
Abstract
Adoptive immunotherapy with genetically modified natural killer (NK) cells is a promising approach for cancer treatment. Yet, optimization of highly efficient and clinically applicable gene transfer protocols for NK cells still presents a challenge. In this study, we aimed at identifying conditions under which optimum lentiviral gene transfer to NK cells can be achieved. Our results demonstrate that stimulation of NK cells with interleukin (IL)-2 and IL-21 supports efficient transduction using a VSV-G pseudotyped lentiviral vector. Moreover, we have identified that inhibition of innate immune receptor signaling greatly enhances transduction efficiency. We were able to boost the efficiency of lentiviral genetic modification on average 3.8-fold using BX795, an inhibitor of the TBK1/IKKɛ complex acting downstream of RIG-I, MDA-5, and TLR3. We have also observed that the use of BX795 enhances lentiviral transduction efficiency in a number of human and mouse cell lines, indicating a broadly applicable, practical, and safe approach that has the potential of being applicable to various gene therapy protocols.
Introduction
However, efficiency of viral gene delivery to NK cells has always proven challenging and less efficient than other cells of the hematopoietic system. In fact, this is not unexpected, since it is well established that NK cells are among the first responders to viral infections (Brandstadter and Yang, 2011) and must have been evolutionarily selected to have high endurance against viral infection (Lanier, 2008). The intracellular antiviral response of NK cells has been studied thoroughly in wild-type virus infections (Seya et al., 2011) but it has been mostly overlooked from a gene therapy point of view whether these responses are still active against viral vectors and have a significant effect in the resistance of NK cells to efficient viral transduction.
To increase the efficiency of viral gene delivery to NK cells we have tried to uncover the mechanisms behind this resistance and focused on antiviral responses resulting from pattern recognition receptor signaling. We have hypothesized that toll-like receptor (TLR) and/or RIG-I-like receptor (RLR) based antiviral responses (Thompson and Locarnini, 2007) are highly active in NK cells and could be tampered with in order to break the resistance. For this purpose, we used an approach of screening inhibitors of receptors that could be involved in the antiviral response. Here, we have successfully identified candidate inhibitors and conditions for optimal stimulation, timing, and enhancement of successful genetic modification by targeted inhibition of antiviral receptor signaling.
Our results bring together a feeder-free, isolation, and culture system of NK cells with an enhanced method for lentiviral gene delivery and present a method that could easily be applied to clinical settings.
Materials and Methods
Cell lines
293FT cells were purchased from Invitrogen (Life Technologies, Grand Island, NY) and maintained in Dulbecco's modified Eagle's medium (DMEM; GIBCO, Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; GIBCO), 0.1 mM nonessential amino acids (Sigma-Aldrich, St. Louis, MO), 6 mM L-glutamine (Sigma-Aldrich), 1 mM sodium pyruvate (Sigma-Aldrich), and 20 mM HEPES (Sigma-Aldrich).
NK92 cells were maintained in CellGro SCGM (CellGenix, Freiburg, Germany) medium supplemented with 20% FBS and 1000 U/ml recombinant human interleukin (rhIL)-2 (Proleukin, Novartis Pharmaceuticals, East Hanover, NJ). K562, U266, RPMI 8226, Jurkat, and CEM cells were maintained in RPMI-1640 medium (GIBCO) supplemented with 10% FBS. B10R cell were maintained in DMEM (GIBCO) supplemented with 10% FBS (GIBCO).
Production of lentiviral vectors
For production of VSV-G pseudotyped lentiviral vectors, 14×106 293FT cells were plated into a poly-D-lysine–coated 150-mm dish (BD Biosciences, San Jose, CA). Next day cells were transfected with 30 μg of lentiviral “gene ontology” (LeGO)-G2 plasmid (courtesy of Prof. Boris Fehse, University Medical Center Hamburg-Eppendorf, Hamburg, Germany), 15 μg of pMDLg/pRRE (Addgene, Cambridge, MA), 10 μg of pRSV-REV (Addgene), and 5 μg of phCMV-VSV-G (Addgene) using calcium phosphate transfection kit (Sigma-Aldrich) in the presence of 25 μM chloroquine (Sigma-Aldrich). Ten hours after transfection, the medium was changed and thereafter virus containing supernatant was collected every 24 hr for 2–3 days and stored at −80°C until further use. A small aliquot from each production was used to determine viral titers by transduction of 293FT cells with serially diluted amounts of virus supernatant.
Primary NK cell isolation and culture
Buffy coats were obtained from healthy donors via the blood bank at the Karolinska University Hospital, Huddinge. The experimental protocols were approved by the local research ethics committee.
The peripheral blood mononuclear cells were isolated by gradient centrifugation, using Lymphoprep (Nyegaard, Oslo, Norway), and washed twice with phosphate-buffered saline (PBS; GIBCO). Cell count and viability were assessed by Türk and Trypan Blue dye exclusion. NK cells were obtained by using NK cell isolation kit (Miltenyi Biotec, Cologne, Germany) and the AutoMACS machine (Miltenyi Biotec) according to manufacturer's instructions. After isolation, NK cells were put into culture at a concentration of 1×106 cells/ml in CellGro SCGM (Cellgenix) supplemented with 10% human AB serum (Lonza, Basel, Switzerland) and 1000 U/ml rhIL-2 (Proleukin). For initial testing of cytokine stimulations prior to transduction (Fig. 1), interleukin (IL)-12 (Peprotech, Rocky Hill, NJ), IL-15 (Peprotech), and IL-21 (Peprotech) were used at a concentration of 20 ng/ml. For the rest of the experiments, only 1000 U/ml rhIL-2 and 20 ng/ml IL-21 were used.
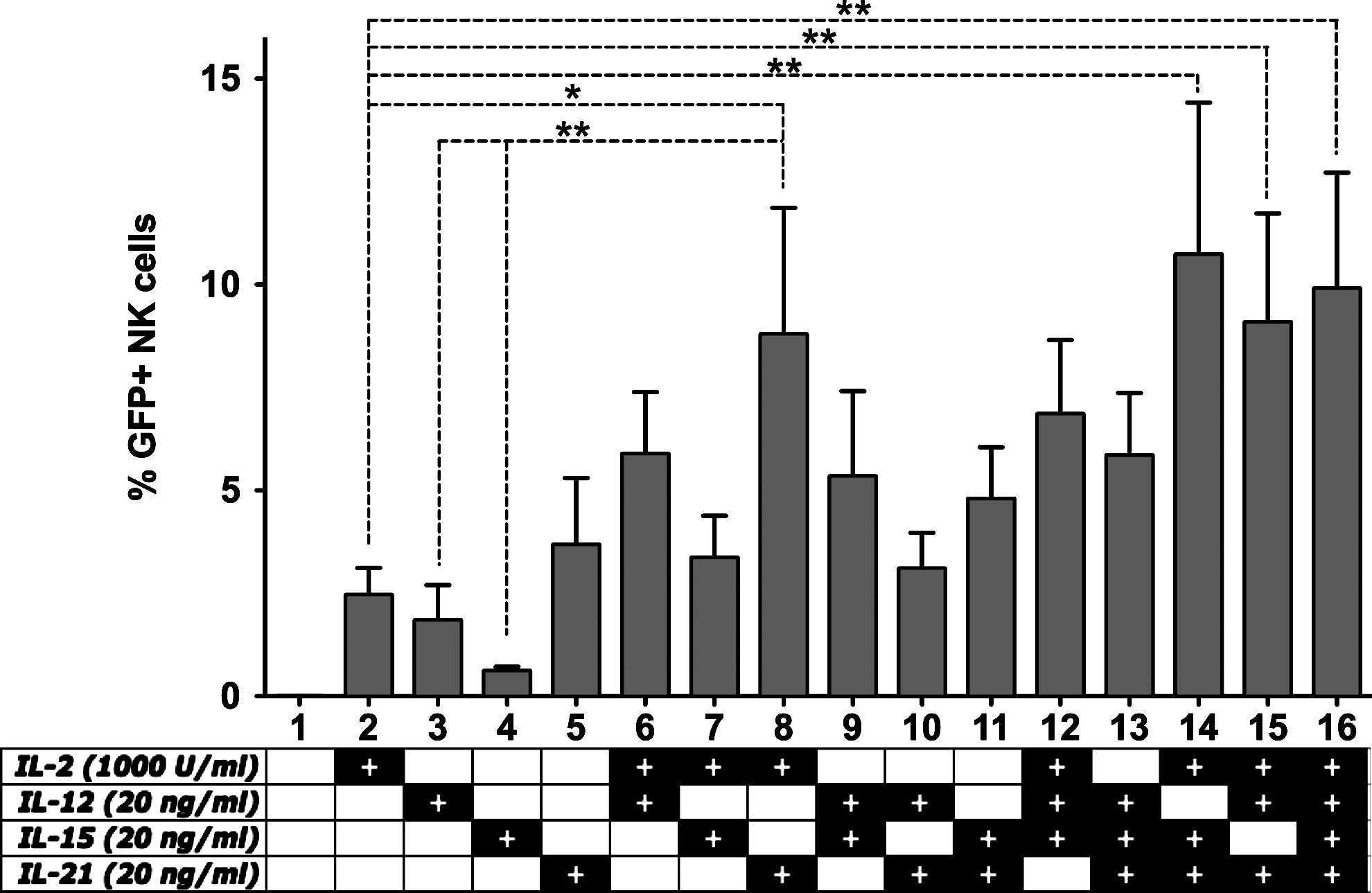
The effect of cytokine stimulation on lentiviral transduction efficiency. Freshly isolated primary natural killer (NK) cells were cultured in CellGro SCGM medium with 10% human serum in the presence of cytokines as indicated in the figure. After 24 hr of stimulation, NK cells were transduced at an multiplicity of infection (MOI) of 20 using the VSV-G pseudotyped lentiviral “gene ontology” (LeGO)-G2 vector. Seventy-two hours post-transduction, acquisition of enhanced green fluorescent protein (eGFP) expression was carried out by flow cytometry. Data from five independent donors. *p<0.05; **p<0.01; one-way ANOVA repeated measures.
Lentiviral transduction of NK cells
For each lentiviral transduction, 0.25×106 NK cells per well were seeded in a 24-well plate (BD Biosciences) and mixed with an appropriate amount of virus supernatant in the presence of 8 μg/ml of protamine sulfate (Sigma-Aldrich) or polybrene (Sigma-Aldrich) in a final volume of no more than 1 ml. The cytokines were replenished and plates were centrifuged at 1000×g for 1 hr at room temperature. After centrifugation, without removing viral supernatants, the plates were incubated at 37°C, 5% CO2 for 4–6 hr. At the end of the incubation, a second centrifugation at 1000×g for 1 hr at room temperature was carried out, after which the supernatants were removed from the wells and 1 ml of fresh NK cell growth medium (CellGro SCGM supplemented with 10% human AB serum) per well was added. The cells were maintained in this medium with daily addition cytokines (IL-2, 1000 U/ml; IL-12, 20 ng/ml; IL-15, 20 ng/ml; IL-21, 20 ng/ml) for at least 3 days before acquisition of enhanced green fluorescent protein (eGFP) expression was carried out. (Combinations of cytokines were used as indicated in Fig. 1. Only IL-2 and IL-21 were used in the rest of the experiments) In indicated experiments, the following inhibitors of TLR and RLR signaling were present during the transduction: 2-aminopurine (InvivoGen, San Diego, CA), BAY11-7082 (InvivoGen), Celastrol (InvivoGen), CLI-095 (InvivoGen), H-89 (InvivoGen), BX795 (InvivoGen), Norharmane (Sigma-Aldrich), and IRAK1/4 inhibitor (Sigma-Aldrich).
Lentiviral transduction of cell lines
For the transduction of cell lines, 2×105 cells per well were seeded into 24-well plates. For B10R cells, treatment with 6 μM BX795 was started 30 min prior to transduction and continued during the transduction process since we observed in preliminary experiments that this increases efficiency of treatment. For the rest of the cell lines, treatment with 6 μM BX795 was initiated simultaneously with the transduction process. Appropriate amount of viral supernatants were added into the wells along with 8 μg/ml polybrene (B10R cells) or protamine sulfate (NK92, Jurkat, CEM, U266, RPMI 8226 cells) and the total volume was adjusted to 500 μl. The plates were centrifuged at 1000×g for 1 hr followed by incubation at 37°C, 5% CO2 for 4–6 hr, after which the virus containing supernatant was removed and fresh growth media was added. The cells were grown for at least 3 days before acquisition of eGFP expression was carried out.
Flow cytometry
All antibody stainings for flow cytometry were done according to the following protocol. For surface stainings, the cells were washed once with PBS and incubated with appropriate amounts of antibody at 4°C for 30 min. The labeled cells were then washed with PBS and fixed in 1% paraformaldehyde prior to data acquisition. Data acquisition was done on a FACSCalibur (BD Biosciences), CyFlow ML (Partec GmbH, Munster, Germany), and LSRII-Fortessa (BD Biosciences) with standard filters. Data were analyzed with the FlowJo software (TreeStar Inc.). The antibodies used for NK cells were CD56 (NCAM16.2), CD56 (B159), CD3 (SK7), CD3 (SP34-2), CD69 (FN50), NKp44 (P44-8.1), CD16 (3G8), CD226 (DNAM-1) (DX11), CD25 (M-A251), and NKG2D (1D11) from BD Biosciences; NKG2A (Z199), CD158a,h (KIR2DL1/S1) (EB6B), CD158b1/b2,j (KIR2DL2/3/S2) (GL183), NKp30 (Z25), NKp46 (BAB281), and CD244 (2B4) (C1.7) from Beckmann Coulter; CD158e1/e2 (KIR3DL1/S1) (DX9), CD62L (DREG-56) from BioLegend; and CD45 (HI30) from Invitrogen.
Evaluation of NK cell-mediated cytotoxicity
The cytotoxic capacities of the final products were evaluated in vitro with a standard 4-hr 51Cr-release assay against K562 cells. In short, K562 target cells were labeled with 100 μCi of 51Cr for 1 hr at 37°C, washed twice with PBS, and resuspended in RPMI medium. A total of 3×104 target cells in 100 μl RPMI medium was placed in triplicates into V-bottomed 96-well plates and incubated for 4 hr with 100 μl of effector cells at appropriate concentrations to obtain effector:target (E:T) ratios from 1:3 to 10:1. Aliquots of supernatants were counted using a Packard Cobra Auto-Gamma 5000 Series Counting System (Meriden, CT). The percentage specific 51Cr release was calculated according to the formula: percent specific release=[(experimental release – spontaneous release)/(maximum release – spontaneous release)]×100.
Analysis of NK cell degranulation
NK cells were co-incubated with K562 target cells at a ratio of 1:1 in a final volume of 200 μl in round-bottomed 96-well plates at 37°C and 5% CO2 for 6 hr. Fluorochrome-conjugated anti-CD107a monoclonal antibody (mAb) or the corresponding IgG1 isotype control was added at the initiation of the assay. After 1 hr of co-incubation, Monensin (GolgiStop, Becton Dickinson) was added at a 1:100 dilution. Surface staining was done by incubating cells with anti-CD3 and anti-CD56 mAbs for 30 min at 4°C. The cells were then washed, resuspended in PBS, and immediately analyzed by flow cytometry.
Statistical analysis
For preparation of graphs and statistical analysis, GraphPad Prism (GraphPad Software Inc. La Jolla, CA) was used.
Results
Cytokine stimulation of primary NK cells prior to lentiviral transduction
Previous studies have demonstrated that the stimulation of cells prior to lentiviral genetic modification presents a chance to boost transduction efficiencies (Costello et al., 2000). Although lentiviral vectors do not require active cell division for successful integration of the viral genome, ongoing cellular activity is critical for efficient genetic modification. Therefore, we primarily aimed at understanding the basic necessary activation signals for short-term induction of NK cells prior to genetic modification. Extrapolating from previous reports of NK cell transduction, we decided to test IL-2, IL-12, IL-15, and IL-21 for this purpose.
Freshly isolated NK cells were cultured overnight in media containing a single cytokine or combinations thereof. After 24 hr, the cells were transduced with the VSV-G pseudotyped LeGO-G2 (Weber et al., 2008) vector (expressing eGFP under the influence of SFFV promoter) at a multiplicity of infection (MOI) of 20, and eGFP expression was acquired 3 days later (Fig. 1).
We observed that, when used as a single cytokine, IL-2 and IL-21 seem to perform better than IL-12 and IL-15. Moreover, the combination of IL-2 and IL-21 gives a significant improvement in transduction efficiency. Although addition of IL-12 or IL-15 to IL-2 seems to improve the transduction efficiency compared to IL-2 alone, the effect is weaker than addition of IL-21 such that even the combination of IL-2/IL-12/IL-15 fails to perform better than IL-2/IL-21. Finally, addition of IL-12 or IL-15 to the IL-2/IL-21 combination does not significantly enhance the transduction efficiency.
Taken together, these results indicate that, of the tested cytokines, a combination of IL-2 and IL-21 is sufficient for optimal stimulation of NK cells prior to transduction.
Inhibition of pattern recognition receptor signaling enhances transduction efficiency in NK cells
We have hypothesized that TLR- or RLR-mediated detection of viral vector components might activate an antiviral response in NK cells, negatively effecting the efficiency of lentiviral transduction. In order to test this hypothesis, we attempted to use small molecule inhibitors of TLR and RLR signaling preceding lentiviral transduction. In addition, we also decided to include the proteasome inhibitor bortezomib due to the documented ability of proteasome inhibitors to increase transduction efficiencies in hematopoietic stem cells and T cells (Santoni de Sio et al., 2008; Leuci et al., 2011).
In initial experiments, in which the treatment with inhibitors was done for 6–12 hr prior to exposure to the lentiviral vector and removed during the transduction, no significant change in transduction efficiency was observed. However, when inhibitors were present throughout the transduction protocol the effects on transduction efficiencies were significantly pronounced (Fig. 2). We have observed significant toxicity to NK cells while using Celastrol, CLI-095, H-89, and Norharmane at recommended concentrations and lost majority of the cells, deeming these inhibitors unfit for use in such an approach. 2-Aminopurine and BAY11-7082 seemed to have a minimal effect in improving transduction, while bortezomib showed an improvement but negatively affected NK cell viability and morphology.
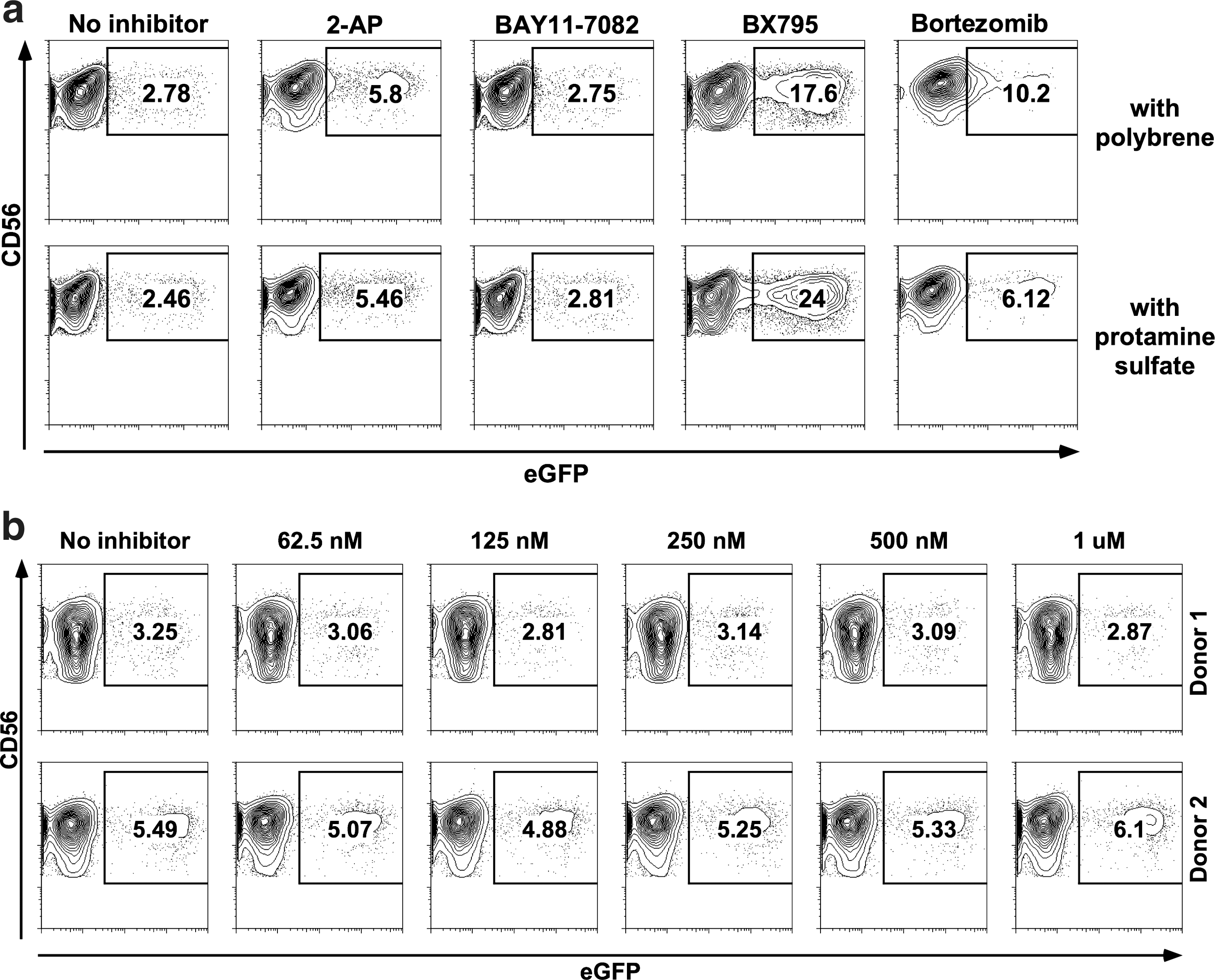
Inhibition of innate immune signaling pathways for enhancement of lentiviral transduction efficiency. NK cells stimulated with interleukin (IL)-2/IL-21 for 2 days were transduced in the presence of various inhibitors.
Most interestingly, the use of BX795 at 2 μM concentration dramatically increased transduction efficiency (Fig. 2a). BX795 is an inhibitor of the TBK1/IKKɛ complex, which acts as a common mediator in the signaling pathways of RIG-I, MDA-5, and TLR3 (Clark et al., 2009). Therefore, it might be possible to state that the lentiviral RNA is recognized by one or more of these receptors and an antiviral response is triggered that can be inhibited by the use of BX795.
Additionally, we have observed in preliminary experiments that the use of BX795 better synergizes with protamine sulfate (8 μg/ml) rather than polybrene (8 μg/ml), although the two compounds seem to perform similarly in the absence of any signaling inhibitors. Therefore, protamine sulfate was preferred instead of polybrene in subsequent experiments.
In order to rule out the involvement of other TLRs, we also undertook transductions with an inhibitor of the IRAK1/4 complex, which is crucial to TLR signaling except TLR3. As shown in Fig. 2b, the use of IRAK1/4 inhibitor had no effect on transduction efficiency at any of the concentrations tested.
Taken together, these results support the hypothesis that during transduction, intracellular antiviral defense mechanisms including one or more of the receptors RIG-I, MDA-5, and TLR3 are activated and contribute significantly to the resistance of NK cells to lentiviral genetic modification.
BX795 has a dose-dependent effect with minimal toxicity to NK cells
Testing different concentrations of BX795 has shown that the inhibitor has a dose-dependent effect on increasing genetic modification efficiency in NK cells (Fig. 3a). Although a significant effect is seen at 2 μM concentration, this effect increases even more up to 6 μM after which it seems to stabilize. Although there's always an interdonor variability regarding NK cell transduction efficiency, the use of BX795 has not failed to increase efficiency in any of the donors tested during our studies. At 6 μM concentration, the effect varies from 1.97-fold to 5.18-fold and averages at 3.8-fold (n=18). Remarkably, the use of BX795 at the indicated concentrations presented no immediate toxic effects on NK cells as determined by Annexin-V/PI staining after BX795 treatment (Fig. 3b). Also of importance, treatment with BX795 increased the efficiency of transduction only when it was present during the transduction and not when the NK cells were pretreated overnight and BX795 was washed away prior to transduction, indicating that the inhibition is reversible (Fig. 3c).
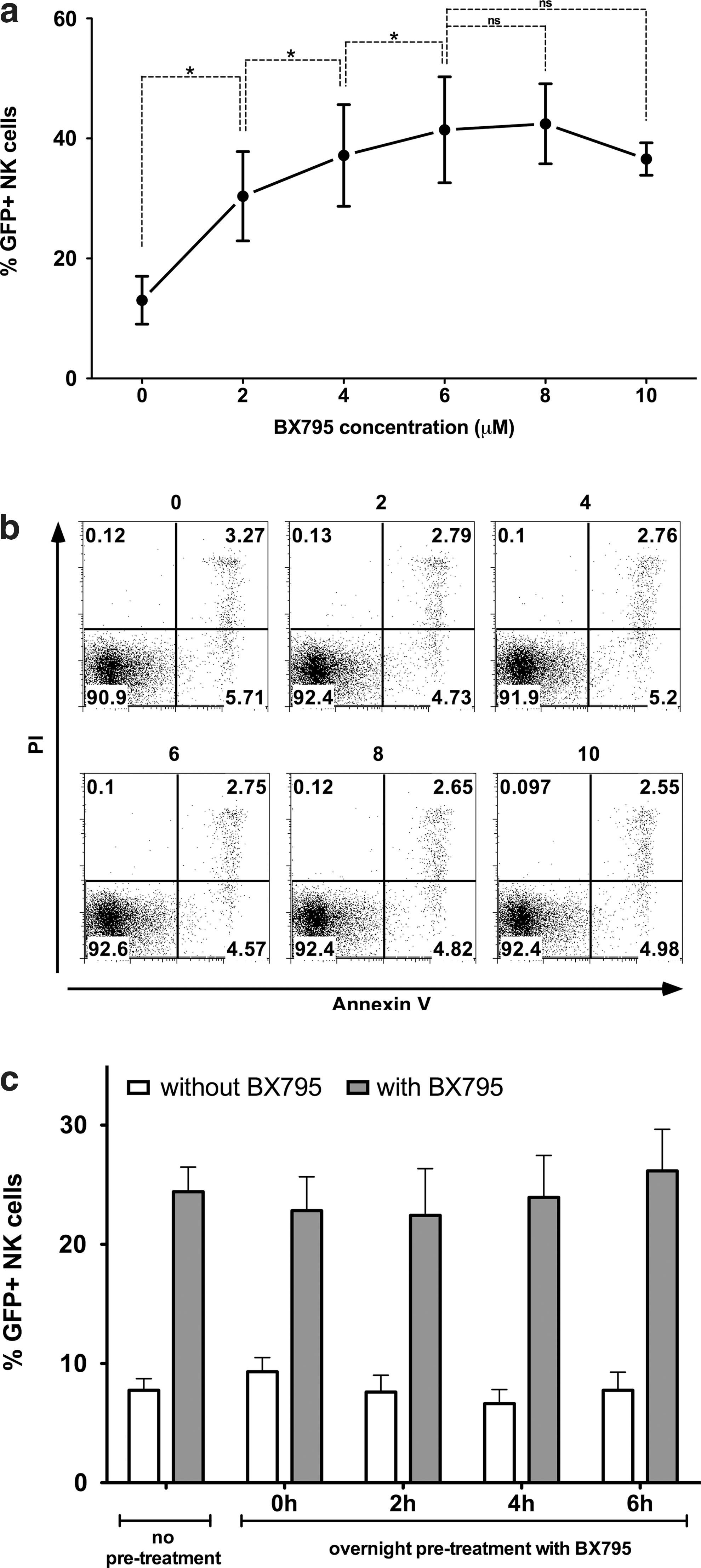
Dose response, nontoxicity, and reversibility of BX795 treatment.
Optimal time of lentiviral transduction
In an attempt to find the optimal balance between time and efficiency, we have investigated how long prestimulation of freshly isolated NK cells would be necessary for efficient transduction using the IL-2/IL-21/BX795 protocol. For this purpose, we carried out transduction of freshly isolated NK cells as well as those that were stimulated with IL-2/IL-21 for 1, 2, 3, or 4 days (Fig. 4). Our results clearly demonstrate that while there was very little transduction of freshly isolated cells, the efficiency started increasing after culture with IL-2/IL-21 and reached its maximum at 2 days. Further culture of the cells prior to transduction did not increase efficiency. These results indicate the importance of prestimulation for efficient genetic modification, while clearly demonstrating that excessive stimulation does little to improve the outcome.
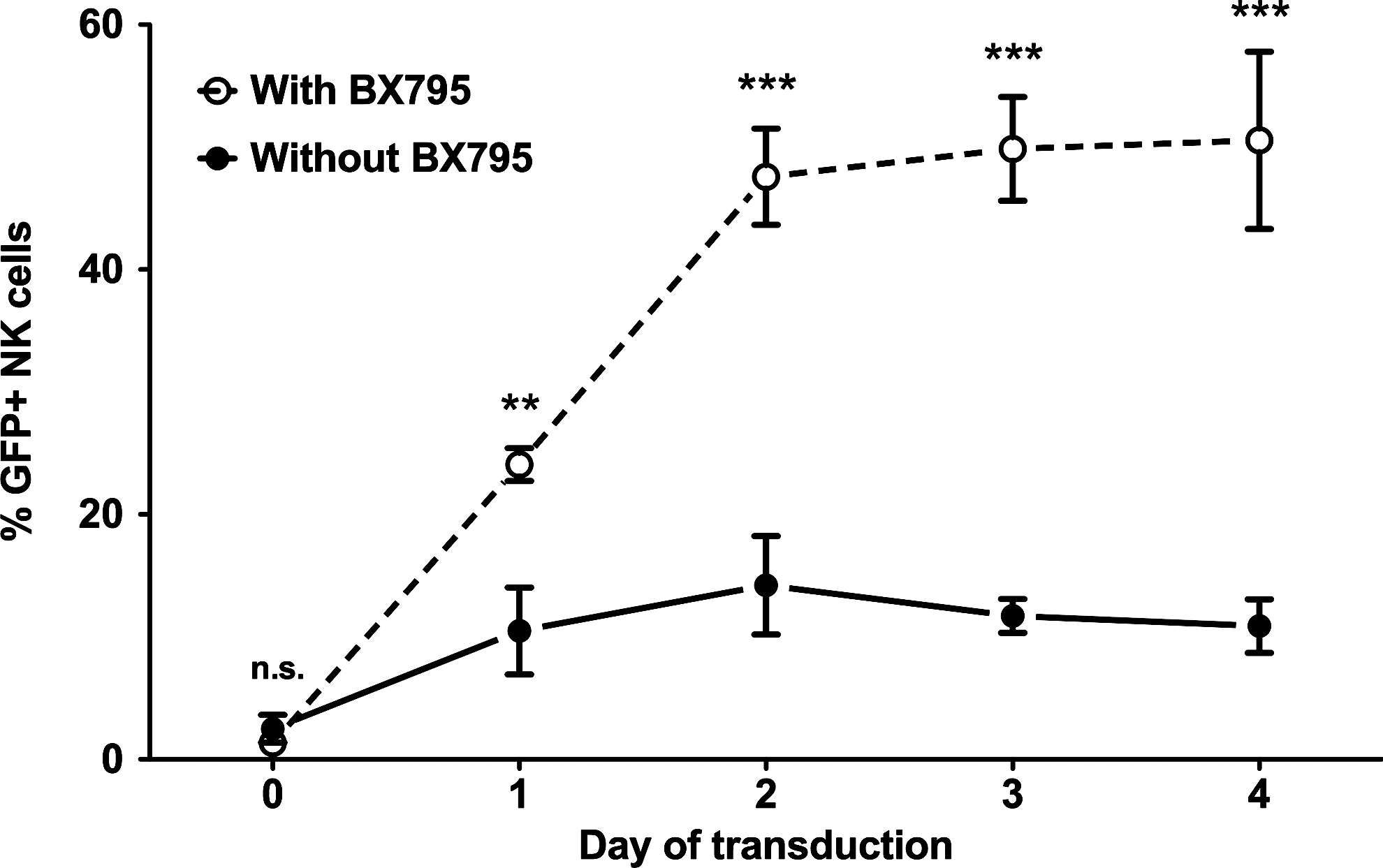
Effect of stimulation time on genetic modification efficiency. Freshly isolated NK cells were either directly transduced (day 0) or stimulated in CellGro SCGM medium with 10% human serum in the presence of IL-2 (1000 U/ml) and IL-21 (20 ng/ml) for 1, 2, 3, or 4 days and subjected to lentiviral transduction in the presence and absence of BX795. For each transduction, eGFP expression was acquired 72 hr later. The efficiency of transduction peaked after 2 days of stimulation and remained more or less stable until day 4. Data from four donors. n.s., not significant; **p<0.01; ***p<0.001; two-way ANOVA repeated measures.
Functional capacity and phenotype of NK cells is not altered by genetic modification
We also investigated whether the process of genetic modification using BX795 along with IL-2/IL-21 stimulation presents any functional or phenotypic concerns regarding NK cell cytotoxic capacity. We did not observe any alteration in cellular cytotoxicity after treatment with BX795 alone or transduction in the presence of BX795 (Fig. 5). The cytotoxic activities against K562 cells were comparable in all groups. Moreover, degranulation assays further proved that both the cytotoxic capacity as shown by PMA/ionomycin stimulation and the rate of degranulation against K562 were comparable, not only between different samples but also within each sample when the GFP+ and GFP− cells were analyzed separately. Finally, phenotypic analysis revealed that the transduction protocol yields polyclonal NK cells without any detectable change in expression levels of cell surface receptors analyzed when compared to nontransduced cells (Fig. 6), forming an expected basis for the observation of similar cytotoxic activities. These results clearly demonstrate that the use of BX795 to enhance lentiviral transduction of NK cells is efficient and safe, providing a novel and significant improvement for further studies on genetic modification of NK cells with transgenes of interest.
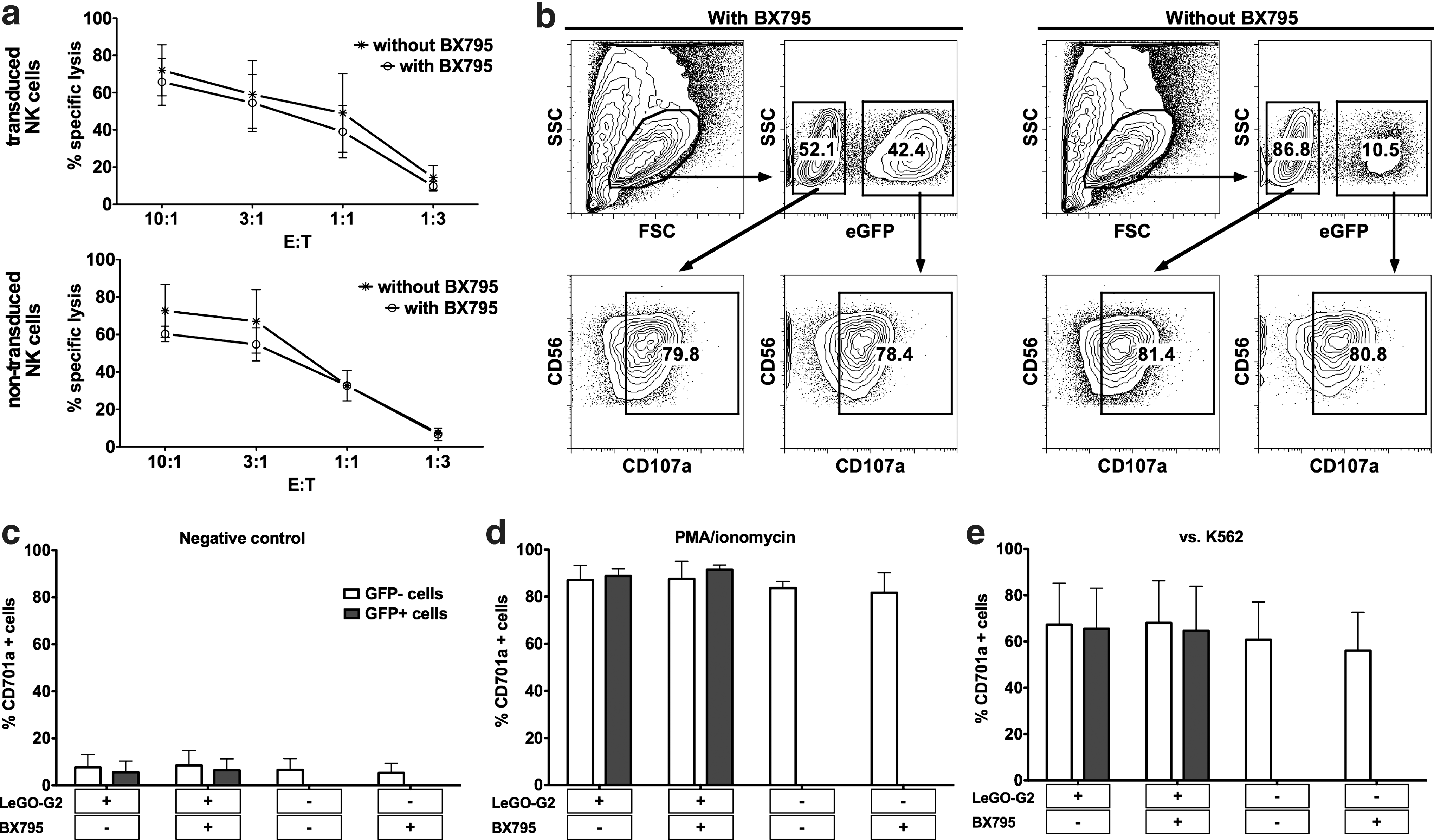
Function of genetically modified NK cells is not altered.

Phenotypic analysis of NK cells. Phenotype of nontransduced NK cells that were cultured
The effect of BX795 on lentiviral transduction of other cell types
Although we observed a clear ability of BX795 to promote transduction efficiency in primary human NK cells, it is possible that this effect is specific to NK cells under these conditions. To determine whether the use of BX795 to inhibit TBK1/IKKɛ signaling promotes transduction efficiency in other species and cell types, we ran a set of experiments in which different human cell lines of hematopoietic origin (NK92, U266, Jurkat, CEM, RPMI8226) as well as the mouse immortalized bone marrow macrophage cell line B10R were transduced with the VSV-G pseudotyped LeGO-G2 vector alone or in the presence of BX795. We observed reproducible increases in the transduction efficiency after BX795 treatment in all tested cell lines except U266 (Fig. 7). The most pronounced effects were observed in the hard-to-transduce NK cell line NK92 and the mouse macrophage cell line B10R. In other cell types in which transduction efficiency was already quite high even in low MOI, a significant increase of GFP fluorescence was observed, indicating the efficient integration of higher number of copies of the viral vector.
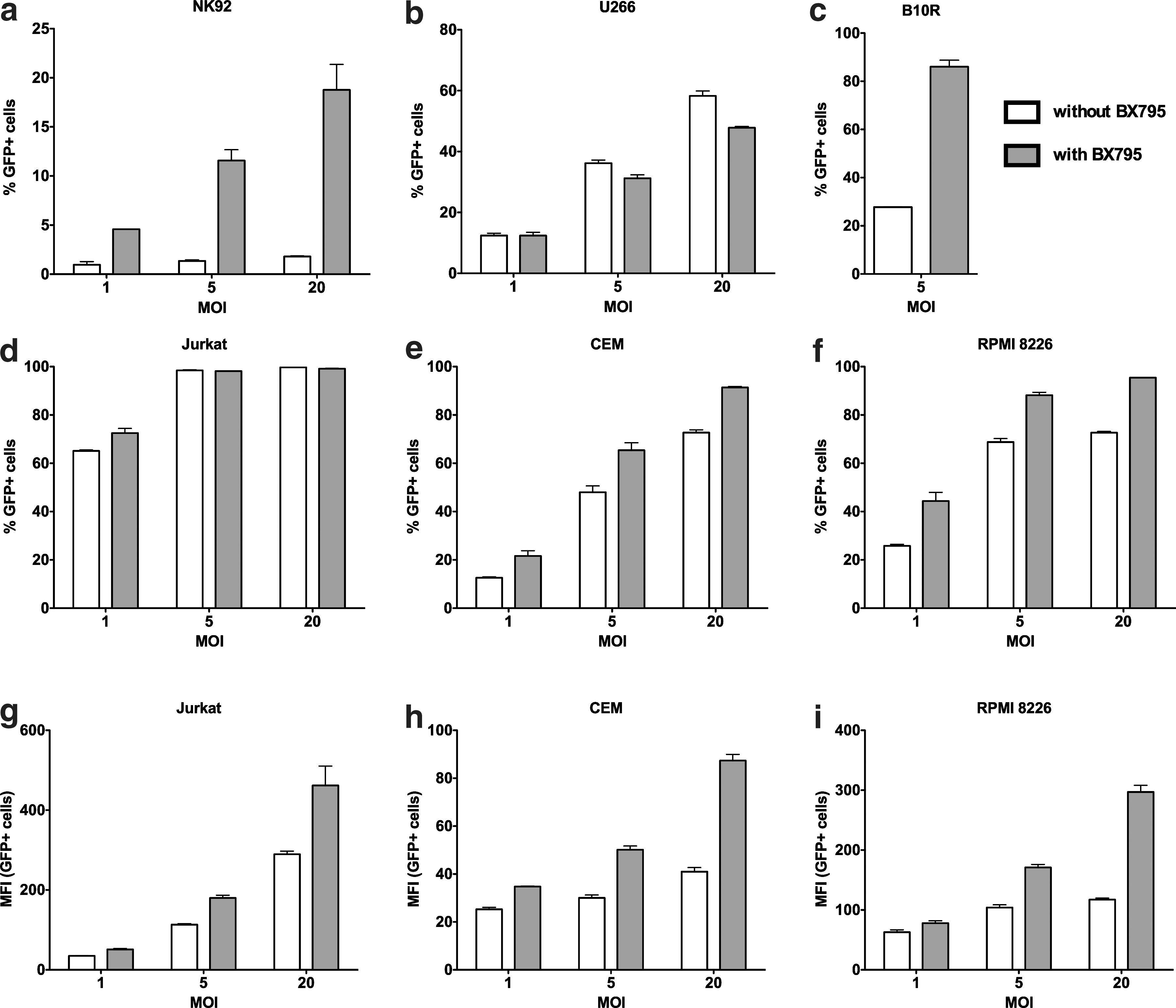
The use of BX795 treatment for enhancing transduction efficiency in other cell types. Human cells lines of hematopoietic origin (NK92, U266, Jurkat, CEM, RPMI 8226) as well as the mouse macrophage cell line B10R were transduced in the absence and presence of 6 μM BX795. An enhancement in transduction efficiency either as elevated number of GFP+ cells
Taken together, these results demonstrate that BX795 works to promote successful lentiviral transductions in different species and is a candidate for common use in gene therapy protocols with a variety of different cell types.
Discussion
Genetic modification of NK cells opens up a potential strategy for patients that can be treated with adoptive cell therapy, while providing invaluable insights into the basic biology of NK cells. However, difficulties in genetically modifying primary NK cells have hampered further development in the field.
Methods of transient genetic modification such as transfection, electroporation, nucleofection, and adenoviral infection are under constant development and have come a long way from applications on NK cell lines to GMP-compatible protocols for primary NK cells (Schroers et al., 2004; Zhang et al., 2004; Grund and Muise-Helmericks 2005; Goding et al., 2007; Schoenberg et al., 2008; Boissel et al., 2009). However, the possibility of stable genetic modification with transgenes enhancing or targeting NK cell cytotoxicity has many advantages. Optimization of viral genetic modification in NK cells presents a multifaceted problem ranging from the source of NK cells to culture conditions, the choice of cytokines and critical viral elements such as envelopes or promoters, and the process of viral infection. Previous reports have included various approaches such as the use of feeder cells (Imai et al., 2005; Micucci et al., 2006; Kruschinski et al., 2008), multiple rounds of transductions (Becknell et al., 2005; Karimi et al., 2005; Micucci et al., 2006) or co-culture with virus producing cells (Nagashima et al., 1998) in an attempt to ensure efficient culture and genetic modification of NK cells.
In this study, we aimed to optimize a protocol for transduction of NK cells with VSV-G pseudotyped lentiviral vectors that does not rely on stimulation with feeder cells or long-term expansion/activation of the cells in an attempt to define a fast and efficient protocol that can both be practically used for experimental studies of NK cell biology and has the potential to be easily scaled-up for future clinical applications. For this purpose, we primarily sought a firm starting point by evaluating different cytokine stimulations prior to viral transduction. Among the cytokines we tested were IL-2 and IL-15, which are commonly used for culture and activation of NK cells, as well as IL-12 and IL-21, which have been previously reported to have a positive effect on genetic modification efficiency of NK cells (Micucci et al., 2006; Figueiredo et al., 2009). Our results showed that stimulation of freshly isolated NK cells with 1000 U/ml IL-2 and 20 ng/ml IL-21 for 24 hr provides greater genetic modification efficiency compared to using any of the mentioned cytokines alone. We observed that the IL-2/IL-15 combination did not provide any improvement over IL-2 alone, while the IL-2/IL-12 combination showed increased transduction efficiency. Since we aimed at optimizing a fast protocol for the transduction of NK cells, we chose the 24-hr time point for comparison of different cytokine stimulations. However, it should be kept in mind that maximum stimulation by different cytokines might be reached at different time points and a comparison at 24 hr does not account for this effect.
Although the positive effect of IL-12 was in line with a previous report by Micucci et al. (2006), the magnitude of the effect was not as strong in our culture system, which could be due to the existence of EBV-infected feeders in the former study, which are known to continuously secrete IL-12 (Gri et al., 1998). Nevertheless, we observed that the IL-2/IL-21 combination presented an even greater enhancement. IL-21 has previously been shown to synergize with IL-2 in activation of NK cells and primarily signal through STAT3 phosphorylation as compared to STAT5 for the rest of the IL-2 family of cytokines which might, at least to some extent, explain the differential effects observed by the addition IL-21 into the stimulation cocktail prior to lentiviral transduction (Gowda et al., 2008; Skak et al., 2008; Liao et al., 2011). Although the addition of a third cytokine to this combination slightly increased transduction efficiencies in some donors, the extent of the increase was not significant enough to justify adding a third or fourth cytokine to the cocktail.
Furthermore, we hypothesized that inhibition of innate immune receptor signaling would contribute to enhanced transduction efficiency. It is well known that TLRs and RLRs play a major role in detection of viral infections and induction of an antiviral state (Kumar et al., 2011; Wilkins and Gale, 2011). Many wild-type viruses have developed elaborate schemes to avoid detection by these receptors and increase their virulence (Randall and Goodbourn, 2008). In the case of viral vectors, the removal of various viral genes that counteract host responses but are dispensable for vector production is often preferred due to safety and practicality considerations. Inevitably, this would render viral vectors more prone to inducing strong innate responses upon target cell infection (Shayakhmetov et al., 2011). Recent data have proven that such responses are indeed triggered by various viral gene delivery mechanisms such as HSV (Olschowka et al., 2003; Suzuki et al., 2007), AdV (Worgall et al., 1997; Liu and Muruve 2003), AAV (Zaiss et al., 2002; Zhu et al., 2009), baculovirus (Hervas-Stubbs et al., 2007), and lentivirus (Brown et al., 2007) based vectors. Moreover, research on genetic modification of T cells using lentiviral vectors has shown convincingly that although they are not necessary for viral packaging, the HIV-1 accessory proteins increase transduction efficiency when present (Chinnasamy et al., 2000; Costello et al., 2000). Despite the necessity of removing these accessory genes for safety issues, to our knowledge, no efficient method of circumventing this loss of infectivity via such an approach as small molecule inhibitors has been reported. As noted before, the information about the role or activity of intracellular antiviral responses to viral vectors is limited. The compelling demonstration of improved transduction in the presence of BX795 supports but does not yet formally prove that antiviral responses are activated by viral vectors and further work on these mechanisms has the potential to drastically improve gene therapy protocols.
We have shown that the use of BX795, an inhibitor of the signaling molecule TBK1/IKKɛ that acts downstream of RIG-I, MDA-5, and TLR3, greatly enhances lentiviral transduction efficiency in primary human NK cells. This effect seems to be dose-dependent and nontoxic to the cells at effective doses. Moreover, the absence of increased transduction efficacy when the inhibitor is used only prior to and not during transduction suggests that such an intervention with small molecules is reversible, thus safer considering a theoretical risk of the adoptively transferred genetically modified cells to encounter wild-type virus infections in vivo.
The data regarding the transduction of other cell types put forward the use of BX795 as a transduction-enhancing agent in a different number of scenarios. Of the tested cell lines, only U266 showed a lack of increased transduction efficiency. Interestingly, the application of BX795 also provided a significant improvement in the lentiviral transduction of the mouse macrophage cell line B10R. Macrophages are difficult to genetically modify using DNA expression plasmids because they express the innate immune receptor Aim2, which detects double-stranded DNA, thus promoting the formation of the Aim2 inflammasome and activating caspase-1–dependent cell death (Desmet and Ishii, 2012). To circumvent DNA-sensing systems in macrophages, researchers have turned to viral transduction systems. However, macrophages also express numerous viral defense systems (Desmet and Ishii, 2012). As with human NK cells, inhibiting TBK1/IKKɛ activity in mouse macrophages leads to significant increases in transduction efficiency. This demonstrates that this approach is a viable method to improve viral transduction of other cell types from other species and suggests that it may have broad technical applications to improve the efficiency of genetic modification.
Our results present a proof-of-principle for feasibility of such approaches for enhancement of gene therapy applications. From our preliminary observations with other cell types, it is clear that not only NK cells, but also cells of various types and species will benefit from this approach. Further characterization of pathways involved in this response and in-depth analysis of the use of such inhibitors is warranted to improve gene therapy strategies.
Footnotes
Acknowledgments
The authors would like to thank Adil Doganay Duru for helpful discussions. This research is funded by VINNOVA Foundation, CliniGene EU Network of Excellence, Radiumhemmet, and the Swedish Research Council.
Author Contributions
T.S. conceived and designed the project; performed virus production, NK cell culture, lentiviral transduction of NK cells and hematopoietic cell lines, flow cytometry and degranulation experiments; analyzed and interpreted data; and wrote the manuscript. S.N. and S.E.A. performed lentiviral transduction of B10R cells and flow cytometry, analyzed and interpreted data, and wrote the manuscript. M.G. and B.S. performed NK cell isolation, culture, cytotoxicity, and degranulation experiments; analyzed and interpreted data; and wrote the manuscript. E.A. designed and supervised the project, interpreted data, and wrote the manuscript.
Author Disclosure Statement
The authors declare no competing financial interests.