that may affect transgene expression following balloon catheter–mediated delivery in dogs. Biomarkers of liver health and pathology, ALT and AST, showed a dramatic short-term increase in all treated dogs (Table 2), signaling inflammation and cell damage. 51,52 ALT and AST values returned to normal within 2 weeks for ALT and a few days for AST (Fig. 3C–F and Supplementary Fig. S2B and C), in agreement with previous results. 41 Evaluation of the liver by ultrasound immediately after infusion and by computed tomography scan 2 days after infusion did not indicate any rupture or other visible injury to the liver. The animals were clinically healthy starting the day after the infusion and continuing through the end of the follow-up, which lasted as long as 5.5 months.
The transient increase of ALT and AST in response to plasmid infusion varied among dogs (Fig. 3C–F and Table 2) and appeared to be inversely associated with cSEAP half-life following a single DNA infusion. The dogs that had the longest sustained cSEAP expression, Dog18-1 and Dog20, had the lowest increases of ALT and AST (8- to 18-fold), while Dog 24-1, which had a relatively shorter cSEAP half-life, exhibited a 44- and 164-fold increase of ALT and AST, respectively (Table 2). These data suggest that liver inflammation and cell damage curtail expression, although the small number of dogs does not support a statistically sound correlation. However, repeat plasmid administration with GdCl3 treatment (Dog18-2, Dog24-2, and Dog26-2) led to overall higher increases of ALT and AST. These observations, together with lack of prolonged transgene expression, despite GdCl3 immunomodulation upon vector re-infusion, also strongly suggest that an increase of hepatocellular toxicity is associated with a loss of immunosuppressive effect. Notably, in Dog9, which did not receive GdCl3, the rate of cSEAP decline was equally rapid after the first and the second infusion, and the respective ALT and AST profiles were similar after both infusions.
SB-mediated expression of therapeutic proteins with and without GdCl3 immunomodulation
To test if immunosuppression would prolong expression of a potentially therapeutic transgene, transposon-plasmid encoding myc-tagged cFIX was infused into GdCl3-treated Dog26, which resulted in a peak level of ∼2 μg/mL, about 40% of the endogenous level of clotting factor. The protein had a half-life of 20 days (Tables 1 and 2 and Supplementary Fig. S2A). Thus, immunomodulation with GdCl3 prolonged cSEAP and cFIX expression in all dogs after a single transposon infusion.
Upon re-infusion of the cFIX-encoding transposon with GdCl3 treatment (Dog26-2), a rapid decline in circulating transgenic protein was observed, which peaked at 20% the endogenous level but became undetectable by 3 weeks post infusion. These results were similar to those obtained with re-infusion of cSEAP transposons. Similarly, repeat plasmid administration led to an overall higher increase of ALT and AST (Supplementary Fig. S2A), supporting the observations that an increase in hepatocellular toxicity is associated with a loss of immunosuppressive effect.
The study further examined whether hydrodynamic delivery of transposons would be therapeutic in dogs for MPS I and VII, as it had previously been demonstrated in mice. 13,18,19 The goal of short-term experiments was to evaluate peak expression levels of transgenic lysosomal enzymes GUSB and IDUA, respectively, as well as transposition in the liver, prior to the onset of adaptive immune response. The transposons were co-delivered with a SB transposase expression cassette that was either on the same or on a separate plasmid, referred to as cis- or trans-configuration, respectively (Fig. 2 and Table 2). The one-plasmid SB configuration ensures co-expression of the transgene and SB transposase in the same cell, thus increasing the chance of transposition. The two-plasmid configuration increases the likelihood of delivery to the nucleus due to a smaller plasmid size and also permits alteration of relative transposon and transposase dosing, which can improve transposition efficiency. 10,12 Two versions of SB transposase were used: SB1146 or SB100X. 47 The latter mediates a 10- to 20-fold higher rate of transposition in mice after hydrodynamic delivery. 13
The transposon plasmid pT2/CAGGS-hGUSB (Fig. 2) that drove strong expression of human GUSB in mice 18 was infused into the whole liver (Fig. 1A) of a young adult normal dog, along with pCMV-SB100X in trans (Dog13). The animal was euthanized 4 days after infusion at which point GUSB activity in plasma was 350 nmoles 4MU/mL/h, about 3.5-fold above the background enzyme activity of an untreated WT dog (Fig. 4A and Table 1). One hundred tissue samples were collected from the treated dog, and all contained measurable levels of transposon DNA that spanned 0.01–0.8 copies/genome equivalent (Fig. 4C and D), with an average of 0.08 copies/genome equivalent. Peak expression in the GUSB-treated Dog13 liver was <5% the peak activity in mice infused with the same transposon: 350 versus 9,400 nmoles 4MU/mL/h.
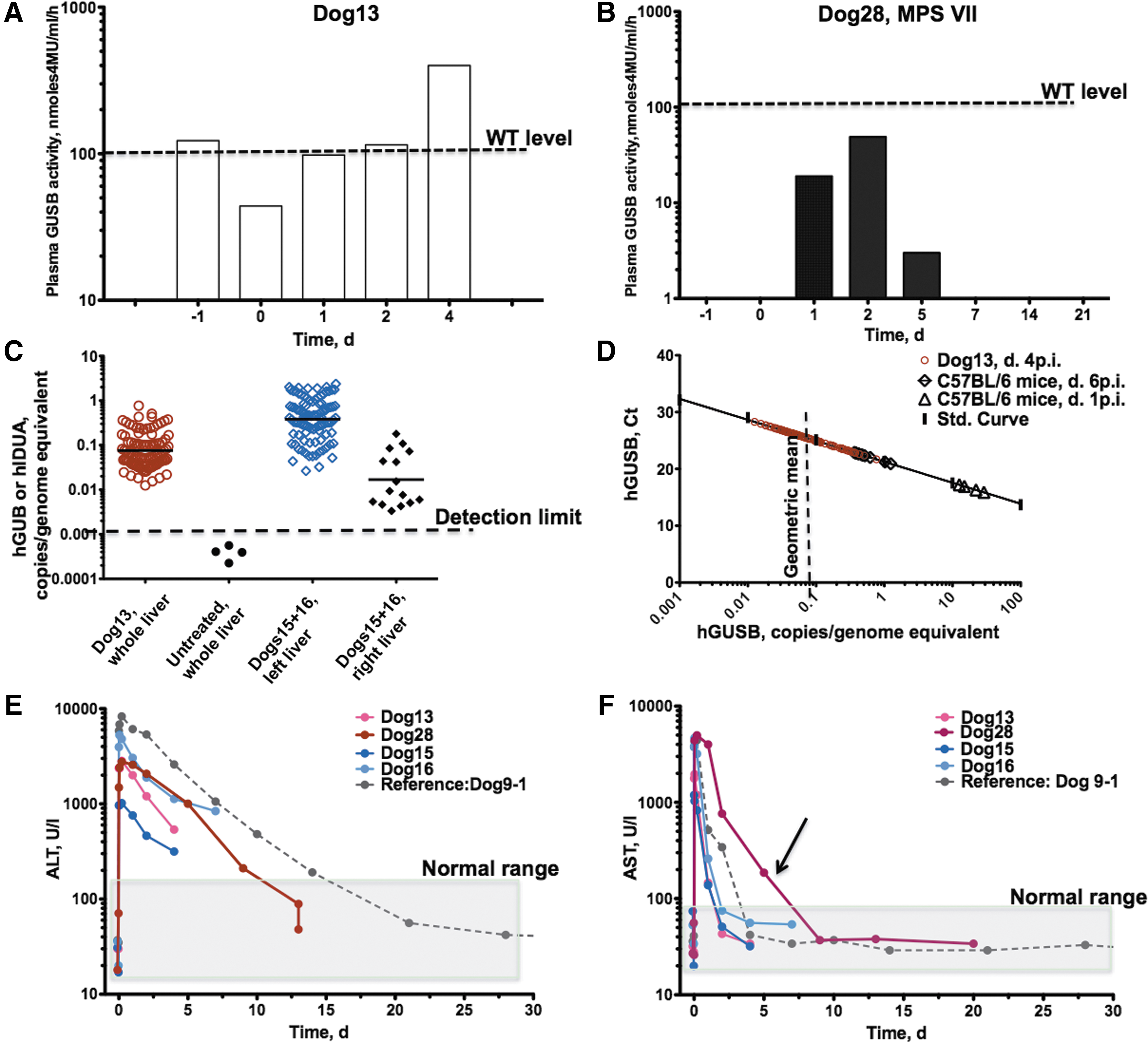
Delivery of GUSB and IDUA to dog liver without GdCl3 immunomodulation resulted in low-level, short-term expression.
Of 100 liver samples, three contained quantifiable levels of transposon-excision product (EP) at about 0.002 copies/genome equivalent, which corresponded to 2–3% EP/transposon molecules (0.002/0.08). This value is within the range previously reported for mouse liver when SB11 was used 19 and implies that transposition was functional in the dog liver, although at about 20 times lower efficiency than in mice treated with SB100X. 13
The next two dogs were treated with the IDUA-expressing transposon configured in cis. This transposon was previously demonstrated to mediate strong long-term expression in male NOD/SCID MPS VII mice. 19 The plasmids were infused into the left portion of the liver. The rationale for targeting only the left portion of the liver was to (1) minimize invasiveness, and (2) achieve a higher ratio of volume of infused solution to liver mass. Following hydrodynamic infusion into the left hepatic vein, normal male dogs (Dog15 and Dog16; Table 1) were sacrificed at days 4 and 7, respectively. Fifty tissue samples were collected from the left lateral liver lobe, and 10 samples were collected from the right liver portion. DNA qPCR confirmed delivery to the left lateral lobe, which was 20-fold higher than that to the right liver samples (p < 0.0001). Focused delivery of hIDUA transgene to the left side of the liver in Dog15 and Dog16 resulted, on average, in 0.4 copies/genome equivalent, a fivefold higher transgene frequency compared to that of hGUSB transgene in Dog13, in which the whole liver was infused (Fig. 4C and D). However, there was no detectable increase in IDUA enzymatic activity above normal levels. These data suggest that if there was any transgenic expression of hIDUA, it was lower than the innate IDUA activity in unaffected Dog15 and Dog16. No excision products were detected.
Catheter-mediated delivery of GUSB-encoding transposons into a canine model of MPS VII was evaluated, 27,28 in which there is <1% of normal GUSB activity due to a missense R166H mutation. This dog model recapitulates many features of MPS VII in humans, including hepatomegaly caused by accumulation of storage material inside the lysosomes of liver cells. MPSVII Dog28 was treated with a triple amount of DNA (7.6 mg of DNA/kg; Table 1) that might approximately compensate for the increased liver volume due to hepatomegaly. To minimize immune responses, transposon pKT2/LSP-cGUSB containing the “self”-transgene canine GUSB and liver-specific promoter ApoEhCRhAAT that directs strong, hepatocyte-restricted transgene expression in dogs 40 and mice was constructed. 13 A similar transposon carrying hIDUA and also co-delivered with pCMV-SB100X mediated sustained expression ≥100-fold WT in immunosuppressed C57BL/6 mice in a year-long study.
Dog28 was treated with GdCl3, but the regimen was incomplete. A week after DNA infusion, the port had to be removed because of infection. In this dog, the veins were hard to access and became unusable after each injection of GdCl3, likely due to its leakage into the tissue surrounding the blood vessel. Of note, the MPS VII dog was the only one in this study where there was difficulty accessing the veins.
Plasma GUSB activity in Dog28 was detectable on days 1–5 post infusion, peaking on day 2 at 50% of activity level in the unaffected sibling (Table 1 and Fig. 4B). GUSB activity was not detectable thereafter, and 3 weeks post treatment, the dog was euthanized. The half-life of hGUSB in plasma was estimated at about 18 h—10 times shorter than cSEAP (T1/2 ≈ 6 days).
Liver samples from Dog28 were collected and assayed for enzymatic activity, GAG accumulation, and GUSB gene frequency. GUSB activity was not detected in either plasma or liver lysates by 4MU-based enzyme assay. Quantitative assay of GAG levels in the treated liver samples showed no reduction in storage. Representative transmission electron micrographs and toluidine blue–stained liver tissue sections exhibited full pathology similar to that of liver samples from untreated animals (Supplementary Fig. S3A and B). However, GUSB-expressing cells were visualized in histochemically stained sections of the left lateral lobe at a frequency of 1:10,000–1:100,000 cells, whereas no stained cells were detected in the untreated control liver tissue (Supplementary Fig. S3C). The low frequency is consistent with the inability to detect cGUSB DNA due to the qPCR assay's sensitivity limit of 1:1,000 genome equivalents. These numbers are also in agreement with the qPCR detection of cSEAP transposon post expression at low but measurable frequencies in all treated dog livers.
Dog28 exhibited transient elevation of transaminases ALT and AST (Fig. 4E and F). While ALT elevation resolved within the time frame of cSEAP- and cFIX-injected dogs, elevated AST levels in the MPS VII dog lasted longer than in all normal dogs. It is presumed that the liver in this dog was more prone to damage from hydrodynamic delivery than was the liver in the WT dogs, possibly due to complications associated with hepatomegaly.
Discussion
This is the first report of prolonged transgene expression of >4 months in large animals after non-viral vector delivery. Previously, infusion in rats achieved maintenance of human FIX for 2 months following a modified hydrodynamic injection. 53 This study reports that expression of reporter cSEAP and therapeutic protein cFIX was extended from 6 to about 24 weeks after a single vector administration, when a transient, 30-day regimen of GdCl3 was administered. Moreover, in one dog, cSEAP expression stabilized at about fourfold greater than the pretreatment level (Dog18-1; Fig. 3A). The lack of an immunosuppressive effect in the MPS VII dog is most probably due to an insufficient dose of GdCl3 caused by problems with accessing blood vessels in the affected dog that may be associated with the tissue properties of the MPS VII animal. Intravenous administration of GdCl3 blocks phagocytosis in the liver and selectively eliminates large Kupffer cells, resident liver macrophages that produce cytokines, and other inflammatory mediators in response to bacterial infection and liver injury. 54 In rats, Kupffer cells repopulate the liver 3–7 days post treatment, and effectiveness of GdCl3 is not diminished by repeated administration if repopulation is complete. 55 However, in GdCl3-immunosuppressed dogs, prolonged transgene expression was observed only after the first DNA infusion. Dramatic loss of expression upon vector re-administration, similar to that observed in dogs, has been previously demonstrated in transposon-treated immunosuppressed mice. 56
Hepatocellular toxicity, while transient, appeared to curtail transgene expression. Thus, following initial vector administration in GdCl3-treated dogs, lower peak levels of liver transaminases ALT and AST were observed in dogs exhibiting a longer duration of transgene expression. However, when vector infusion and GdCl3 treatment were repeated, transgene and ALT and AST profiles were similar to those observed in dogs that were not administered GdCl3.
Nine infusions of cSEAP transposon resulted in up to an approximately 200-fold increase in the pre-infusion level of heat-stable alkaline phosphatase, and two infusions of cFIX transposon attained a level of clotting factor that was up to 40% of the endogenous level of canine FIX. These results confirmed the reproducibility of the catheter-mediated, non-viral delivery of SB transposons to dogs. The shorter half-life of GUSB compared to cSEAP in dogs treated without immunosuppression is almost certainly due in part to rapid uptake of the lysosomal enzyme via mannose-6-phosphate-mediated endocytosis. 57 Avid cellular uptake is documented in patients with lysosomal storage diseases treated with bone-marrow transplantation. For example, in MPS I patients, IDUA activity in plasma is below the detection limit, 58 even after full bone-marrow engraftment, and it takes >2 years to reach 35–40% of the normal control value. 59
In dogs, transgene expression regulated by the CAGGS promoter achieved its peak level in plasma at 2–4 days post infusion compared to 1 day post infusion in mice. The reason for the difference in appearance of peak expression is unclear. In GUSB- and IDUA-treated dogs, the detected number of transgenes per liver cell was 20–100 times lower than that in mice. This lowered efficiency of gene transfer/cell in the dog probably explains the shorter duration of detectable transgene expression 1 week post infusion, since in NOD/SCID mice, expression levels in the plasma drop 5- to 10-fold before stabilization, 19 which would render expression undetectable were it to happen in dogs.
On the other hand, the long-term experiments with both immune-suppressed and NOD/SCID mice treated with the same transposons as dogs showed that while extended expression drops 5- to 10-fold below the day +1 level, the transgene copy number drops 30- to 40-fold. 13,19,60 This suggests that 70–80% of plasmids in the liver are lost, but those that enter nuclei and are expressed are retained at a higher rate. In immunocompetent mice, further loss of activity/transfected cells occurs with the onset of adaptive immune responses. These observations emphasize the importance of finding effective immunosuppression for dogs that are hydrodynamically infused with naked DNA.
As has been previously demonstrated in mice, in the absence of immune responses, transposition is a critical factor that affects the longevity of expression. 13 Excision products that are the prime evidence of transposition were detected in liver samples from a normal dog infused with hGUSB-transposon, which was co-administered with pCMV-SB100X plasmid. In contrast, in dogs infused with hIDUA-encoding transposon with the 10- to 20-fold weaker SB11 transposase, 13 excision products were not detected, even though the IDUA transposons and transposase SB11 were on the same plasmid. Apparently, to achieve detectable transposition of therapeutic SB transposons into canine hepatocyte genomes, the strongest SB transposase, SB100X, must be used. Inclusion of the SB transposon and transposase expression cassettes on the same plasmid assures that both components of the transposon system are present in successfully transfected cells. While cis and trans SB delivery yielded comparable transposition frequencies in mice, the cis configuration may be preferable for treating dogs because delivery is roughly 100-fold lower. More work on this aspect of transposon delivery is needed.
Previously, the kinetics of loss underlying SB-mediated hIDUA expression were analyzed in mice treated using many different conditions, such as ubiquitous CAGGS versus liver-specific ApoEhCRhAAT promoter, SB11 versus SB100X transposase, cis versus trans SB transposon configuration, different DNA doses, ±immunosuppression, and so on. 13 It was found that many combinations of factors affecting transgene expression lead to only a limited number of post-treatment scenarios. Without immunosuppression, in mice, expression was lost rapidly, independently of transposition (e.g., half-life of hIDUA was 1–2 days). This is similar to what was observed in dogs in the absence of immunosuppression. However, when mice were immune suppressed, stable expression was attained due to transposition, and stabilized expression from episomes at levels deemed therapeutic for MPS I was never observed. Nevertheless, episomal expression in immune-suppressed mice could continue for months. 13 This explains prolonged declining expression in cSEAP-treated dogs, which is predominantly episomal.
In viral and non-viral liver-directed gene therapy for systemic disease, such as hemophilia or MPS I and VII, the levels of sustained therapeutic protein secreted from the liver into the blood determine the extent of correction. 19,61 –63 Hemophilia A and B correction requires a relatively low continuous presence of normal protein in the circulation. Thus, Cantore et al. 64 showed that liver-directed lentiviral vector–mediated gene therapy of canine hemophilia B dogs provided stable, long-term FIX activity up to 1% of normal with therapeutic benefit. Moreover, in a human clinical trial, Nathwani et al. achieved stable therapeutic expression of FIX at around 5% of the normal level lasting >4 years after a single i.v. administration of an adeno-associated virus vector encoding FIX. 65 Higher transgene expression will be required for MPS. Bigg et al. 33 monitored animals after i.v. injection of gamma-retroviral vector into neonatal MPS VII dogs for 11 years and reported that sustained serum GUSB at 41% of WT had beneficial effects on heart valves and prolonged life-span. Thus, stable activity at about 40% WT levels represents a reasonable target for treatment of human patients with MPS I and MPS VII, which is also comparable to the amount of enzyme delivered by enzyme replacement therapy. Moreover, mouse studies in MPS IIIA indicate the superiority of continual slow infusion versus repeated bolus injection of the recombinant enzyme in ameliorating MPS IIIA. 66
This study is a proof of principle that immune suppression can improve maintenance of the active transgenic protein in serum. Potent immune suppression strategies are currently being explored for translation of preclinical studies to large animal models and humans that also permit re-dosing in such common scenarios as liver growth, vector silencing, and loss of transduced liver cells. 35,67 –70 In future studies, it will be important to determine which types of immune response cause the decline in transgene expression in order to find effective immune suppressive therapy. However, hepatic delivery remains the Achilles heel in non-viral gene therapy studies aiming at clinical trials. 42 –44 It remains to be seen whether the SB transposons can be used in the in vivo approach as successfully as they are being used to treat blood cancers in human patients. 71
Footnotes
Acknowledgments
We thank our colleagues on the Program Project Grant for Gene Therapy for Metabolic Disorders and members of the Center for Genome Engineering for advice and critiques, Dr. Kathy Ponder for the gift of canine GUSB cDNA, and Dr. Mark Kay for pAAV-hFIX16 plasmid containing the liver-specific promoter. Breeding stock for the MPS VII dog in this study was derived from a colony supported by the NIH grant NS085381 to Patricia I. Dickson. This work was supported by NIH grants R01DK082516 and P0HD32652 to the University of Minnesota and R44HL072539 and R41DK081249 to Discovery Genomics, Inc.
Author Disclosure
P.B.H., K.A.H., E.R.O., and R.S.M. have equity in Immusoft, Inc. E.R.O. and R.S.M. are employees of Immusoft, Inc. (formerly Discovery Genomics, Inc.). E.L.A, B.C.H, J.B.B, M.U.R., D.W.H., and N.M.E. have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.