
Abstract
Null mutations in the UGT1A1 gene result in Crigler–Najjar syndrome type I (CNSI), characterized by severe hyperbilirubinemia and constant risk of developing neurological damage. Phototherapy treatment lowers plasma bilirubin levels, but its efficacy is limited and liver transplantation is required. To find alternative therapies, we applied AAV liver-specific gene therapy to a lethal mouse model of CNSI. We demonstrated that a single neonatal hUGT1A1 gene transfer was successful and the therapeutic effect lasted up to 17 months postinjection. The therapeutic effect was mediated by the presence of transcriptionally active double-stranded episomes. We also compared the efficacy of two different gene therapy approaches: liver versus skeletal muscle transgene expression. We observed that 5–8% of normal liver expression and activity levels were sufficient to significantly reduce bilirubin levels and maintain lifelong low plasma bilirubin concentration (3.1±1.5 mg/dl). In contrast, skeletal muscle was not able to efficiently lower bilirubin (6.4±2.0 mg/dl), despite 20–30% of hUgt1a1 expression levels, compared with normal liver. We propose that this remarkable difference in gene therapy efficacy could be related to the absence of the Mrp2 and Mrp3 transporters of conjugated bilirubin in muscle. Taken together, our data support the concept that liver is the best organ for efficient and long-term CNSI gene therapy, and suggest that the use of extra-hepatic tissues should be coupled to the presence of bilirubin transporters.
Introduction
T
In CNSI, enzyme replacement therapy represents a possible alternative strategy, and gene therapy offers a tool to achieve this therapeutic possibility. Over the past 20 years, a number of ex vivo and in vivo gene therapy protocols have demonstrated efficacy when applied to the Gunn rat [see (Miranda and Bosma, 2009) for a detailed review], but none of them has yet arrived to the clinic, suggesting that a more throughout understanding of the molecular correlates of the CNSI pathology is needed.
Liver has unique features that render it an attractive organ for gene therapy: (1) it is the largest organ in the body; (2) it has a dual circulation systems and it is highly vascularized, increasing the possibility to transduce the organ with higher efficiency; (3) it has a dense net of ducts that could potentially clear the production of toxic products, the bile canaliculi. Moreover, as the skeletal muscle, liver has a very low cell turnover (Sell, 2003) but can regenerate following various types of insults, and (4) above all, the liver is the main tissue of UGT1A1 expression (Tukey and Strassburg, 2000; Buckley and Klaassen, 2007).
Besides liver, skeletal muscle was proposed as a surrogate organ to express the therapeutic protein both for CNSI and other liver genetic defects such as hemophilia (Miranda and Bosma, 2009; High, 2011; Chuah et al., 2012; Pastore et al., 2012). Skeletal muscle is an attractive alternative tissue because of several advantages it might offer. First, the muscle is a very abundant tissue, accounting for approximately 40% of the total body mass. Second, it is highly vascularized and easily accessible by intramuscular injection. Third, viral vectors are currently available that transduce skeletal muscle fibers at very high efficiency. Fourth and final, AAV-mediated gene transfer to human skeletal muscle persists and is transcriptionally active for a period of at least 10 years (Buchlis et al., 2012), and probably longer periods. Because of these favorable characteristics, it is not surprising that several of the clinical trials for liver metabolic diseases performed to date have entailed intramuscular gene delivery (Mingozzi and High, 2011). In particular, various gene therapy approaches have been performed in the animal models of CNSI by targeting skeletal muscle, including injections of naked plasmid DNA and the use of AAV vectors (Danko et al., 2004; Jia and Danko, 2005; Bortolussi et al., 2012; Pastore et al., 2012). In the former case, a rapid drop of the therapeutic effect was observed, which was associated with the loss of UGT1A1 protein expression after a couple of weeks postinjection of the plasmid DNA (Danko et al., 2004; Jia and Danko, 2005). In the latter case, involving AAV vectors, efficacy was more evident (approximately 50% reduction compared with untreated controls) and long lasting (Bortolussi et al., 2012; Pastore et al., 2012). We have recently shown that neonatal gene transfer of AAV9-CMV-hUGT1A1 to the skeletal muscle can rescue bilirubin-induced lethality in a lethal murine model of CNSI we developed (Bortolussi et al., 2012).
In both AAV approaches targeting skeletal muscle in rats and mice, UGT1A1 expression levels in this tissue were comparable to those of wild type (WT) liver (Bortolussi et al., 2012; Pastore et al., 2012). However, they were accompanied by a less than ideal reduction of total bilirubin (TB) values, suggesting that other factors are necessary to attain therapeutic success.
Based on these considerations, in this study we tested the therapeutic efficacy of liver-specific transduction in the mouse model of CNSI. Delivery of the UGT1A1 cDNA was achieved with an AAV serotype 8 (AAV8) vector in which transgene expression was controlled by a liver-specific promoter, carrying the enhancer element of the ApoE gene and the minimal promoter region of α-1-antitrypsin (AAT) (Mingozzi et al., 2003).
We demonstrate that neonatal gene transfer was successful and the therapeutic effect lasted up to 17 months postinjection. Moreover, we compared efficacy of this liver-specific gene therapy with the results obtained after skeletal muscle transduction. We showed that, despite that the transduced liver expressed 26 times less hUGT1A1 than the transduced muscle, the levels of total plasma TB were significantly lower in the former treatment. Our results revealed that less than 5–8% of normal UGT1A1 liver expression was sufficient to maintain life-long low TB levels, while much reduced efficiency was obtained with about 20–30% of Ugt1a1 expression in the skeletal muscle.
This striking difference in the therapeutic effect between liver and skeletal muscle apparently resides in the liver-specific expression of Mrp2 and/or Mrp3 transporters (also known as Abcc2 and Abcc3, respectively), which extrude conjugated bilirubin from the hepatocyte to the bile and blood, respectively (Kamisako et al., 2000). Taken together, our data strongly support the concept that the liver is the most suitable target organ for efficient CNSI gene therapy and suggest that the potential use of extrahepatic tissues should be directly related to the presence of bilirubin transporters.
Materials and Methods
Animals
Ugt1 mutant mice have been described previously (Bortolussi et al., 2012). WT littermates were used as a control. Mice were housed and handled according to institutional guidelines, and experimental procedures approved by International Centre for Genetic Engineering and Biotechnology (ICGEB) board. Animals used in this study were at least 99.8% C57Bl/6 genetic background, obtained after more than 9 backcrosses with C57Bl/6 mice. Mice were kept in a temperature-controlled environment with a 12/12 hr light–dark cycle. They received a standard chow diet and water ad libitum.
Production, purification, and characterization of the rAAV vectors
The AAV-hUGT1A1 vector used in this study is based on AAV type 2 backbone in which the inserted human UGT1A1 cDNA is under the transcriptional control of either cytomegalovirus (CMV) immediate early promoter as previously described (Arsic et al., 2004) or the enhancer element of the ApoE gene and the minimal promoter region of α-1-antitrypsin (AAT) as previously described (Mingozzi et al., 2003). Infectious vectors were prepared by the AAV Vector Unit at ICGEB Trieste (
Gene transfer procedure: PT treatment
For the AAV gene transfer procedure, pups at postnatal day 4 (P4) were intraperitoneally injected with a single dose of AAV8-AAT-hUGT1A1 or AAV9-CMV-hUGT1A1 (3.2×1011 viral particles, 1.3×1011 vpg/g). Newborns were exposed to blue fluorescent light (20 μW/cm2/nm; Philips TL 20W/52 lamps; Philips) for 12 hr/day (synchronized with the light period of the light/dark cycle) up to 10 days after birth and then maintained under normal light conditions. Intensity of the blue lamps was monitored monthly with an Olympic Mark II Bili-Meter (Olympic Medical).
Bilirubin measurements
Blood samples were collected at the indicated time points postinjection in mutant and WT littermates by decapitation or facial vein exsanguination or cardiac puncture. Bilirubin determination in plasma was performed as previously described (Bortolussi et al., 2012). Gall bladder was dissected upon sacrifice of the mice and bile fluid was collected by centrifugation.
Concentrations of biliary UCB and bilirubin-glucuronic acid conjugates in 2-month-old mice were determined by HPLC as previously described by Zelenka et al. (2008) and by Spivak and Carey (1985), respectively.
Quantitative determination of UCB in tissues of 2-month-old mice was performed as previously described by Zelenka et al. (2008).
Albumin determination in plasma
Albumin determination in plasma samples was performed with the Bromocresol Green method as previously described by Rodkey (1965), adapting the method to use minimal volumes (2 μl of plasma). In each test a standard curve was performed by dilution of a stock solution (10 mg/ml) of mouse albumin (Sigma) in water. Absorbance values at 630 nm were obtained by using a multiplate reader (Perkin Elmer Envision Plate Reader)
Liver histology
Liver biopsies from AAV-treated animals and their WT littermates were extracted and fixed with 4% paraformaldehyde in PBS overnight at 4°C. After cryoprotection in 20% sucrose in PBS and 0.02% sodium azide, specimens were frozen in optimal cutting temperature compound (BioOptica) and 14 μm slices were obtained in a cryostat. Masson trichrome staining was performed as previously described (Bortolussi et al., 2012).
For immunofluorescence, liver specimens (14 μm) were stained with Hoechst (10 μg/ml) and mounted with Mowiol 4–88 (Sigma). Images were acquired on a Nikon Eclipse E-800 epi-fluorescent microscope with a charge-coupled device camera (DMX 1200F; Nikon). Digital images were collected using ACT-1 (Nikon) software.
Rotarod analysis
The coordination and balance ability on a rotating cylinder was assessed 1 month postinjection with an accelerating apparatus as previously described (Bortolussi et al., 2012).
Viral genomes determination
Total DNA from liver and skeletal muscle was extracted using the Wizard SV Genomic DNA Purification System (Promega) according to manufacturer's instructions. The vector genome copy number was quantified by real-time PCR using specific primers for AAT or CMV promoter. Real-time PCR was performed using the following primers—for AAT: pGG2-906 DIR and pGG2-1051 REV; for CMV: pZac DIR and pZac REV (see Supplementary Table S1; Supplementary Data are available online at
Southern blot analysis
Low-molecular-weight Hirt DNA was extracted from liver samples taken at different time points after AAV transduction as previously described (Davidoff et al., 2003), with a minor modification of the method: samples were pulverized in liquid nitrogen instead of using a Dounce homogenizer. Undigested or SpeI-digested Hirt DNA (15 μg) was run in a 0.7% agarose gel, blotted onto a nylon membrane (Z-Probe GT Genomic membrane; Bio-Rad), and hybridized with a α-P32-labeled 1622 bp fragment containing the hUgt1a1 cDNA expression cassette. After being washed, the membrane was exposed using a Cyclone phosphor-screen (Packard Bioscience), and the radioactive signal was detected using Cyclone Storage phosphor-imager (Packard Bioscience).
High-molecular-weight DNA was extracted using the Wizard SV Genomic DNA Purification System (Promega) according to manufacturer's instructions. About 5 μg of undigested or digested (with EcoRV or XhoI/NotI) total genomic DNA was run in a 0.7% agarose gel and subjected to Southern blot analysis as described above.
Preparation of total RNA, RT-PCR, and real-time PCR analysis
To reduce variability that could be generated by uneven distribution of the AAV vectors in the analyzed tissue, the complete organ was reduced to powder with a mortar and liquid nitrogen, and the sample was then aliquoted to analyze proteins and mRNA expression and viral genome copies. Total RNA from mouse liver and skeletal muscle was prepared using EuroGOLD Trifast (Euroclone) according to manufacturer's instructions. About 1 μg of total RNA was reverse-transcribed using M-MLV (Invitrogen) and oligodT primer according to manufacturer's instructions. Total cDNA (1 μl) was used to perform either RT-PCR or qPCR using specific primers listed in Supplementary Table S1. qPCR was performed using iQ SYBR Green Supermix (Bio-Rad) and a C1000 Thermal Cycler CFX96 Real-Time System (Bio-Rad).
For qRT-PCR gene expression analysis in different tissues, the study of a reference gene with stable mRNA transcription levels is required. To this purpose, we tested six housekeeping genes directly comparing their CT values between liver and skeletal muscle: glyceraldehyde 3-phosphate dehydrogenase (Gapdh), β-actin (β-act), TATA-box binding protein (TBP), succinate dehydrogenase subunit A (Sdha), α-tubulin (Tub), and hypoxanthine-guanine phosphoribosyltransferase (HPRT) (data not shown). Among the analyzed housekeeping genes, α-tubulin was expressed at analogous CT values between liver and skeletal muscle and was therefore used to compare hUGT1A1 expression between both tissues. Expression of the gene of interest was normalized to a house-keeping gene (Gapdh or Tub). Real-time PCR data were analyzed using the ΔΔCt method.
Preparation of total protein extracts and Western blot analysis
Liver and skeletal muscle tissues were homogenized in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS, 50 mM Tris HCl, pH8, 2× protease inhibitors) and analyzed by Western blot analysis as described previously (Bortolussi et al., 2012). Primary antibodies used were as follows: anti-human UGT1 rabbit polyclonal antibody, anti-Mrp2 and anti-Mrp3 (Santa Cruz Biotechnology), and anti-β-tubulin mAb E7 (Developmental Studies Hybridoma Bank).
UGT1a1 activity determination
Liver microsomes (6-, 11-, 18-, 30-, and 60-day-old mice) of each genotype were prepared as described previously (Bortolussi et al., 2012). Protein concentration was determined by the bicinchoninic acid assay (Smith et al., 1985).
Glucuronidation assay was performed as described previously (Nguyen et al., 2008) with minor modifications. Briefly, assays were performed after 1 hr of enzymatic reaction using the following conditions: 10 mM MgCl2×6H2O, 50 mM Tris-HCl (pH 7.7 at 37°C), 10 μg/ml phosphatidycholine, 15 μM bilirubin (bilirubin was previously dissolved in DMSO at a concentration of 0.33 μg/μl and diluted 1:10 in DMSO), 1 mM uridine diphosphate-glucuronic acid, and 1 μg/μl microsomal proteins (previously incubated for 1 hr at 4°C with digitonin at a concentration of 0.35 mg/ml of microsomes) (Gordon et al., 1984) in a total volume of 50 μl. Reactions were stopped with 50 μl of methanol with 0.02% of butylated hydroxytoluene. Samples were centrifuged at 10,000 g for 10 min at 4°C, and supernatants were collected for HPLC-MS analysis (LC-MS) as described previously (Nguyen et al., 2008) and adapted to our instrumentation.
Supernatant was transferred into a conical vial for injection into the LC-MS system. The HPLC used was a Surveyor Thermo Finnigan system with pump autosampler and diode array detector (Thermo Finnigan). Bilirubin and its glucurono-conjugated species were injected and separated on a Kinetex C18 column (150×4.6 mm; 5 μm particle size; Phenomenex) with a cartdrige with the same stationary phase (Security Guard ULTRA; Phenomenex). The mobile phase A was 1 mM ammonium formate in water, and the mobile phase B was 1 mM ammonium formate in methanol. Separation was achieved using a linear gradient of 70% B to 95% B in 3.75 min at a flow rate of 0.9 ml/min. After 0.75 min, the column was re-equilibrated to initial conditions over 4.5 min, stopping the runs at 15 min. The absorbance of the eluted pigments was monitored at 455 nm with 195 nm as a reference wavelength.
Mass spectrometry characterization and detection of bilirubin and the mono- bilirubin glucuronide conjugates (BMG) formed were performed using an LCQ Deca XP Plus model (Thermo Finnigan), utilizing a standard electrospray ionization source operated in positive mode and with an ion trap detector.
BMG and bilirubin peaks integration was performed with XCalibur Thermo Finnigan software version 1.4, and the amount of BMG produced by each reaction was calculated with the following equation: AreaBMG/(AreaBMG+Areabilirubin).
Statistics
Results are expressed as mean±SD. The Prism package (GraphPad Software) was used to analyze the data. Values of p<0.05 were considered statistically significant.
Results
Liver-directed UGT1A1 gene therapy efficiently rescues neonatal lethality
We previously demonstrated that neonatal transfer of hUGT1A1 cDNA under the control of the CMV promoter was active in skeletal muscle but not in liver (Bortolussi et al., 2012). Thus, to promote liver-specific expression of the therapeutic gene, we cloned the hUGT1A1 cDNA under the control of the enhancer element of the ApoE gene and the minimal promoter region of the AAT gene (Mingozzi et al., 2003). In addition, to achieve a higher efficiency in liver-specific transduction, the viral genomes were packaged in AAV8 (Davidoff et al., 2005; Nakai et al., 2005).
Because of the early lethality of mutant mice, the gene therapy approach was combined with PT treatment (12 hr/day, 20 μW/cm2/nm) since birth up to P10 after birth, as already described (Bortolussi et al., 2012). Despite PT treatment, mutant mice appeared visibly jaundiced compared with WT littermates at postnatal day 4 (P4) (Fig. 1A). At that age, mutant mice were treated with a single intraperitoneal (IP) injection of AAV8-AAT-hUGT1A1 (∼3.2×1011 vpg/mouse). About 48 hr after vector administration (P6), they were indistinguishable from their WT littermates, suggesting that the viral genomes reached the liver and efficiently expressed the hUGT1A1 protein at therapeutic levels. At that age, plasma bilirubin determination revealed that mutant mice treated with AAT-hUGT1A1 had bilirubin levels 8.5 times lower than mutant mice treated only with PT (0.5±0.2 mg/dl AAV+PT-treated vs. 4.5±0.2 mg/dl PT-treated at P6, p≤0.001, ANOVA test; Fig. 1B) and that those levels were not statistically different from WT values (0.2±0.1 mg/dl WT).
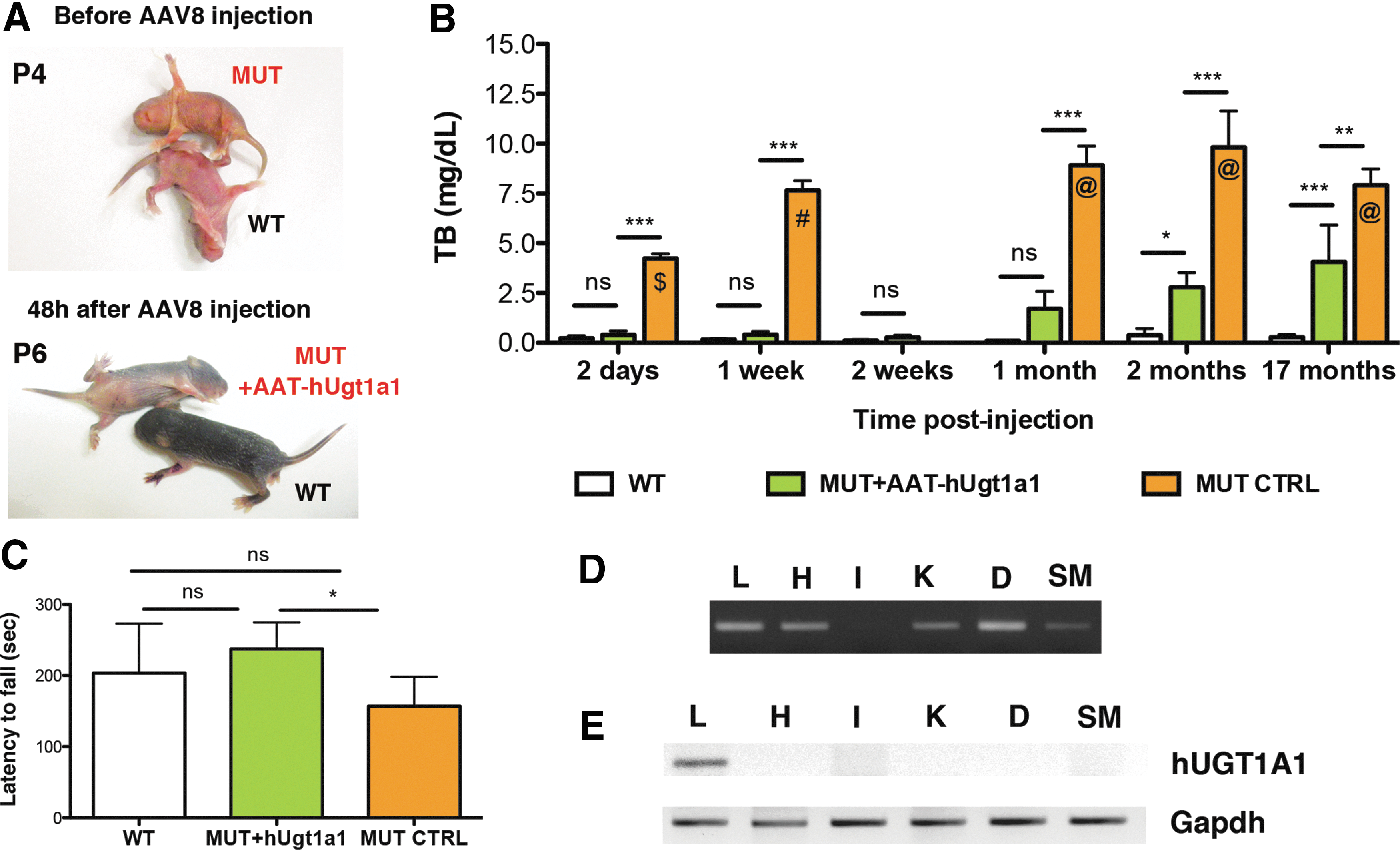
Gene therapy rescues neonatal lethality of Ugt1 mutant mice.
Pups were maintained under PT treatment up to postnatal day 10 and then kept under normal light/dark conditions. All gene therapy-treated mutant mice survived and reached adulthood without any coordination and balance dysfunction, as assessed by the rotarod test, suggesting no obvious neurological damage (Fig. 1C).
Because of the early lethality of the mutant mice, we lacked the adult negative control (adult mutant mice without AAV) necessary to estimate the real drop in bilirubin levels consequent to the gene therapy treatment. We have previously shown that PT treatment alone extends the lifespan of mutant mice up to day 20 after birth (Bortolussi et al., 2012). Since the decreased efficacy of PT could be caused by fur growth, we shaved mutant mice every third day aiming to improve PT efficacy. Shaved mutant mice were kept up to 20 days after birth under PT treatment and then maintained in normal light conditions. All shaved mutant mice survived and reached adulthood (data not shown), and were used as uninjected controls. This result confirmed that the most critical period is the first 20 days of life, and that shaving of the coat allowed the blue light to reach the skin capillaries, reducing plasma bilirubin to life-compatible levels. Determination of plasma bilirubin levels in shaved mutant controls was performed at P30, 10 days after discontinuation of PT, when TB values were considered stable (Fig. 1B).
Plasma samples from WT, AAT-hUGT1A1-treated, and uninjected control mutant mice were collected at different time points (2 days, 1 and 2 weeks, and 1, 2, and 17 months postinjection) and TB levels determined. Bilirubin levels in AAT-hUGT1A1-treated mice were similar to those of WT littermates during the first month after injection (Fig. 1B). Seventeen months postinjection, AAT-hUGT1A1-treated mutant mice still showed 50% less total plasma bilirubin than uninjected control mutant mice (Fig. 1B; p<0.01). These levels were well below the risk of neurological damage. Plasma albumin concentration was comparable among all groups for the time points tested (Supplementary Fig. S1A). Seventeen months postinjection, mice were sacrificed and liver tissue samples were subjected to histological analysis. Masson's trichromic staining of the liver sections showed normal histology without any fibrosis-rich area in AAT-hUGT1A1-treated mice or in uninjected control mutant mice (Supplementary Fig. S1B).
We next determined the genome viral distribution by PCR. Viral genome copies were detected in liver, heart, kidney, diaphragm, and skeletal muscle except intestine (Fig. 1D). RT-PCR analysis of the same tissues confirmed that the expression of the therapeutic gene was restricted to liver (Fig. 1E).
Persistence of transgene expression and hUgt1a1 conjugation capacity following neonatal gene transfer
We next investigated the persistence of hUGT1A1 transgene after neonatal gene transfer. Livers from AAT-hUGT1A1-treated mice were collected at different time points (2 days, 1 and 2 weeks, and 1 and 2 months postinjection) and vector copies per diploid genome were determined by real-time PCR.
We observed that the value of vgp was very high 2 days after viral delivery, but it rapidly declined within 1 week (Fig. 2A; p<0.001), concomitantly with the rapid growth of both the body and the liver (Supplementary Fig. S2). Following this initial decline, vgp levels remained stable over time, up to 17 months after injection (Fig. 2A).
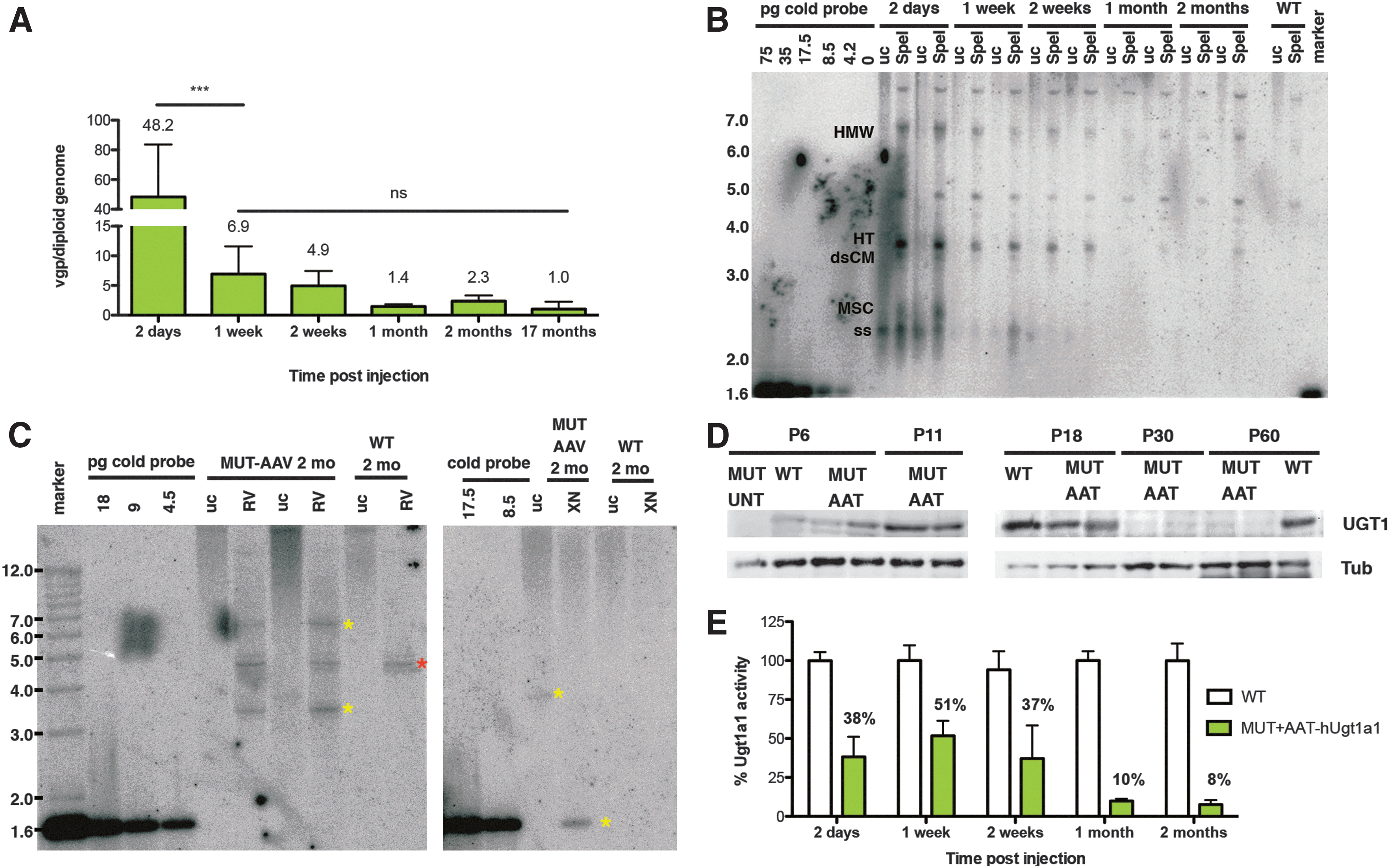
Persistence of hUgt1a1 transgene expression in liver following neonatal gene transfer.
To better understand the mechanism responsible for the long-term therapeutic efficacy of the gene transfer, we purified low-molecular-weight Hirt DNA from the liver of these mice and performed Southern blot analysis of undigested and SpeI-digested DNA (Fig. 2B). At early stages (2 days postinjection), we observed a high proportion of viral genomes in single-stranded (ss) conformation and monomeric supercoiled circular form. To determine the presence of transcriptionally active double-stranded (ds) viral genomes, we digested the Hirt DNA preparation with the SpeI restriction enzyme, which cuts only once in the vector genome and results in a single 3.3 kb band of the monomer. By 2 weeks postinjection, the ss forms were converted into ds genomes, which appeared to be also present in concatameric conformation (Fig. 2B; HT and HMW, respectively).
To further confirm that the long-term therapeutic effect was mediated by episomal ds vectors, we prepared high-molecular-weight genomic DNA from 2-month-old AAT-hUGT1A1-treated mutant mice and WT littermates, digested it with EcoRV (single cutter) or a combination of XhoI and NotI to release the hUgt1a1 cDNA from the expression cassette, and performed a Southern blot analysis. Similarly to the results observed with Hirt DNA using an enzyme that cuts only once in the viral genome, we obtained a band of 3.3 kb, corresponding to the linearized viral genome. When the DNA was cut with enzymes that release the hUGT1a1 cDNA, we observed a single band of 1.6 kb (Fig. 2C). The intensity of the 1.6 kb XhoI–NotI band was similar to that obtained after SpeI digestion, suggesting that most of the viral genome was episomal.
To determine the changes in UGT1a1 expression and enzyme activity in the early phases of rapid liver growth of the AAV-transduced pups, we performed Western blot analysis of liver proteins and determined Ugt1a1 bilirubin-glucuronidation activity in liver microsomes. Ugt1 protein levels were very high up to P18, and suffered an important reduction at P30 (Fig. 2D). Ugt1a1 bilirubin-glucuronidation activity paralleled Western blot results. In fact, we observed high bilirubin-glucuronidation activity during the first 2 weeks after transduction, reaching 50% of the activity of WT littermates at 1 week posttransduction. We observed a reduction in enzyme activity 1 month after injection, which remained stable at 2 months (Fig. 2E).
These results support the hypothesis that the long-term efficiency of the gene therapy treatment was mediated by transcriptionally active ds episomal genomes.
Liver hUGT1A1 is more efficient in lowering plasma bilirubin than skeletal muscle hUGT1A1
hUGT1A1 mRNA expression levels were determined 17 months post-AAT-hUGT1A1 injection by semi-quantitative RT-PCR. The reaction was performed using specific primers able to amplify the human UGT1A1 mRNA but not the endogenous mouse version. As expected, WT and mutant PT-treated liver samples did not express the human UGT1A1 mRNA (Fig. 3A), while AAT-hUGT1A1-treated mutant mice expressed detectable levels of hUGT1A1 mRNA. As a positive control, we used skeletal muscle of a 5-month-old CMV-hUGT1A1-treated mutant mouse (Bortolussi et al., 2012). Interestingly, skeletal muscle of the CMV-treated mutant mice expressed much higher hUGT1A1 mRNA levels than the liver of AAT-hUGT1A1-treated mutant mice. To obtain more accurate data of the observed differences, hUGT1A1 expression levels of AAT-hUGT1A1-treated liver and CMV-hUGT1A1-treated skeletal muscle were compared by real-time RT-PCR in a new group of mutant animals, 2 months after AAV injection. Despite that mutant mice treated with AAT-hUGT1A1 gene therapy showed a stronger reduction in bilirubin levels than CMV-hUGT1A1-injected mice (3.1±2.6 vs. 6.4±2.8 mg/dl, respectively; p=0.02; Fig. 3B), the liver from AAT-hUGT1A1-treated mutant mice expressed 26 times less hUGT1A1 than skeletal muscle of CMV-hUGT1A1-treated mutant mice (26=4.72; Fig. 3B) and contained more viral DNA (2.3±0.9 vs. 0.8±0.6 viral genomes/diploid genome, liver vs. skeletal muscle, respectively; p≤0.01).
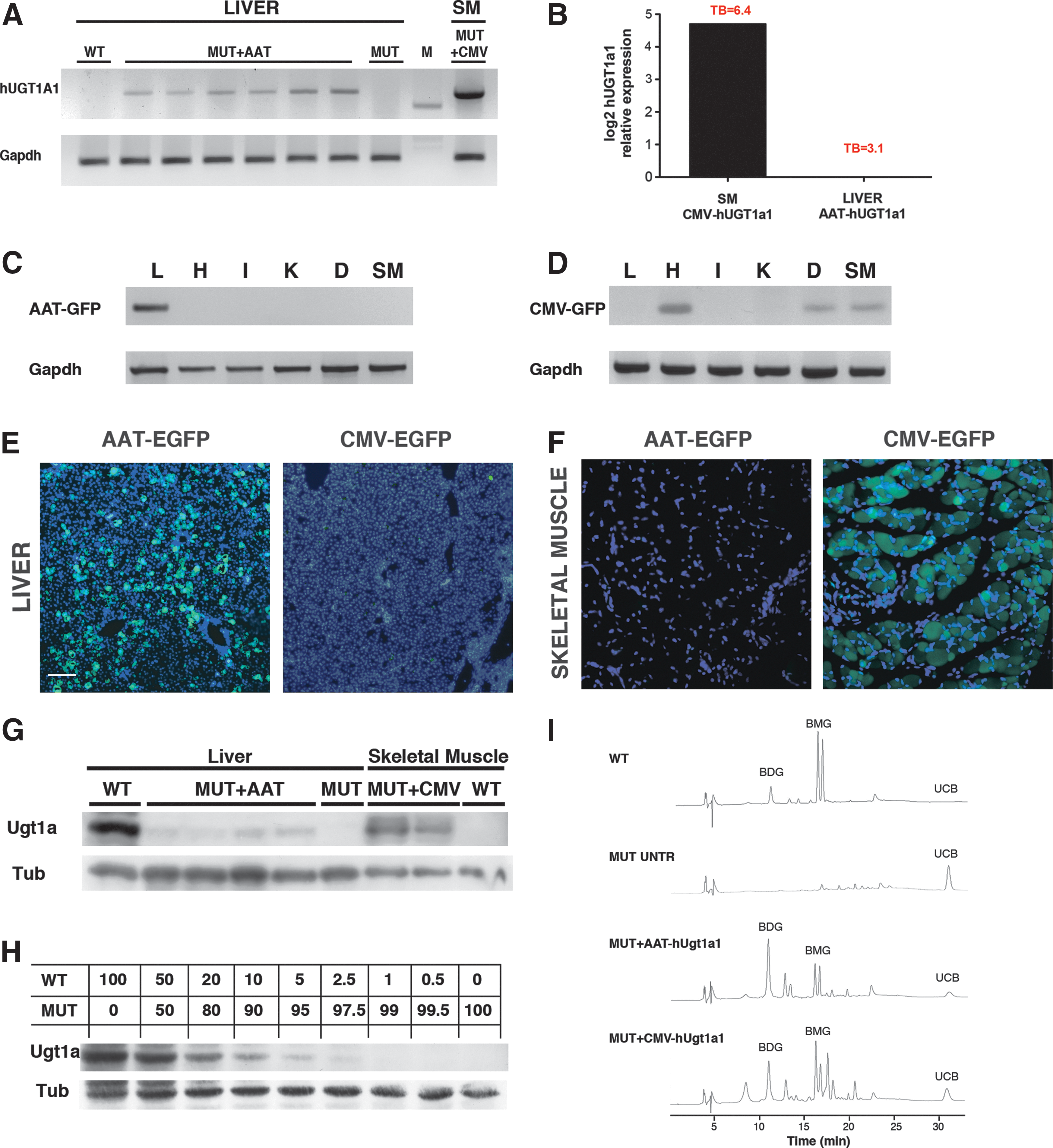
Liver and skeletal muscle hUGT1A1 expression and bilirubin pigments in bile samples.
The expression patterns of the two promoters were confirmed using AAV-EGFP-reporter vectors (Fig. 3C–F). WT mice were injected at P4 with a single dose of either AAV8-AAT-EGFP or AAV9-CMV-EGFP and sacrificed at P13. We observed a strong EGFP liver expression (but no expression in skeletal muscle, as expected) using the AAT promoter (Fig. 3C and E), while liver expression was almost undetectable using the CMV promoter at both mRNA and protein levels (Fig. 3D and E), corroborating our previous findings that showed transcriptional silencing of the CMV promoter in liver over time (Bortolussi et al., 2012). Moreover, we confirmed that in our system CMV-EGFP expression was restricted to muscles such as heart, diaphragm, and skeletal muscle (Fig. 3D and F).
In line with the qRT-PCR results, Western blot analysis using a polyclonal antibody recognizing both mouse and human UGT1 proteins confirmed that the liver of the AAT-hUGT1A1-treated mutant mice expressed lower levels of hUGT1A1 compared with the same amount of total protein extract from skeletal muscle of the CMV-treated animals (Fig. 3G).
To roughly estimate the levels of UGT1 expression, we prepared a calibration curve of the Ugt1 protein by mixing liver protein extracts from WT and mutant mice at different proportions (Fig. 3H). Thus, by interpolating the signal obtained in the AAT-hUGT1A1-treated mutant mice with that of the calibration curve, we estimated that about 5% of the WT Ugt1a1 levels were present in the livers of AAT-hUGT1A1-treated mutant mice and that this amount of enzyme was sufficient to maintain bilirubin levels below the threshold of neurotoxicity risk. On the contrary, interpolating the signal obtained by CMV-hUGT1A1-treated mutant mice, we estimated that similar amounts of total protein extract of skeletal muscle expressed about 20–30% of the WT Ugt1a1 levels.
Reduced expression of bilirubin transporters in the skeletal muscle correlates with the moderated efficiency of skeletal muscle-directed gene therapy
These findings prompted us to investigate more in detail the reasons why skeletal muscle, which apparently had a very strong hUGT1A1 expression, was not as efficient as the liver in lowering plasma bilirubin levels.
In WT animals, bile samples collected at 2 months postinjection showed two prominent peaks corresponding to isomers of bilirubin monoglucuronoside (BMG, C8, and C12 glucuronides), one peak corresponding to bilirubin diglucuronoside (BDG), and almost undetectable levels of UCB (Fig. 3I). On the contrary, bile from mutant, untreated animals (PT-Shave treated up to 20 days, analyzed 40 days after the end of PT treatment) contained only UCB. In both AAV-AAT-hUGT1A1- and AAV-CMV-hUGT1A1-treated mutant animals, we observed a clear increase in conjugated bilirubin pigments (BDG and BMG) in the bile samples compared with untreated mutant controls (Fig. 3I).
One of the key factors in bilirubin conjugation is the availability of UDP-glucuronate. Two enzymes drive the production of the active form of glucuronic acid: UDP-glucose pyrophosphorylase (Ugp) and UDP-glucose dehydrogenase (Ugdh). We investigated, by semiquantitative RT-PCR analysis, whether these two enzymes were differentially expressed in the liver and skeletal muscle. We observed that both tissues expressed the two enzymes at high levels (Fig. 4).
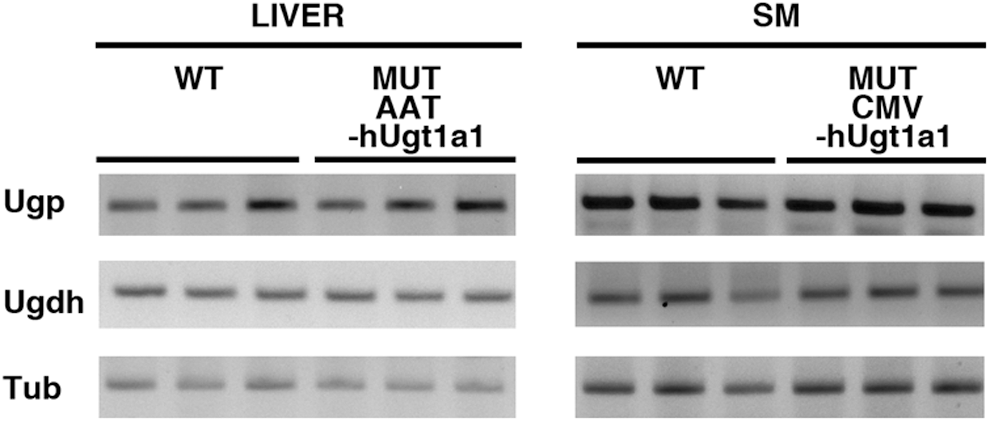
UGT1A1 glucuronosidation enzymes. RT-PCR of total liver (left panel) and skeletal muscle (right panel) mRNA from WT, AAT-hUGT1A1 (MUT+AAT), and CMV-hUGT1A1 (MUT+CMV) mutant mice at 2 months of age. Expression analysis of mouse UDP-glucose pyrophosphorylase (Ugp) and UDP-glucose dehydrogenase (Ugdh). Mouse α-tubulin (Tub) was used as endogenous control.
We then evaluated bilirubin accumulation in liver and skeletal muscle of the transduced mice by the Zelenka method (Zelenka et al., 2008). We observed that AAT-hUGT1A1-treated mice accumulated much less bilirubin in all tissues analyzed (liver, skeletal muscle, cerebellum, and forebrain) than aged-matched untreated mutants (p≤0.01, AAT-hUGT1A1-treated mice vs. PT-treated mutant mice; Fig. 5B–E). Surprisingly, CMV-treated mutant mice accumulated the same amounts of bilirubin in tissues as age-matched untreated mutants, indicating a constraint of skeletal muscle-gene therapy to efficiently clear tissue bilirubin (p≤0.01, AAT-hUGT1A1-treated mice vs. CMV-hUGT1A1-treated mice; Fig. 5B–E). Moreover, bilirubin content in forebrain and cerebellum revealed that liver-directed gene therapy was much more effective than skeletal muscle-directed gene therapy in preventing bilirubin accumulation in the central nervous system (p=NS, AAT-hUGT1A1-treated mice vs. WT mice; Fig. 5D and E).
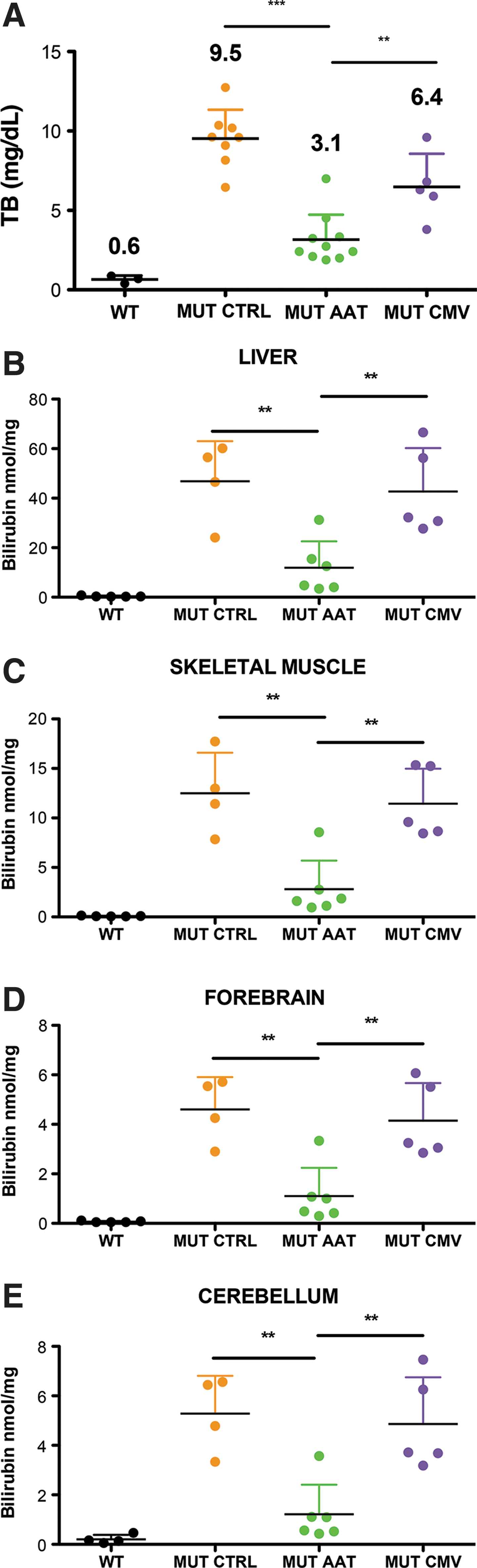
Tissue bilirubin accumulation.
We reasoned that bilirubin, once entered the muscle and conjugated, has to reach the bloodstream to be eliminated in bile. Normally, these steps are performed by active transporters in the liver (Kamisako et al., 2000; Thomas et al., 2008). Thus, we evaluated, by semiquantitative RT-PCR, the expression of different transporters known to export conjugated/unconjugated bilirubin from hepatocytes (Fig. 6A). We observed that skeletal muscle did not express mRNA of Mrp2, Mrp3, Oatp2, and OAtp1b2, but it expressed Mrp1, Mrp4, and Mrp5 that may secrete conjugated bilirubin at lower efficiency rates. Since Mrp2 and Mrp3 are the two most efficient exporters of conjugated bilirubin, we determined their protein levels by Western blot analysis. We confirmed that Mrp2 and Mrp3 were not detectable in skeletal muscle extracts from WT or AAV-CMV-hUGT1A1-treated mutant animals, but were present in liver protein extracts from WT and AAV-AAT-hUGT1A1-treated mutant mice (Fig. 6B).
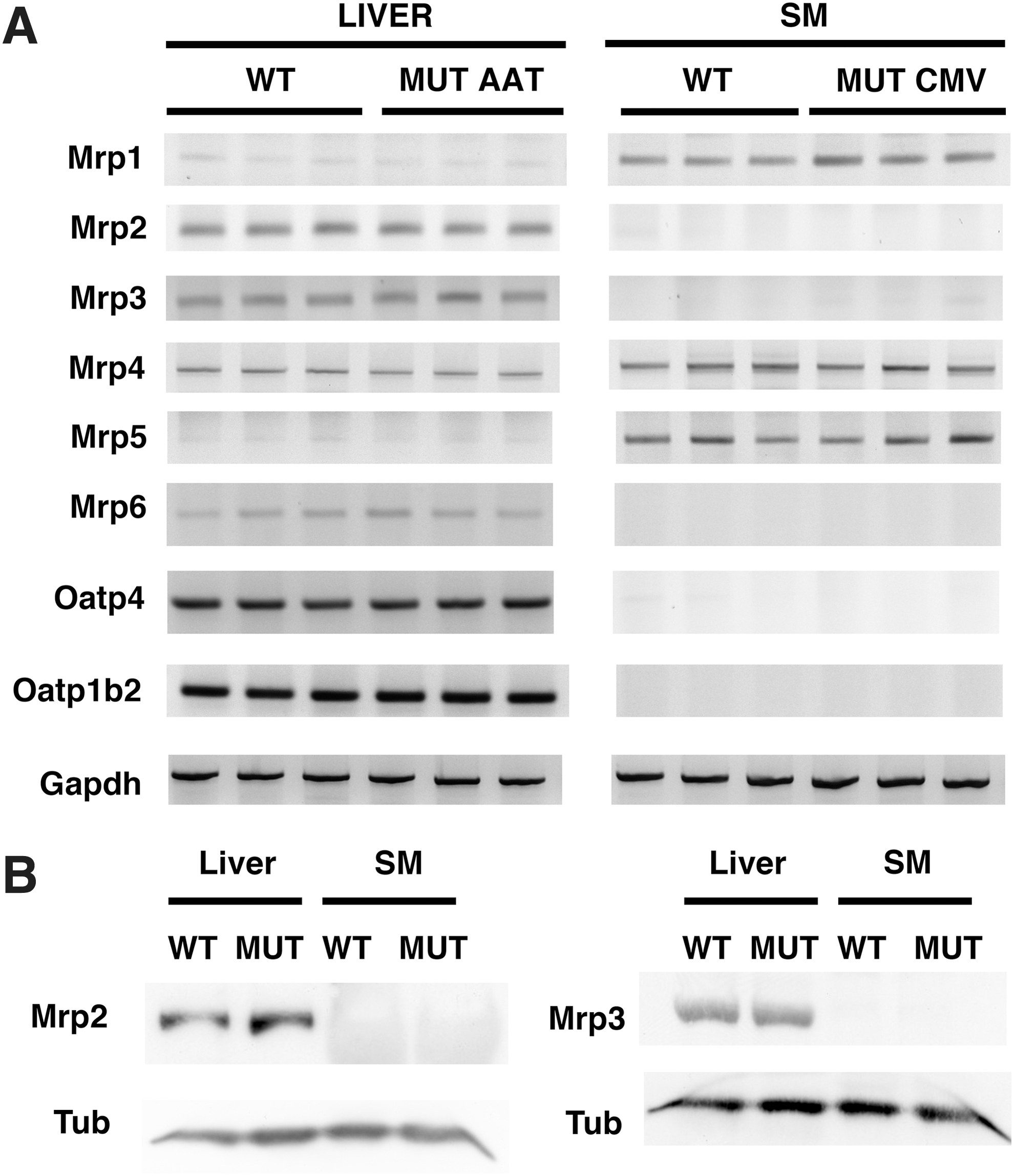
Tissue distribution of bilirubin transporters.
Discussion
Gene therapy is a promising and attractive therapeutic approach for metabolic diseases affecting the liver. Successful approaches of AAV-mediated gene therapy of coagulation factor IX deficiency have recently been performed in hemophilia B patients (Nathwani et al., 2011), paving the way to the treatment of other monogenic liver diseases, such as CNSI.
In the present work, we have shown long-term correction of a mouse model of CNSI (Bortolussi et al., 2012), by means of a single neonatal IP injection of an AAV vector, serotype 8, expressing hUGT1A1 in the liver. AAV8 was selected because it is the most effective one in liver transduction (Nakai et al., 2005). After neonatal AAV transduction, all AAT-hUGT1A1-treated mutant animals survived showing an important and clinically relevant reduction in plasma bilirubin during the first months of treatment (70–80% reduction), which was maintained up to the end of the experimental protocol (50% reduction 17 months postinjection). Histological and functional features of the AAV-treated mice were normal, in contrast to what observed by Seppen et al. (2006), who found that rat Gunn livers treated with liver-specific AAV8-Ugt1a1 vectors presented large nodules resembling fat deposits.
Therapeutic hUGT1a1 glucuronidation activity was already effective in lowering bilirubin as early as 48 hr after viral transduction, reaching ∼38% of WT levels, suggesting a rapid conversion of the viral genome into dsDNA and the consequent activation of transgene transcription. Rapid conversion from ssDNA to dsDNA was also observed in liver (Davidoff et al., 2005; Cunningham et al., 2008) and skeletal muscle (Vincent-Lacaze et al., 1999) transduced with eGFP-, FIX-, and EPO-expressing AAVs.
We observed a rapid loss of hUGT1a1 expression and activity in the liver after neonatal transduction, which was probably because of the degradation of viral genomes during liver growth and cell division, as also reported (Wang et al., 2005; Cunningham et al., 2008). Our data suggest that transgene expression was mediated by transcriptionally active ds episomal genomes, as evidenced by the presence of ds circular DNA genomes in low-molecular-weight Hirt DNA preparations in mutant adult mice. The residual episomal genomes were sufficient to guarantee plasma bilirubin levels below the threshold for the risk of developing brain damage and kernicterus (Ostrow et al., 2004). However, the observed increase in plasma bilirubin levels in the aged animals, associated to a reduction in the viral genome copies in hepatocytes, suggests that further optimization of the therapeutic protocol is still necessary.
Therefore, as UGT1A1 is expressed at high levels in the liver but it is also expressed at lower levels in other organs such as intestine and kidney (Buckley and Klaassen, 2007), we considered the alternative possibility of expressing UGT1A1 in a surrogate tissue such as skeletal muscle, with the aim of improving the efficiency of the therapy.
As mentioned above, targeting the skeletal muscle for gene therapy offers a series of advantages over a liver-directed gene therapy approach (Mingozzi and High, 2011; Buchlis et al., 2012). However, when we treated in parallel mutant mice with CMV-hUGT1A1 and AAT-hUGT1A1 AAV vectors, plasma bilirubin levels were much higher in CMV-hUGT1A1-treated mice than in AAT-hUGT1A1-treated ones, despite of the 4–6-fold higher muscle expression of hUGT1a1. This result is in line with that observed in previous attempts to treat animal models of CNSI by skeletal muscle-directed gene therapy, which have produced partial success, with reduction of plasma bilirubin levels up to 50% of untreated controls, despite moderated to elevated levels of UGT1A1 expression (Danko et al., 2004; Jia and Danko, 2005; Bortolussi et al., 2012; Pastore et al., 2012).
These results suggest that one or more steps in the bilirubin-conjugation pathway may be limiting or missing in skeletal muscle. We detected the presence of mRNA of the two enzymes responsible of the generation of glucuronic acid in skeletal muscle, Ugp and Ugdh, indicating that the missing/limiting step may not be linked to the generation of the UGT1A1 substrate, as previously proposed (Pastore et al., 2012). In line with this conclusion, we found bilirubin mono- and di-glucuronosides in the bile of skeletal muscle-treated mice, as previously observed in muscle of plasmid-treated Gunn rats (Danko et al., 2004), suggesting that the glucuronosylation reaction was also functional in mouse skeletal muscle.
We next focused our attention on bilirubin transporters. We found that the main bilirubin transporters of conjugated bilirubin Mrp2 and Mrp3 (Jedlitschky et al., 1997) were expressed at undetectable levels in skeletal muscle, in addition to the lack of other transporters reported to have a role in bilirubin and bile acid uptake, such as Oatp4 and Oatp1b2 (Wagner et al., 2005; Chiou et al., 2013). On the contrary, skeletal muscle had higher levels of Mrp1, reported to transport conjugated bilirubin out from cells, although at lower rates than Mrp2 (Jedlitschky et al., 1997; Rigato et al., 2004) and Mrp4, the latter also reported to have a role in bile acid export (Rius et al., 2006). These results were consistent with the observation that, despite the presence of high levels of UGT1A1 in skeletal muscle, we observed accumulation of bilirubin in this tissue in the AAV-CMV-hUGT1A1-treated animals, supporting the hypothesis that the limiting step may be the export of conjugated bilirubin from the skeletal muscle fiber. We also observed that tissue bilirubin in brain, cerebellum, and liver of AAV-CMV-hUGT1A1-treated animals reached the levels of untreated animals, and was significantly higher than in the corresponding organs of the animals transduced in the liver, reinforcing the conclusion that muscle-directed gene therapy is less efficient than liver-directed gene therapy for CNSI.
An additional consideration in favor of the superior efficacy of liver-directed gene therapy is that only a fraction of UGT1A1 activity is needed to obtain complete normalization of the missing function, similar to what happens for other enzymes produced by the liver [e.g., FIX (Nathwani and Tuddenham, 1992)]. In hepatocyte-transplanted patients, it was shown that about 5% of normal liver activity was enough to lower plasma bilirubin to safe levels (Fox et al., 1998). Transplantation experiments in the Gunn rat showed that about 12% of liver mass significantly reduced serum bilirubin to normal levels (Asonuma et al., 1992). These results are in agreement with those obtained in our study, which showed that approximately 5–8% of UGT1A1 enzyme in the liver (compared with WT levels, as analyzed by WB and bilirubin-glucuronidation activity) was sufficient to produce a significant drop in plasma bilirubin levels, while much higher amounts were needed after skeletal muscle transduction. The higher efficiency of the liver in conjugating bilirubin is even more evident if we consider that skeletal muscle is the most abundant tissue, reaching about 35–40% of the total fat-free body mass (Heymsfield et al., 1990), while the liver represents only 3–4% of total body weight (Mouse Phenome Database,
To summarize, IP injection of AAV-mediated liver-specific expression of UGT1A1 into mutant mouse pups resulted in life-long reduction of plasma bilirubin and protection from bilirubin-induced brain damage.
Our results indicate that the liver has to be considered as the main target to direct the efforts in the development of efficient gene therapy protocols to cure CNSI.
Footnotes
Acknowledgments
This work was supported by Telethon (GGP10051), by Friuli-Venezia Giulia Regional Grant, and by Beneficentia Stiftung to A.F.M. (ICGEB); by AXA Research Fund to G.B. (ICGEB); by intramural research grant of Fondazione Italiana Fegato to C.B. and C.T.; and RVO VFN64165 from the Czech Ministry of Health to L.V. The authors thank Prof. Fatima Bosch and Dr. Eduard Ayuso, who kindly provided the plasmid carrying the ApoE/AAT-EGFP; M. Dapas and M. Zotti for help with AAV preparation; M. Rossi for technical assistance; M. Sturnega and S. Artico for help with animal care; E. Tongiorgi for microscopes resources; and the integrants of the Mouse Molecular Genetics Group for critical reading of the article.
Author Disclosure Statement
The authors have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.