
Abstract
Synthetic splice-switching oligonucleotides (SSOs) target nuclear pre-mRNA molecules to change exon splicing and generate an alternative protein isoform. Clinical trials with two competitive SSO drugs are underway to treat Duchenne muscular dystrophy (DMD). Beyond DMD, many additional therapeutic applications are possible, with some in phase 1 clinical trials or advanced preclinical evaluation. Here, we present an overview of the central factors involved in developing therapeutic SSOs for the treatment of diseases. The selection of susceptible pre-mRNA target sequences, as well as the design and chemical modification of SSOs to increase SSO stability and effectiveness, are key initial considerations. Identification of effective SSO target sequences is still largely empirical and published guidelines are not a universal guarantee for success. Specifically, exon-targeted SSOs, which are successful in modifying dystrophin splicing, can be ineffective for splice-switching in other contexts. Chemical modifications, importantly, are associated with certain characteristic toxicities, which need to be addressed as target diseases require chronic treatment with SSOs. Moreover, SSO delivery in adequate quantities to the nucleus of target cells without toxicity can prove difficult. Last, the means by which these SSOs are administered needs to be acceptable to the patient. Engineering an efficient therapeutic SSO, therefore, necessarily entails a compromise between desirable qualities and effectiveness. Here, we describe how the application of optimal solutions may differ from case to case.
Introduction
S
The success of the early DMD exon-skipping trials has also inspired a number of other therapeutic applications of SSOs, foremost among them ISIS-SMNRx for the treatment of spinal muscular atrophy (SMA). This SSO has rapidly progressed to phase 2 clinical trials for type I SMA in infants because of the promising data from the initial dose-ranging study (Swoboda et al., 2013). Type I SMA leads to a severely reduced life span of less than 2 years, and there are currently no other treatment options. More therapeutic applications are still in the translational stages with some in vivo proof-of-principle data available.
Here, we briefly analyze clinical developments and the various available oligonucleotide chemical modifications. It appears that toxicity of SSOs is largely determined by these chemical modifications, with sequence-dependent toxicity being less of an issue (Aartsma-Rus and Muntoni, 2013).
Lessons learned in these early clinical trials will be applicable to the further development of therapeutics still in the translational stage and, it is hoped, lead to a shortened and simplified clinical approval pathway. However, it is becoming clear that the lessons learned from the special case of DMD, where the aim is to cause exon skipping in a low-expressed dystrophin pre-mRNA, may not be entirely typical. We propose that there is a relationship between target pre-mRNA expression levels and required oligonucleotide concentration in the nucleus for effective splicing manipulation and discuss the ensuing necessity for tissue-specific delivery reagents in more detail.
Clinical Development of SSOs to Treat Duchenne Muscular Dystrophy
DMD is an X-linked, inherited, and progressive muscle-wasting disease afflicting 1 in 3500 newborn boys, typically diagnosed between the ages of 3 and 5 years. It is caused by specific gene mutations in dystrophin, an essential part of the dystrophin-associated glycoprotein complex that connects the actin cytoskeleton to the surrounding extracellular matrix via the cell membrane, providing vital structural support (Cohn and Campbell, 2000). Loss of dystrophin function results in muscle degeneration and replacement with fibro-adipose tissue, leading to severe disability, loss of ambulation, and eventually an early death due to respiratory or cardiac failure.
Dystrophin gene mutations cause mostly deletions of certain exons resulting in frameshifts in the exons that follow, premature termination, and thus loss of protein function. SSOs can restore the open reading frame by skipping additional exons to get back into frame. This leads to the expression of internally truncated but mostly functional dystrophin protein, similar to the isoforms found in the milder Becker muscular dystrophy (Koenig et al., 1989).
The clinical development of eteplirsen and drisapersen has been well reviewed (Arechavala-Gomeza et al., 2012; Kole and Leppert, 2012; Koo and Wood, 2013) and therefore we present only the most up-to-date data available, which has not been covered in these sources. GSK (Brentford, UK) and Prosensa (Leiden, The Netherlands) reported that the primary end point of improvement in the 6-minute walk test (6MWT) was not met in a 2-year phase 3 study (NCT01254019;
Sarepta Therapeutics reported 96-week data from their phase 2b open-label extension study of eteplirsen (NCT01540409). These demonstrated stabilization in the 6MWT with a difference of 68 m between the combined 30- and 50-mg/kg/week treatment groups and the placebo/delayed treatment group, which had shown a decline up to 36 weeks after the trial started. The boys in the delayed treatment group were crossed over into the treatment groups at week 24 and showed stabilization of 6MWT values at week 36 (Mendell et al., 2013). Further data at 120 weeks demonstrate that this stabilization still persists (Sarepta Therapeutics, 2014a). Accelerated U.S. Food and Drug Administration (FDA) approval on the back of these data was denied because of concerns about the disconnection between increases in dystrophin levels and clinical outcome in the drisapersen trial and the lack of a validated quantitative assay for dystrophin. Another concern was the low number of trial participants (only 12 were enrolled in total). The FDA also expressed concern about the 6MWT as an appropriate end point and stated their belief that a placebo-controlled trial would be necessary to prove efficacy. However, the FDA provided a guidance letter suggesting that they would consider accelerated approval under certain conditions and Sarepta Therapeutics has stated its intent to submit a New Drug Application by the end of 2014 (Sarepta Therapeutics, 2014b).
Taken together, these setbacks may have negative implications for the whole field of therapeutic SSOs, even though Prosensa has reiterated its commitment to exon-skipping therapies for DMD and Sarepta Therapeutics is continuing development of eteplirsen.
Clinical Development of SSOs to Treat Spinal Muscular Atrophy
On the other hand, an SSO for the treatment of spinal muscular atrophy (SMA) has generated positive data in a phase 1 clinical trial. SMA, an autosomal recessive disease affecting 1 in 6000 newborn children, is caused by mutations in the survival of motor neuron gene 1 (SMN1) and results in progressive loss of motor neurons in the spinal cord and subsequent atrophy of voluntary muscles. Clinical severity depends on naturally occurring exon 7 inclusion levels in the almost identical SMN2 gene and the number of copies of SMN2. In SMN2, a C>T transition in an exon 7 splicing enhancer site normally results in an unstable exon 7-deleted isoform and insufficient levels of full-length isoform (Khoo and Krainer, 2009; Fig. 1A). However, effective SMN2 exon 7 inclusion can be achieved by blocking an intronic splicing silencer in the 5′ region of intron 7 (ISS-N1; Singh et al., 2006; Hua et al., 2008) with SSOs. On the basis of successful mouse studies (Hua et al., 2010, 2011; Passini et al., 2011) a candidate SSO was quickly moved into a phase 1 clinical trial by Isis Pharmaceuticals (Carlsbad, CA), because validation of pharmacological efficacy in nonhuman primates was not possible as they lack SMN2.
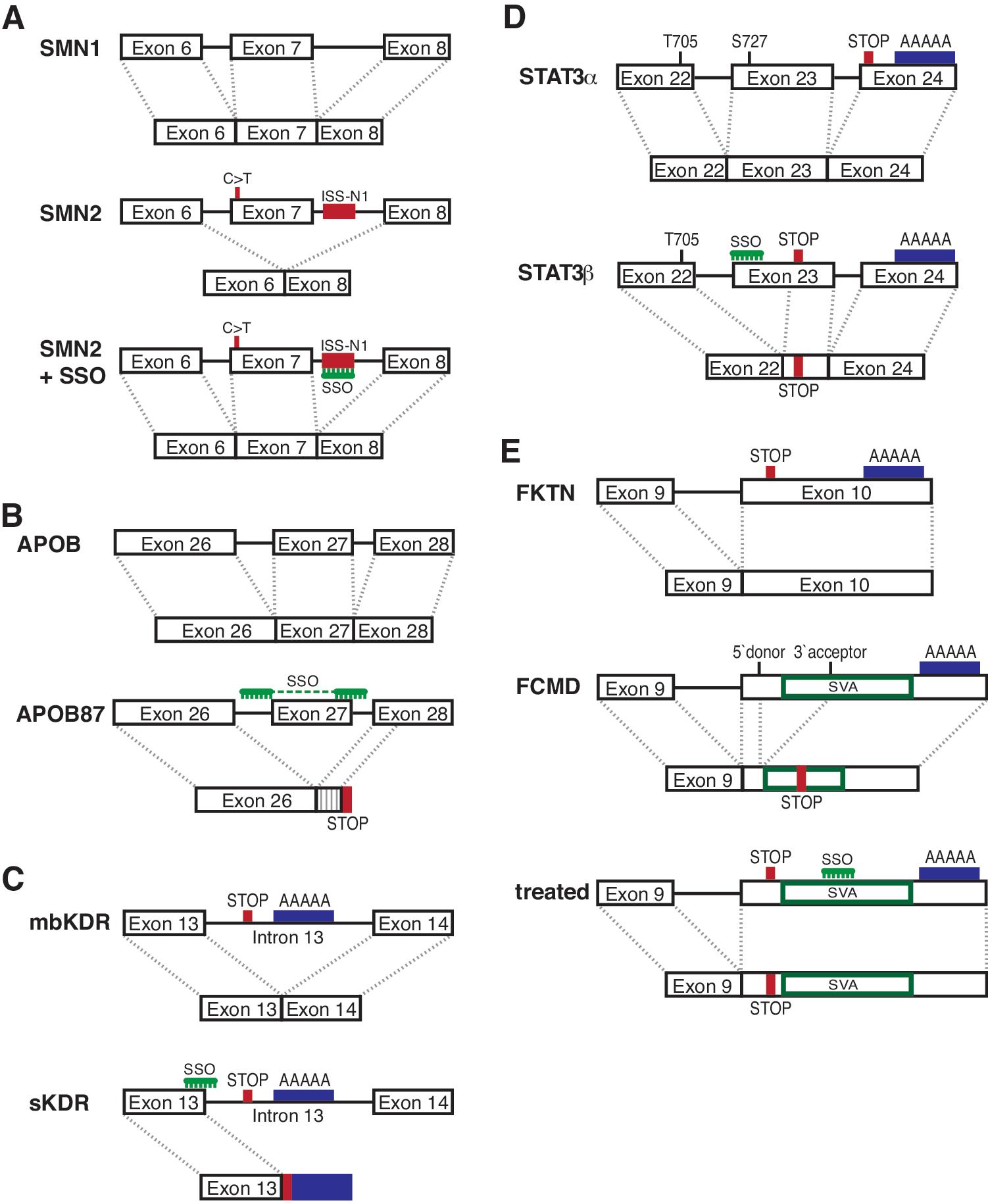
Mechanisms of splice modulation.
In the phase 1 trial (NCT01494701), direct injection of a single 9-mg dose of ISIS-SMNRx into the intrathecal space resulted in broad distribution in the central nervous system and some motor function improvement in children with SMA (Swoboda et al., 2013). Preclinical and mouse data (Hua et al., 2010) on SSO persistence and continued efficacy in the spinal cord suggest that infrequent dosing every 6–9 months may be enough for a sustained therapeutic effect. However, systemic SSO delivery may be needed to alleviate cardiac pathology and to increase peripheral motor neuron function and consequently survival (Hua et al., 2011; Porensky et al., 2012). Thus, it may be necessary to administer the SSO both systemically and directly into the intrathecal space, with the former at more frequent intervals. Moreover, in mice there is a tight therapeutic window (≤28 days from birth) during which SMN expression is required to overcome early death (Foust et al., 2010; Le et al., 2011). As yet, it is unclear how long such a therapeutic window would be in humans, but the data from mouse studies certainly emphasize that early treatment is better.
Taking this into account, together with the promising results from the first SMA clinical study, Isis Pharmaceuticals has already initiated a phase 2 clinical trial (NCT01839656) in infants with type I SMA, the most severe form of the disease; patients have an expected life span of less than 2 years. The study is expected to run for 1 year with three intrathecal injections at two different dosing levels and will enroll 20 infants up to 210 days of age. All enrolled patients will be treated with active drug. Interim data from this trial and the phase 1 extension study are extremely promising and show dose- and time-dependent improvements that correlate with increased levels of SMN protein (Isis Pharmaceuticals, 2014a,b). Given the disease severity and absence of other treatment options, it is hoped that this SSO will be successful and thereby also provide a much-needed boost to the field of therapeutic splicing modulation following the setbacks in the DMD field.
Additional Therapeutic Applications
As enumerated in Table 1, several additional SSO therapeutics have progressed into the in vivo proof-of-principle stage, validating diverse concepts of splicing modulation induced by oligonucleotides. The number of such published studies is increasing each year, and thus we make no claim to be exhaustive. Here, we concentrate on select in vivo studies that we find particularly innovative, as information about other in vitro and in vivo studies can be found in van Roon-Mom and Aartsma-Rus (2012) or Havens and colleagues (2013).
2′-Me, 2′-O-methyl oligoribonucleotide; 2′-MOE, 2′-O-methoxyethyl oligoribonucleotide; AAV, adeno-associated virus; APOB, apolipoprotein B; BP, branch point sequence; ESE, exonic splice enhancer; ISE, intronic splice enhancer; KDR, kinase insert domain receptor/vascular endothelial growth factor receptor 2; LDL, low-density lipoprotein; LMNA, lamin A; PMO, phosphorodiamidate morpholino oligonucleotide; PPT, polypyrimidine tract; PS, phosphorothioate; SSOs, splice-switching oligonucleotides; STAT, signal transducer and activator of transcription; VEGF, vascular endothelial growth factor; Vivo-PMO, octaguanidine-conjugated PMO.
A novel therapeutic concept in this domain is to generate a desirably functional protein isoform by splice switching. We have used this method in apolipoprotein B (APOB) to treat familial hypercholesterolemia, a genetic disease characterized by high levels of low-density lipoprotein (LDL; Disterer et al., 2013; Khoo et al., 2007). In this case, skipping of exon 27 results in a premature stop codon in exon 28, thus leading to a truncated protein, APOB87 (Fig. 1B). This imitates natural shortened APOB isoforms found in familial hypobetalipoproteinemia, where LDL levels are low because of increased LDL clearance and reduced LDL assembly. In hypercholesterolemic human APOB transgenic mice, generation of relatively low levels of skipped APOB87 (6.8%) was shown to reduce LDL cholesterol by 34–51% (Disterer et al., 2013).
A related option is to alter the balance between a full-length isoform and a naturally occurring splice form with a different function. One way to achieve this is to induce alternative polyadenylation via inclusion of an intron containing polyadenylation sites into the mRNA. For example, the kinase insert domain receptor (KDR; Uehara et al., 2013) is a membrane-bound vascular endothelial growth factor (VEGF) receptor with a soluble isoform that lacks an intracellular tyrosine kinase domain. This is due to inclusion of intron 13, which contains a stop codon and two polyadenylation sites that are subsequently used and thus result in a shortened mRNA with exons 14–30 excluded (Fig. 1C). The soluble isoform naturally occurs in human cornea, still binds VEGF, but is functionally inactive, and has been shown to prevent lymphangiogenesis in this tissue. Uehara and colleagues show that a phosphorodiamidate morpholino oligonucleotide (PMO) increased the soluble isoform in vitro and suppressed hemangiogenesis and lymphangiogenesis after laser photocoagulation or corneal suture as well as reducing graft rejection after transplantation in mouse models.
This principle is also applicable to other receptor tyrosine kinases (Vorlová et al., 2011). For instance, Fms-related tyrosine kinase 1 (FLT1) is another membrane-bound vascular endothelial growth factor with similar structure. Again, PMOs can be used to influence the ratio of the membrane-bound and soluble isoforms through inclusion of intron 13 into the mRNA (Owen et al., 2012). Increases in the soluble FLT1 isoform are shown to decrease choroidal neovascularization after laser injury. This also induced tumor regression in a human breast adenocarcinoma xenograft mouse model. Soluble FLT1 and KDR therefore represent important drug targets in cancer metastasis.
Another important target in cancer is the signal transducer and activator of transcription 3 (STAT3) protein. Here, two isoforms exist. The full-length α form can be activated by phosphorylation at two key amino acids (tyrosine-705 and serine-727), causing dimerization and subsequent translocation to the nucleus, where binding to its target promoter sequences induces gene expression. The truncated STAT3β isoform is generated when an alternative acceptor site in exon 23 is used and thus lacks the C-terminal transactivation domain, including the serine-727 phosphorylation site necessary to induce transcription, but can still bind to its target promoters (Fig. 1D). Redirection of splicing from the STAT3α to the STAT3β isoform prompts apoptosis in cancer cells and inhibits tumor growth in a xenograft model (Zammarchi et al., 2011).
SSOs can also be used to reduce aberrant splice forms, preferentially expressing an isoform that diminishes damage done by loss of the correct full-length isoform due to a splice site mutation (Gedicke-Hornung et al., 2013). The authors described an mRNA variant (variant 4; Var-4) of cardiac myosin-binding protein-C (Mybpc3), with in-frame deletions of exons 5 and 6. Redirection of splicing away from the three aberrant mRNAs, missense Mut-1 and out-of-frame Mut-2 and −3, toward increased expression of the Var-4 RNA isoform was shown to restore cardiac function and prevent left ventricular hypertrophy in a mouse model of hypertrophic cardiomyopathy. For efficient delivery to the heart, the authors developed an adeno-associated virus serotype 9 (AAV9) vector with the splice-switching oligonucleotides embedded in modified U7 small nuclear RNA (U7 snRNA) (Goyenvalle et al., 2004). The modification directs the normally nonspliceosomal U7 snRNA to the spliceosome, thus delivering the inserted oligonucleotide to the proper subcellular localization.
Another innovative application of splicing modulation addresses the problem of exon trapping caused by inherited SINE-VNTR-Alu (SVA; SINE, short interspersed elements; VNTR, variable number of tandem repeats) retrotransposon insertions (Taniguchi-Ikeda et al., 2011; Fig. 1E). This retrotransposon contains a strong 3′ splice site. Taniguchi-Ikeda and colleagues demonstrated that insertion of this retrotransposon was responsible for abnormal splicing in the fukutin (FKTN) gene leading to Fukuyama muscular dystrophy. Correction of the anomalous splicing pattern, using SSOs targeted to the 3′ splice site within the SVA, partially rescued normal fukutin expression and function in a mouse model and also in patient-derived cells. In addition, they showed that exon trapping by an SVA is also present in patients with autosomal recessive hypercholesterolemia or neutral lipid storage disease. Insertion of an SVA has also been implicated in hereditary elliptocytosis, X-linked agammaglobulinemia, neurofibromatosis type 2, and X-linked dystonia-parkinsonism, suggesting that SSOs would be appropriate therapeutic reagents for treatment of these diseases (Taniguchi-Ikeda et al., 2011).
More straightforward correction of aberrant splicing patterns has been described in the Usher syndrome 1C (USH1C; Lentz et al., 2013) and laminin A (LMNA; Osorio et al., 2011) genes, both examples caused by mutations that lead to activation of cryptic splice sites. At present, there is no cure for either Usher syndrome or Hutchinson-Gilford progeria, rare but devastating genetic disorders. The outcome of these studies bodes well for future clinical development, particularly with the increased funding and interest in treatment of rare diseases.
Measuring the Efficiency of Splice-Switching Oligonucleotides
It is of concern, however, that the majority of publications rely on quantitative measurement of splice forms using reverse transcription and PCR. It remains common practice to use one primer set to amplify both mRNA variants of interest, even though it is well established that shorter PCR products amplify better than longer products in the same reaction (Walsh et al., 1992). Hence, as the skipped or truncated isoform is generally shorter, the efficiency of splice modulation will be overestimated. Also, relative quantification of end-point PCR products is not appropriate, as the distinct amplification efficiencies between long and short products result in reaching the plateau phase at different times. Therefore, relative quantification of the two isoforms will vary depending on the amount of template input and PCR cycle number. Using radioactive PCR may improve the relative quantification of two bands on a gel compared with ethidium bromide-based detection, but does not address the underlying problem. To overcome these problems, we have developed a fully validated real-time quantitative PCR (RT-qPCR) assay and illustrate its value in measuring SSO-mediated truncation of APOB100 (Fig. 2). Here, the exon 27-skipping efficiency for SSO 3B, using a radioactive end-point PCR assay (Khoo et al., 2007), is significantly higher than when measured by the validated RT-qPCR assay (Disterer et al., 2013), indicating that the earlier assay was positively biased in its assessment of the skipped isoform.
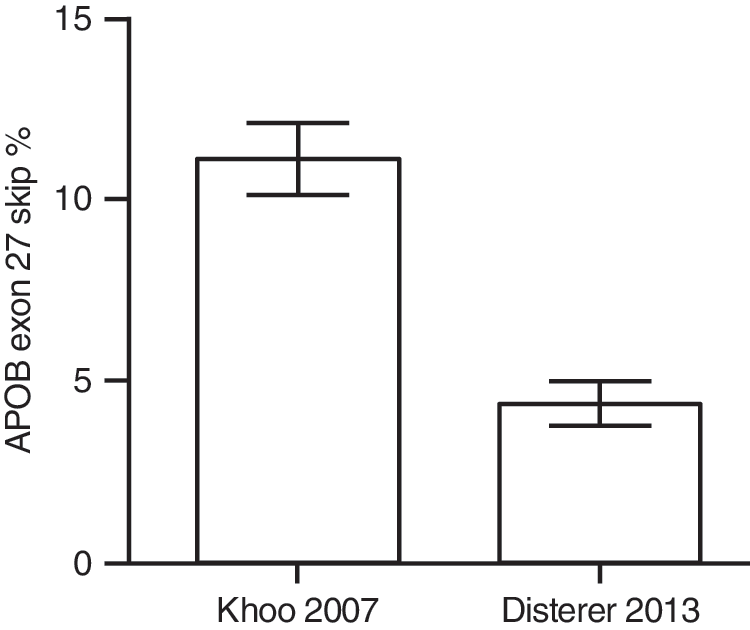
Comparison between radioactive end-point PCR and validated RT-qPCR assay. In both cases, HepG2 cells were transfected with 250 nM SSO 3B (SSO 1 in Disterer et al., 2013) for 48 hr, using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. Khoo and colleagues (2007) reported a skipping percentage of 11.13±0.97% (n=3), using a radioactive end-point PCR assay, whereas Disterer and colleagues (2013) achieved 4.33±0.61% (n=15) with a fully validated RT-qPCR assay. Downstream RNA purification, reverse transcription, and PCR conditions varied slightly, but should not have caused such a consistent difference. Values represent means±SEM from independent experiments as calculated with GraphPad Prism 5 (GraphPad Software).
To date, in other splice-switching studies where RT-qPCR is described, only one nonvalidated reference gene has been used and requisite minimal essential information (Bustin et al., 2009) is lacking. However, accurate relative quantification by RT-qPCR can easily be achieved with careful primer design and separate PCRs for each isoform, as long as a common standard curve is used, derived from a plasmid carrying both PCR amplification products (Fig. 3; Disterer and Khoo, 2012; Disterer et al., 2013).
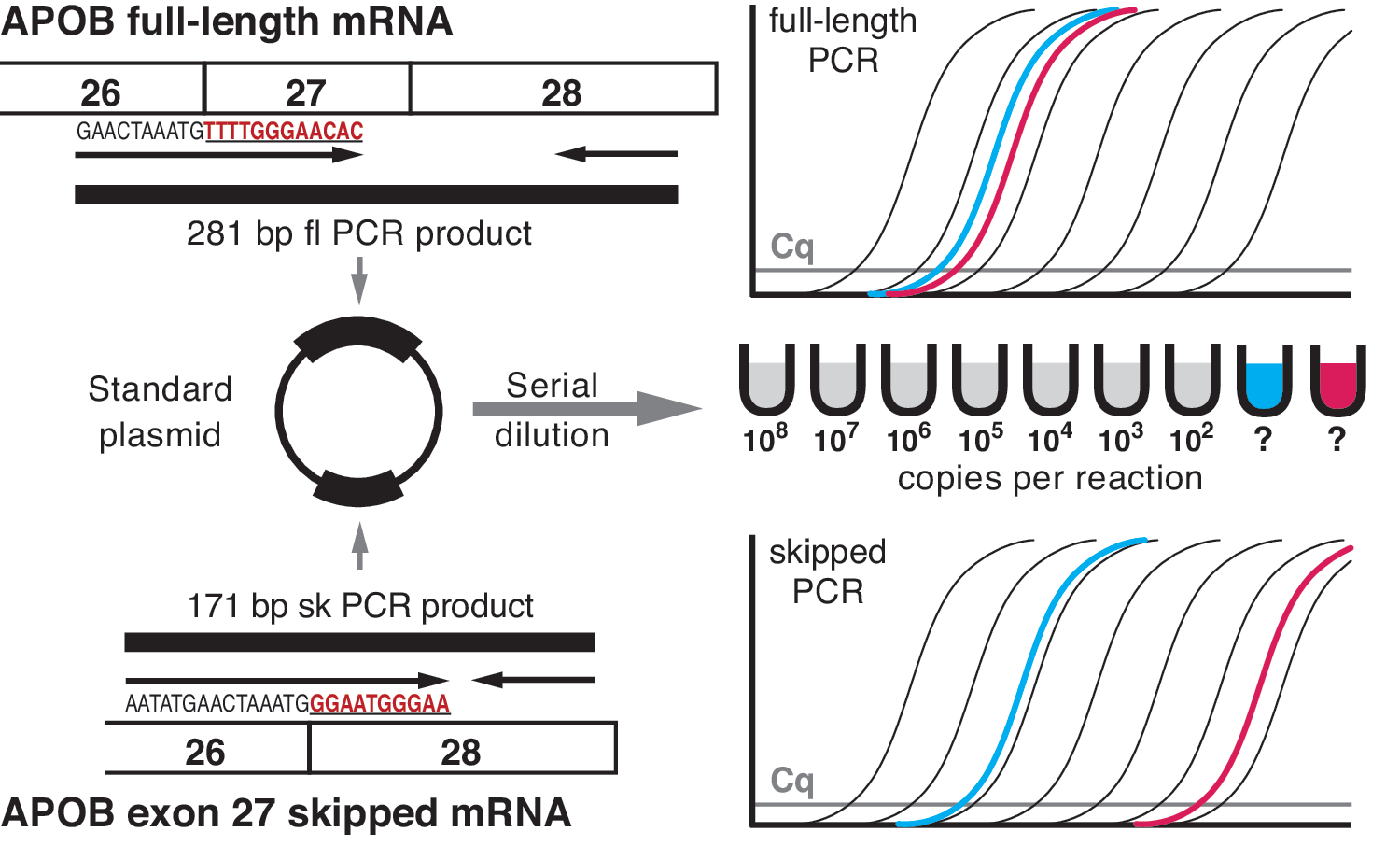
RT-qPCR assay for accurate determination of skipping efficiency. Specific primers for the detection of apolipoprotein B (APOB) full-length and exon 27-skipped mRNA isoforms are designed (left, top and bottom; exon 27- and exon 28-specific sequences are in red and underscored). The resulting full-length (fl) and skipped (sk) PCR products are cloned into a plasmid (left, middle), which is then serially diluted to produce one standard curve (right, middle). This standard curve is used in both the full-length and skipped PCRs to quantify the copy number per reaction for both isoforms (right, top and bottom). As the standard plasmid contains only one copy each of the full-length and skipped PCR products, copy number per reaction results obtained from the two qPCRs can be directly related to each other without the need for reference gene normalization. Thus, the efficiency of skipping can be expressed as a percentage, using the following calculation: sk/(fl+sk)×100. For example, for the blue unknown sample (left thick line in the PCR amplification plots), if copy number per reaction for the full-length PCR is 8×106 and for the skipped it is 2×106, then the skipping percentage would be 20%. The red unknown sample (right thick line in the PCR amplification plots) represents an untreated control with skipping percentage<0.01 (fl=4×106; sk=4×102). Cq, quantification cycle. Color images available online at
SSO Toxicity, Chemistry, and Sequence Selection
Two publications have reviewed SSO chemistry in detail (Saleh et al., 2012; Jarver et al., 2014), so here we provide only a short summary and then focus on a synergistic view of the interplay between sequence selection, chemistry, and toxicity. As is apparent from Table 1, the most commonly used SSO chemical modifications are 2′-O-methyl RNA with phosphorothioate substitutions on the ribose backbone (2′-Me-PS) and phosphorodiamidate morpholino oligonucleotides (PMOs). Oligonucleotides with 2′-Me-PS modifications are commercially available from several oligonucleotide manufacturers, whereas PMOs are sold exclusively by Gene Tools (Philomath, OR) and limited to a maximum length of 30 bases. Two other modifications in common use are 2′-O-methoxyethyl oligoribonucleotides (2′-MOE-PS) and locked nucleic acids (LNAs). Use of the former is limited by availability, as Isis Pharmaceuticals owns the technology and currently no commercial supplier has a licence for production. LNAs are available from Exiqon (Vedbaek, Denmark), but if the Exiqon proprietary algorithm for optimal positioning of the LNA-modified bases within the SSO is requested their location is not transparent to the user.
Toxicities associated with these chemical modifications are well characterized by now. Subcutaneous administration of unprotected 2′-Me-PS causes injection site reactions, proteinuria, elevated urinary α1-microglobulin levels (Goemans et al., 2011), and, in some cases, transient thrombocytopenia (Aartsma-Rus and Muntoni, 2013). The eteplirsen phase 2 trial extension suggests a better safety profile for PMOs, with no serious side effects reported in comparison with placebo, but the small number of participants makes evaluation difficult (Mendell et al., 2013). Sarepta Therapeutics states that no serious side effects were seen in more than 400 patients treated with PMOs as part of their antiviral program. However, although so far there are no dose-limiting toxicities in patients treated with PMO, studies comparing rodents, nonhuman primates, and humans suggest that the same PMO (mg/kg) dose is needed for each, making patient therapy exceedingly expensive.
Oligonucleotides with 2′-MOE-PS modifications are variously reported to be more effective with fewer proinflammatory consequences than 2′-Me-PS (Hua et al., 2010; Yang et al., 2013), but even so, clinical trials with oligonucleotides containing 2′-MOE-PS modifications such as mipomersen have shown that injection site and possibly influenza-like reactions remain a problem (Raal et al., 2010). Long-term studies in DMD using 2′-Me-PS SSOs have shown little evidence of inflammation, suggesting that preclinical toxicology may pick up species-specific sensitivities that may not apply to humans (Frazier et al., 2013). LNA-containing SSOs were found to be more effective than 2′-MOE-PS-modified oligonucleotides (Swayze et al., 2007), but are also associated with significant liver (Swayze et al., 2007) and kidney toxicity (van Poelgeest et al., 2013). Progress in elucidating toxicity mechanisms has been made by reducing the length of LNA oligonucleotides (Seth et al., 2009).
Although the case can be made that toxicity of SSOs is driven mostly by their chemistry, for RNase H-activating LNA antisense oligonucleotides there is an association between particular sequence elements and toxicity (Hagedorn et al., 2013). Clearly, it is important to know whether there is sequence-dependent toxicity, because of its influence on SSO target design, although the extent to which SSO sequences can be modified is restricted by the availability of susceptible pre-mRNA target sequences. A second and not so obvious issue is that if toxicities are generic to oligonucleotide chemistry, it would be much easier to develop SSO-based therapies as regulatory authorities might be persuaded to approve new therapies on the basis of previous preclinical toxicology using older therapies. In this respect, we would encourage companies involved in developing splice modulation therapies to publish more of their preclinical data, at least regarding chemistry and sequence-associated toxicities of oligonucleotides that were not taken into the clinic.
Software prediction tools such as RNAstructure, mfold (Zuker, 2003), and Human Splicing Finder (Desmet et al., 2009) can be incredibly useful in designing SSOs with a high likelihood of effectiveness. General recommendations for SSO sequence design and a step-by-step guide for exon-internal SSOs are shown in Fig. 4 (adapted from Aartsma-Rus, 2012). Although targeting exonic splice enhancer sites close to the 3′ splice site seems the most promising avenue, this strategy is not always successful in the context of other exons such as APOB exon 27 (Khoo et al., 2007; Disterer et al., 2013). The 3′ and 5′ splice sites should not be overlooked and are often used successfully (Table 1). It is also worth considering bipartite (Khoo et al., 2007; Peacey et al., 2012) or tripartite SSOs (Incitti et al., 2010; Disterer et al., 2013; P. Disterer et al., unpublished data) that combine several intronic and/or exonic target sites as these can markedly increase the efficiency of exon skipping.
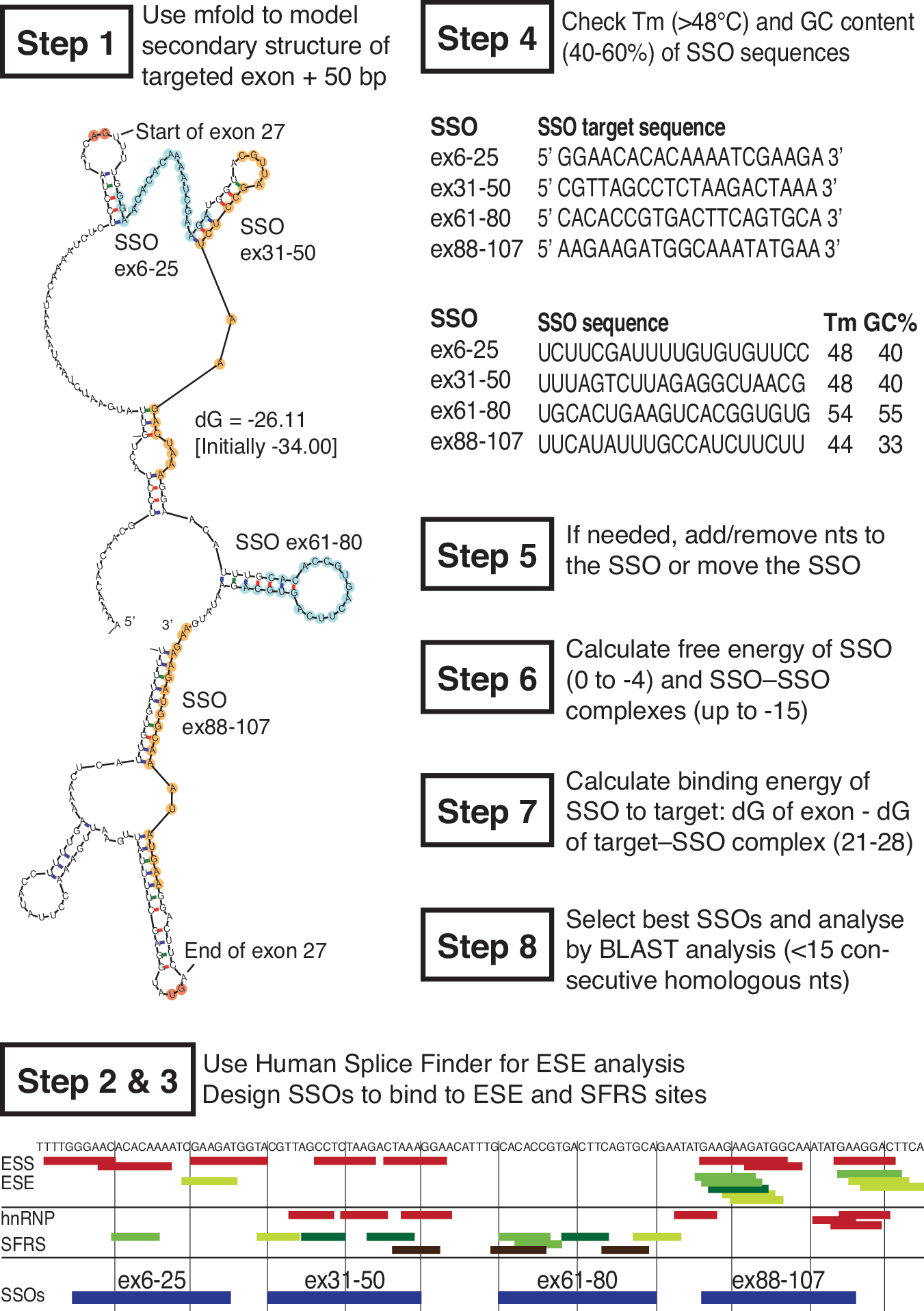
Development of candidate SSOs: step by step (adapted from Aartsma-Rus, 2012). The following general rules increase the chances of successful SSO development. Step 1: Model the secondary structure of the target exon including ∼50 nucleotides upstream and downstream, using mfold (here APOB exon 27). Choose the two or three energetically most stable secondary structures or use the ss-count output to detect partially closed or accessible regions of ∼20–30 nucleotides. Steps 2 and 3: Use Human Splice Finder to analyze exonic splice silencer (ESS, red) and exonic splice enhancer (ESE, green, darker shades=stronger sites) clusters. Binding sites of serine/arginine-rich splicing factors (SFRS) such as SRp55 (light green), SF2/ASF (medium green), SC35 (dark green), or SRp40 (black) are good targets, whereas heteronuclear ribonucleoprotein (hnRNP) suppresses splicing. Design ∼20-mer SSOs, preferably at the 5′ end of the exon, without stretches of three or more Gs or Cs that bind to these regions. Step 4: Analyze the melting temperature T
m (>48°C) and GC content (40–60%) of proposed SSOs (reverse complement of the target sequence). Step 5: If either value is too low or too high, increase or decrease the SSO length. Step 6: Calculate the free energy (dG) of the proposed SSO (for 20-mer SSOs it should be 0 to −4) and SSO–SSO homodimers (up to −15), using RNAstructure. Step 7: Calculate the dG of SSO bound to the target exon (between 21 and 28). Step 8: Analyze proposed SSO, using BLAST; stretches of 15 or more consecutive homologous nucleotides should be avoided. Color images available online at
Sometimes it is necessary to go even further afield from the actual exon to be excluded or included. For example, exon inclusion of SMN2 exon 7 was most efficient with SSOs targeting an intronic negative splice regulator sequence (ISS-N1), which is located 10 bases downstream of the SMN2 intron 7 5′ splice site (Singh et al., 2006; Hua et al., 2008). Targeting intronic structures formed by long-distance interactions up to 300 nucleotides from the exon-intron junction can also be a successful strategy (Singh et al., 2013). Moreover, it is not yet clear whether there are separate design rules or optimal target regions for constitutively included exons, compared with exons that naturally undergo some degree of alternative splicing.
In light of these data, we recommend initial screening of SSOs designed according to the guidelines proposed by Aartsma-Rus (2012), with some additional SSOs targeted to the splice donor and acceptor sites. If these SSOs do not result in highly efficient splicing, combining partially effective oligonucleotides into bipartite or tripartite SSOs should be considered. If this still does not yield efficient splice modulation, exploration of the intron sequence and structure context can be attempted; however, this is time-consuming and technically challenging.
Another important point when designing SSOs is that of sequence specificity and potential off-target effects. Intronic splice site sequences are more similar across all genes than exonic sequences, but all candidate SSOs should be evaluated for off-target binding by performing a BLAST search during the design stage and, after selection of leads, by experimental assays such as exon arrays or RNA-seq (P. Disterer et al., unpublished data). It is also noteworthy that theoretically SSOs targeting exon-only sequences can be sequestered by cytosolic mRNA molecules, especially if the target gene is highly expressed. If so, a higher dose will be required to induce splice switching in nuclear pre-mRNA.
The previous considerations assume, of course, that SSOs bind only to completely matched antisense sequences. An early study found that exon skipping was completely aborted when using mismatch-containing 2′-Me-PS SSOs, but this was not invariant as PMO-modified SSOs with or without two mismatches to the target sequence had equal efficiencies (Heemskerk et al., 2009). Permissiveness is also reported for SSOs modified with LNA (Aartsma-Rus et al., 2004). In addition, Fragall and colleagues (2011) found that a 2′-Me-PS SSO perfectly matching the genomic DNA sequence in control cells showed paradoxically increased skipping efficiency in patient cells containing a single base insertion in the genomic sequence. However, this increase in efficiency could reflect escape of the skipped patient mRNA from nonsense-mediated decay due to the now-corrected reading frame. It appears, therefore, that the type of SSO chemical modification may influence off-target effects during later stages of therapeutic development. In particular, the drive to use high-affinity chemistries such as LNA may in fact enable off-target effects because of their ability to hybridize even with mismatches, and this issue should be borne in mind when selecting the chemistry to be used.
Pharmacokinetics of SSOs
Extended pharmacokinetics is a desirable quality for SSO therapeutics, especially where chronic treatment is envisaged, but varies widely between SSOs constructed on the basis of different chemistries. Phosphorothioate oligonucleotides are characterized by their binding to serum proteins, particularly albumin and α2-macroglobulin. This leads to an extended half-life as renal elimination is reduced. Distribution to tissues is also rapid, with half-lives typically <1 hr. Once distribution is complete, the terminal elimination half-life is extended to 40–60 hr or longer and is largely determined by degradation of the oligonucleotides within the tissue (Crooke, 2004). 2′-MOE-PS-based oligonucleotides are similarly rapidly distributed to tissues and then have extended terminal elimination half-lives of 20–34 days (Yu et al., 2009). On the other hand, the neutral backbone of PMOs precludes binding by serum proteins, and they partition into tissues to a lesser extent than 2′-Me-PS SSOs (Heemskerk et al., 2009). “Plain” PMOs are eliminated relatively quickly with half-lives of 1–20 hr as PMOs are unable to bind serum proteins and are therefore eliminated by renal filtration (Amantana and Iversen, 2005). However, if PMOs are able to distribute into tissues, for example by coupling to cell-penetrating peptides, their elimination half-life is greatly extended as PMOs themselves are extremely stable biologically (Amantana et al., 2007). Therefore, three key determinants of SSO pharmacokinetics emerge, namely, (1) their ability to persist in the circulation (largely determined by their resistance to renal elimination), (2) their ability to partition rapidly into tissues, and (3) their resistance to degradation in peripheral tissues, as opposed to plasma. These factors influence the selection of chemistries for different SSOs.
SSO Chemistry and Tissue Delivery
Oligonucleotide chemistry is also associated with slightly different tissue tropism when systemic delivery is used. For example, LNAs distribute mainly to the kidney, liver, colon, and small intestine (Roberts et al., 2006). PMO SSOs do not bind to plasma proteins and as a result tend to be eliminated quickly through renal filtration, although they can distribute to most tissues in the body (Sazani et al., 2002). As indicated previously, 2′-Me-PS- and 2′-MOE-PS-modified SSOs bind to plasma proteins through their negative charge and thus accumulate mainly in the kidney and liver, with low levels partitioning into other tissues such as spleen and muscle (Sazani et al., 2002; Heemskerk et al., 2010). However, 80% of SSO uptake in the liver is mediated by Kupffer reticuloendothelial cells and only 20% reaches the hepatocytes.
Widespread tissue distribution of SSOs after systemic administration may result in undesirable effects if the target pre-mRNA is also expressed in other tissues. For example, the full-length APOB gene is expressed in liver to produce APOB100 protein, whereas in the intestine the RNA-edited isoform APOB48 is expressed to initiate chylomicron synthesis for the transport of dietary fat and fat-soluble vitamins. Interference with chylomicron assembly, by inhibitors of microsomal triglyceride transfer protein, results in fat and vitamin K malabsorption (Cuchel et al., 2013). This undesirable side effect was evident when using small interfering RNA (siRNA) to knock down APOB mRNA and reduce plasma LDL, as chylomicron levels were reduced by 50% (Soutschek et al., 2004), but is not an issue when using SSO to generate APOB87 mRNA, as this can also undergo intestinal RNA editing to allow normal APOB48 expression (Disterer et al., 2013).
Our own data suggest a correlation between target expression levels, the requisite SSO dose, and efficient delivery to the nucleus for effective splicing manipulation. For instance, we were unable to achieve significant exon 27 skipping of hepatic human APOB in transgenic mice with systemic delivery via tail vein injection at doses up to 25 mg/kg (Disterer et al., 2013). Hydrodynamic injections (25 mg/kg) of fluorescence-labeled SSO increased hepatic delivery slightly, but led to only marginal increases in skipping efficiency (<1.6%). In this mouse model, expression of the human APOB transgene is about five times higher than that of murine Apob, which itself is already expressed at a high level (19710.48 units according to BioGPS; Wu et al., 2012). Significant skipping efficiency was achieved only with the SSO complexed to a liver-directed delivery reagent, Invivofectamine 2.0 (IVF2.0; Life Technologies) (Disterer et al., 2013), which significantly increased delivery of fluorescent SSO. This effect was independent of the SSO sequence (Disterer et al., unpublished data). These findings are comparable to those of Yilmaz-Elis and colleagues (2013), who reported 90% exon skipping of interleukin-1 receptor accessory protein (Il1rap) after a single tail vein injection of a 21-mer SSO complexed to IVF2.0 (10 mg/kg per mouse), whereas four daily injections of SSO alone at 100 mg/kg each were needed to induce ∼50% exon skipping. The relative BioGPS expression value is 8.63 for Il1rap in murine liver, indicating that high doses of SSOs are still required even for genes expressed at relatively low levels. The systemic delivery of SSO leading to SMN2 exon 7 inclusion in a mouse model of SMA resulted in up to 70% efficiency in liver (Hua et al., 2011). However, two subcutaneous injections of up to 160 mg/kg, a total dose of 320 mg/kg, were necessary to achieve this efficiency. Like Il1rap, Smn is expressed at low levels in mouse liver (BioGPS expression value, 21.74).
The emerging evidence suggests that when targeting low-expressed RNAs, systemic delivery of high doses is sufficient to induce effective splice modulation, at least in an easily accessible tissue such as the liver. However, splice modulation of highly expressed pre-RNAs or in a difficult-to-access tissue would require prohibitively expensive and potentially toxic SSO doses to achieve high efficiency when using systemic delivery. Effective and selective SSO delivery to the nucleus of the tissue of interest is of paramount importance to increase potency and to reduce possible adverse effects. This may well necessitate the use of delivery reagents in various forms, including conjugates with cell-targeting ligands, cell-penetrating peptide conjugates, and nanoparticle carrier formulations. These have been developed mostly for siRNA applications, but can be equally useful for SSO delivery (Juliano et al., 2008). It is clear that future SSO therapies will need to use both specific and tailored delivery technologies to obtain the key benefits of increased potency and reduced toxicity.
Conclusions
Up to 50% of human genetic diseases are caused by mutations that affect splicing, and therefore therapeutic modulation of the process, using synthetic SSOs, is emerging as a promising alternative to conventional gene addition technologies. Like gene therapy and antibody- or antisense-based treatments before it, there is a certain boom and bust cycle to the development of splicing therapeutics. Today, perhaps, the field is in a low period with the failure of the drisapersen phase 3 trial and the 2013 FDA rejection of accelerated approval for eteplirsen. However, new guidance from the FDA regarding the possibility of accelerated approval for eteplirsen and the success of ISIS-SMNRx in early trials gives hope for another upward swing and conceivably presages a sustained period of notable progress on several fronts. Solving effective targeted delivery is currently the most important challenge facing the field of splicing modulation and, we suspect, most likely underlies the drisapersen failure.
Footnotes
Acknowledgments
P.D. and B.K. are supported by a British Heart Foundation Project Grant (PG/12/20/29469), A.K. by an ERASMUS scholarship, and Y.E.B. by a BSGCT/Nuffield Foundation Undergraduate Research Bursary.
Author Disclosure Statement
B.K., J.S.O., and P.D. are coinventors of U.K. Patent GB1117880.3 with J. Paul Simons.