
Abstract
The United States’ current list-based approach to biodefense is limited because it considers only known biological agents. Alternatively, developing and adopting a system based on agent-agnostic signatures would enable detection and characterization of both known and novel agents, thereby engendering greater adaptability in the face of an evolving threat landscape. Machine learning (ML) could aid in such a transition, as it can recognize and encode highly complex patterns from multiple input data modalities and has already demonstrated success in many healthcare and defense applications. Functionalizing ML for environmental biodetection requires understanding current technical capabilities. In this article, we provide a systematic review of existing ML platforms and discuss anticipated development efforts needed to achieve effective ML-enabled, agnostic biodetection.
Introduction
T
Machine learning (ML) systems could enable real-time, multiscale, multidimensional agnostic assessment of a perturbation’s nature and source. ML models can recognize and encode highly complex patterns from multiple data modalities, including images, text, and biological/chemical/physical spectra, and can execute assessments that traditionally require human operation. In healthcare, ML models aid diagnosis of clinical threats, including infection, cancer, and strokes,4-6 though greater, more diverse training data and strategies for handling missing data are needed to improve reliability and efficacy. Moreover, ML advances could support more efficient raw data analysis. Recently, large language models have been deployed for biological function prediction.7-9
Interest in environmental biosurveillance is increasing across application spaces and sampling media, including wastewater,10,11 drinking water, 12 and aerosols.13-15 Expansion of antibiotic resistance has amplified interest in surveillance of nonclinical settings. 16 While ML is used broadly in the clinical sciences, its role in environmental biodetection is currently limited. Functionalizing ML-based strategies for environmental detection of potential biological threats requires an understanding of the existing technical landscape. In this review, we systematically evaluate existing ML platforms applied in clinical and nonclinical contexts, focusing on workflows with potential for biosurveillance in nonclinical environments surrounding human populations, and we discuss development efforts needed for effective ML-enabled, agnostic biodetection.
Platform Assessments
Pathogens are organisms that cause disease in their host. 17 Existing environmental biodetection systems rely largely on defined pathogens’ nucleic acid-based signatures via quantitative polymerase chain reaction (qPCR) or next-generation sequencing. Methods for profiling pathogens’ physicochemical properties, including mass spectrometry and Raman spectroscopy, have also been evaluated. Adopting an agent-agnostic paradigm requires less reliance on static, predefined signatures. We hypothesize that features in raw datasets could distinguish health or environmental threats and that analyzing data at its source could promote “reference-independent” systems—self-contained platforms not requiring external databases. While such platforms would still require existing datasets for model training, inference could be performed without access to these datasets/references. Further, if generalizable features for pathogenic function are identified in raw signal, their discriminatory capacity may demonstrate greater extensibility to novel threats.
Direct Processing of Raw DNA Signal
Raw instrument output obtained prior to DNA base assignment could contain unique optical, pH/chemical, or electrical signals and facilitate direct aerosol biodetection.18,19 During nanopore sequencing, for example, electrical current is assigned to nucleotides passing through the nanopore. The raw electrical signal (“squiggle”) carries information beyond the base sequence. 20 Biological sample classification from squiggles was explored using deep learning (DL),20,21 probabilistic models, 22 and reference conversion. 23 A convolutional neural network (CNN) called SquiggleNet, developed using multiple human and bacterial reference DNA datasets and 4,500 electrical signals from 2 million sequence reads, 20 achieved 75% to 95% accuracy when predicting whether DNA sequences were human or microbial. Nanopore sequencing instruments have a small footprint and could be a useful, rapid diagnostic tool. 24
Other platforms include alternative deep neural networks (DNNs) for selective sequencing applications21,25; lightweight platforms for rapid analysis 26 ; and platforms leveraging graphics processing unit (GPU)-based computing architecture,27,28 gene-level assessments, 29 and algorithm-architecture codesign. 30 SquiggleNet and other nascent models demonstrate proof-of-concept for ML-based detection from raw output (Figure 1).
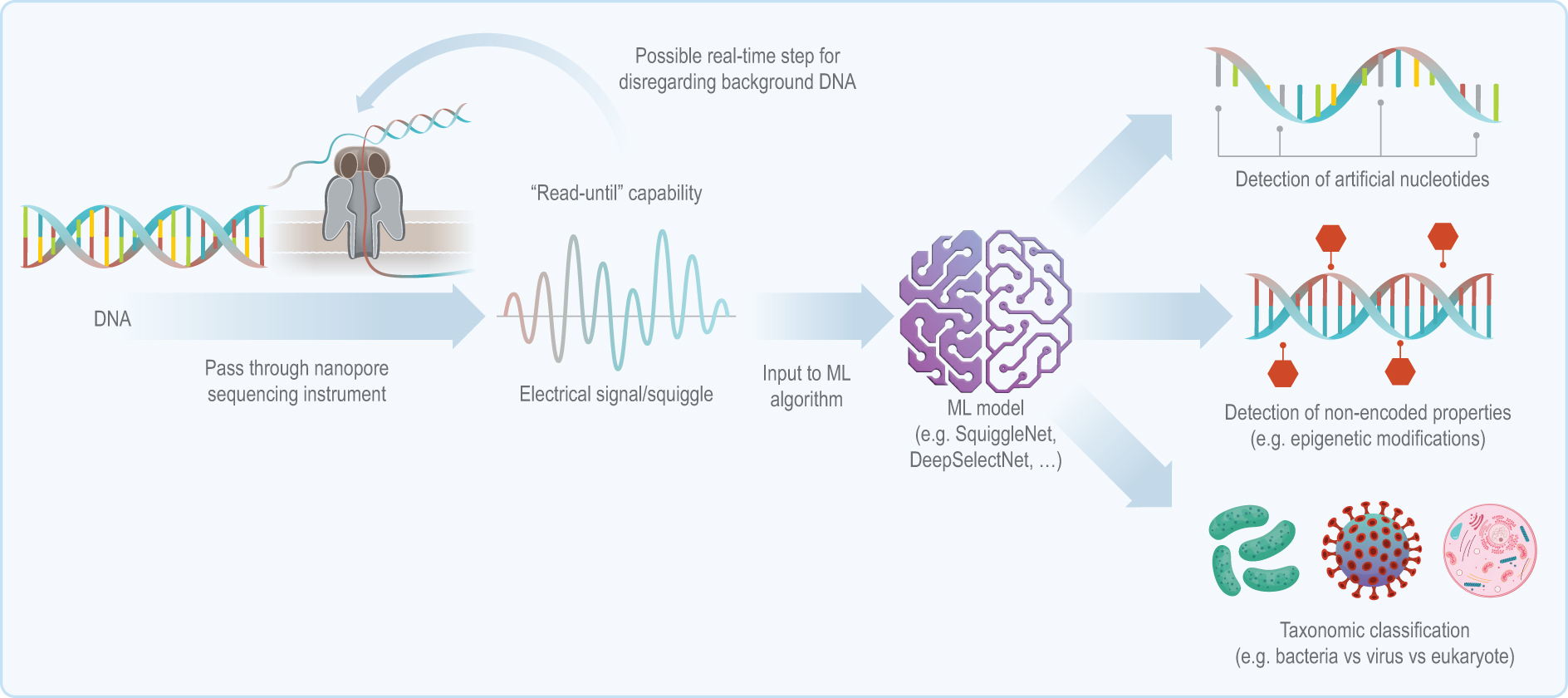
Schematic of the integration of nanopore sequencing and ML, including some potential applications. A sample of DNA is supplied to the nanopore sequencer, which outputs an electrical “squiggle” based on the underlying sequence of nucleotides. ML models trained on such squiggles could be used to detect artificial nucleotides or epigenetic modifications or to classify the sample’s taxonomy. Abbreviation: ML, machine learning.
Analysis speed is a potential advantage of using raw DNA-derived data, as human vs bacterial ML classifiers require only about 1 second of sequencing data and limited memory. These systems could theoretically detect artificial nucleotides and nonencoded properties, like DNA methylation, using random forest and support vector machine models via multiple instance learning. 31 Thanks to the “read-until” ability 32 of nanopore sequencing, such platforms already serve as a preprocessing step for eliminating nontarget (eg, human) DNA from environmental samples. 33
These platforms’ long-term biodetection utility depends on generating test and training datasets for feasibility testing. Future evaluation could use simulated data 34 to assess whether raw data enables categorization of microbes into functionally useful bins.
ML for Gene Virulence Prediction
Determining the taxonomic identity and function of genetic sequences is critical for biodetection. SeqScreen 35 characterizes short gene or protein sequences by predicting taxonomic and functional labels. SeqScreen’s developers created 32 custom functions of sequences of concern (FunSoCs) to describe microbial pathogenesis functions encoded in viral and bacterial sequences. 36 SeqScreen’s pipeline takes protein sequences of at least 50 nucleotides as input, aligns them to UniProt entries, 37 and assigns FunSoC labels through ensemble learning, after representing each sequence as a high-dimensional feature vector (Figures 2 and 3A). A feature vector, depicted in Figure 2, is an ordered list of numbers that describes an object’s real-world properties and that can be used as input for ML models. Feature vectors often serve as input to ML models, algorithms that are trained to identify patterns in existing data and make predictions for unseen data.
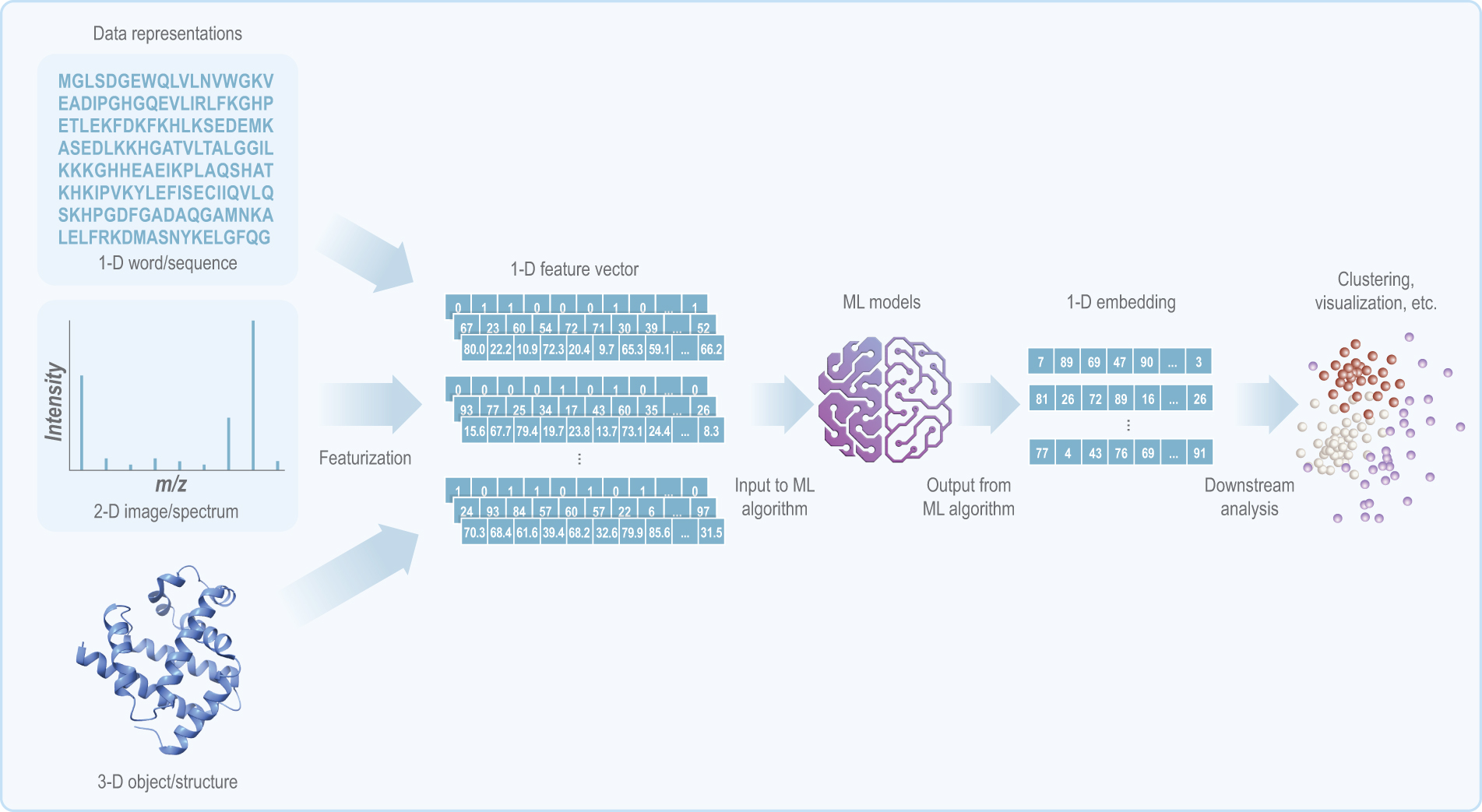
Schematic of a generalized ML workflow and its main components: data representations, feature vectors, models, and embeddings. Biological data can take many forms, including as a 1D word or sequence, a 2D image or spectrum, or a 3D object or structure. Regardless of its dimensionality, input data can be re-represented as a feature vector, an ordered list of numbers that describes an object’s real-world properties. The different types of numbers shown in the feature vectors in the figure are meant to illustrate that there are multiple ways of encoding the same data. In this schematic and those following, numeric values in feature vectors and embeddings are randomly assigned. Abbreviation: ML, machine learning.
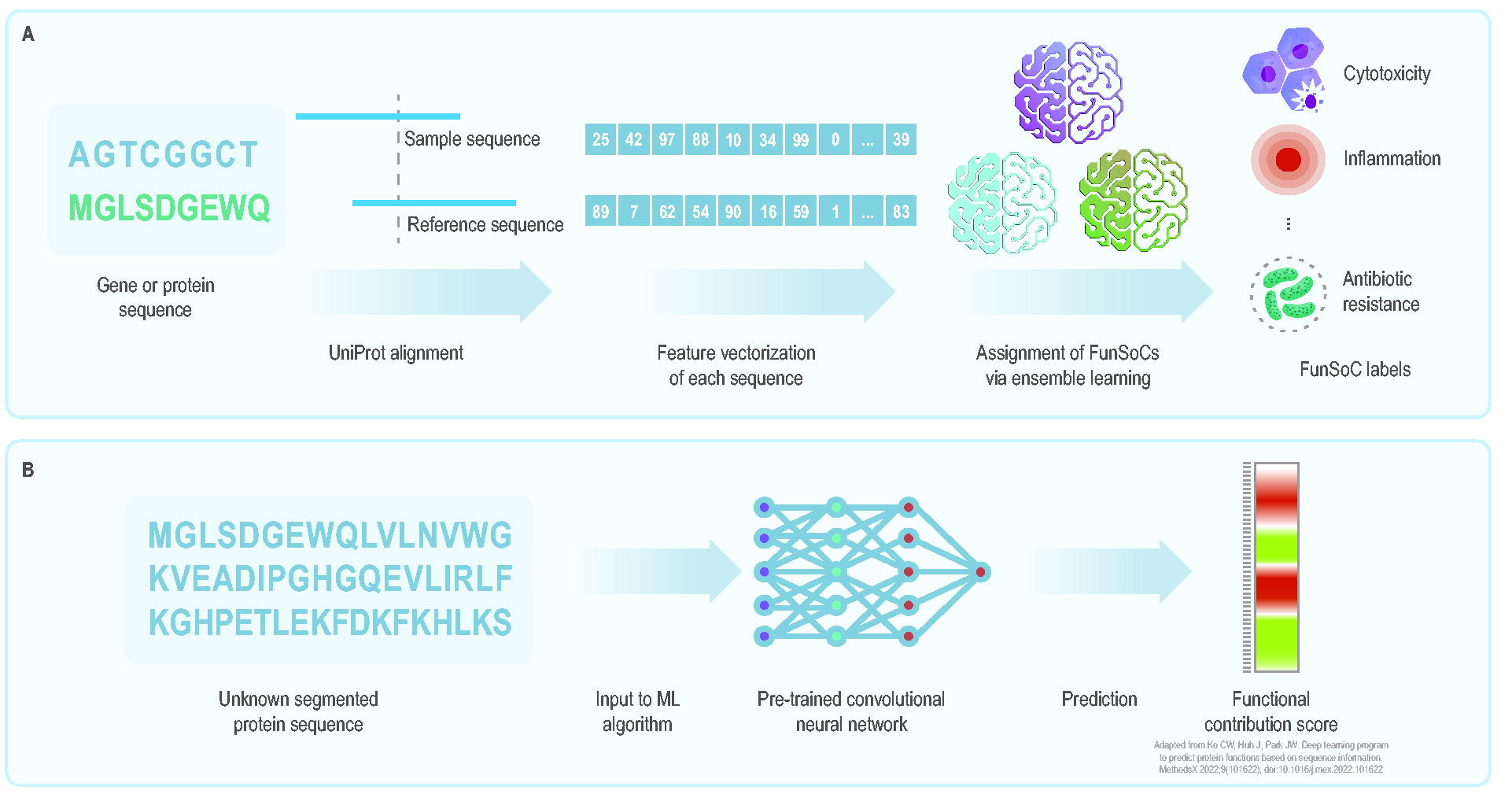
(A) Schematic of the SeqScreen pipeline. 35 The pipeline takes a sample gene or protein sequence as input, aligns it with reference sequences in UniProt, produces a feature vector of the sequence, and predicts with which microbial pathogenesis functions it might be associated. (B) Schematic of the FUTUSA pipeline. 39 The pipeline is designed to take an unknown, segmented protein sequence and, using a convolutional neural network, predict the functional impact of each amino acid. By substituting point mutations into the input sequence, FUTUSA could be used to predict the functional impacts of sequence variations. Abbreviations: FunSoCs, functions of sequences of concern; FUTUSA, function teller using sequence alone; ML, machine learning.
Three ML models, selected because they handle class imbalances (negative samples outnumber positive samples), participate in majority voting to decide labels. SeqScreen training data (98,283 samples) included manually curated virulence-positive sequences, labeled with FunSoCs, and virulence-negative sequences from SwissProt. 38 The majority vote classifier outperformed individual classifiers, achieving high precision (0.90) and recall (0.82) values, with 1.0 indicating full precision or recall.
Pathogens from different taxonomic kingdoms sometimes use similar infection mechanisms. ML approaches that predict virulence could aid taxonomic-agnostic pathogen detection. However, SeqScreen’s taxonomic gene classifiers generated more false positives when specific pathogen data were removed from the reference database. Moreover, because many nonpathogens contain threat-associated genes and SeqScreen assigns threat scores to individual sequence reads, an abundance of false positives can arise from nonthreat organisms. Further refinement is needed for practical application to real-world metagenomic samples. Applicability could be expanded by incorporating more fine-grained virulence types, including FunSoCs for fungal sequences, training on multiorganism samples, and reducing reliance on sequence databases, perhaps by adapting current models to make predictions directly from sequence data.
DL Embeddings for Prediction of Protein Characteristics and Function
Case Study: DL Classification of Function From Protein Sequence
Protein structural data often aids function prediction, but most proteins lack such data. Methods that predict protein function from sequence data alone would therefore be valuable for agnostic biodetection. The developers of FUTUSA (function teller using sequence alone) 39 evaluated function prediction by inputting whole and segmented protein sequences, finding that segmentation produced optimal results (Figure 3B). Using data on oxidoreductases, acetyltransferases, and demethylases, they tested how simulated point mutations affect function prediction. For segmented sequences, mutations had the largest impact on function prediction scores for segments overlapping important functional regions. Scores for unsegmented input sequences remained unchanged, even with mutations in critical regions. FUTUSA, based on segmented sequences of size 64 amino acids, demonstrated improved performance over Protein Basic Local Alignment Search Tool (BLASTP) in all categories for at least 1 of 5 assessed metrics.
Another DL model, 40 ProtCNN, assigns existing Pfam annotations to full-length protein domains, which could aid in predicting Pfam class, inferring coverage, and discovering protein families. The DL study used unaligned domain amino acid sequences from 17,939 classes from Pfam as input, added annotations to approximately 6.8 million sequence regions, and predicted the function of 360 human reference proteome proteins with no previous Pfam annotation. Other promising DL models included ProtENN and ProtREP.
DL models can predict function directly from raw, unannotated protein sequences. Advantages include reduced need for structural information, greater availability of training data, and prediction of functionally essential protein regions using simulated mutations. Limitations include not capturing hierarchical relationships of functional assignments and ignoring prior knowledge of segmented regions and their importance in protein function.
Assessing differences between expected and observed contributions to whole protein function and multicall classification could improve the model. Finally, applying the Pfam annotation platform for biodetection would require further development to categorize additional pathogenic classes.
Case Study: ProteinBERT
The applicability of DL models for proteins extends beyond function-from-sequence predictions. ProteinBERT, 7 based on bidirectional encoder representations from transformers (BERT), 41 is a deep language model (DLM) for learning global and local feature vector representations of protein sequences of any length, which could be used for predicting protein characteristics, such as posttranslational modifications, structure, or protein–protein interactions. With an attention-transformer architecture, ProteinBERT was pretrained on a large unlabeled dataset (106 million proteins from UniRef9037 and 8,943 Gene Ontology annotations) and then fine-tuned. During pretraining, a percentage of input sequences and Gene Ontology annotations were “corrupted” to ensure the model could recover the “uncorrupted” versions. Smaller and faster than models with more parameters, ProteinBERT achieves comparable accuracy.42,43 Current applications include providing input to protein–protein interaction graph neural networks (NNs), 44 predicting protein toxicity, 45 and designing proteins 8 and antibodies. 46
ML models with an attention or transformer architecture can effectively predict protein function but can require extensive design. They are portable and can be fine-tuned for specific tasks after 1 pretraining step. Ideally, the subjects of the original and new tasks would be similar enough that knowledge gained from the original task would also be relevant for the new task. However, fine-tuned models could be prone to poor performance if transferable information is insufficient. Designed for sequence-like data, these models are often alignment independent. Uses could include identifying remote homology and predicting function for distantly related sequences.
Like the DL models described earlier, ProteinBERT does not capture the hierarchical nature of Gene Ontology functional annotations. Additionally, applying DLMs to larger contexts is challenging; so-called “language of life” tasks are currently nonviable. Predicting gene clusters, metabolic networks, protein complexes, and pathway functions would represent a breakthrough for DLMs. Current efforts to predict pairwise protein interactions 44 and immune system gene function 47 seek to address this gap.
Case Study: Deep Embeddings to Understand Microbial Protein Space
Some ML algorithms learn and output “embeddings” (Figure 2). Embeddings are low-dimensional, ordered lists of numbers that reflect key (though often abstract) qualities of the original objects, such as proteins. ML algorithms determine embeddings from the input data in such a way that the more similar 2 objects are with respect to those key qualities, the more similar their embeddings. Embeddings can then be used in downstream analysis, such as clustering or visualization.
For proteins, embeddings can describe characteristics related to protein structure and function in entire microbiomes. For instance, a 3-layer bidirectional long short-term memory (BiLSTM) model was trained with microbial protein sequences from the Unified Human Gastrointestinal Genome catalog 48 and used to generate embeddings for nontraining set proteins. 49 Fed an input protein sequence, the model outputs a vector representation that summarizes the sequence and encodes structure and function features.
The model was validated using bacterial SwissProt database proteins 38 to ensure it could recapitulate known protein properties and relationships, with deep embeddings scoring as well as, or better than, other methods. On label recovery tests, the F1 score for deep embeddings ranged from approximately 0.5 to 0.9, compared with approximately 0.1 to 0.95 for other methods. Model developers visualized SwissProt bacterial protein dataset embeddings using the Uniform Manifold Approximation and Projection method and found that embeddings clustered around labels defined by the Kegg Orthology ID. 50
Embeddings obtained from the BiLSTM model accurately describe the 3D structure and function of microbial proteins, and the model could apply to a broader protein set if trained on a more comprehensive dataset. By avoiding sequence alignments, this DL approach overcomes a limitation of traditional homology-based methods51,52 and enables inference for novel sequences. Functional clustering in the Uniform Manifold Approximation and Projection visualizations could reveal how novel and known proteins relate functionally. Though initial model training is computationally expensive, creating and analyzing new embeddings is fast and efficient.
DNNs for Taxonomic Classification of DNA Sequences
BERT is useful for proteins 7 and DNA. BERTax, 53 based on BERT, 41 contains additional NN layers and can classify DNA sequences’ taxonomic superkingdom and phylum using natural language processing. Because it does not rely on reference genomes, BERTax can make predictions for sequences without close relatives in existing databases. BERTax performs comparably to current methods when related sequences are in the training data, and it outperforms on novel sequences.
BERTax was trained with unsupervised pretraining to learn “DNA language” structure, followed by fine-tuning to learn to predict taxonomic classes. The pretraining data included approximately 2.5 million genomic fragments of 1,500 nucleotides from across the 4 superkingdoms with a sequence similarity constraint of at least 80%.
BERTax is reference-independent, making it inherently pathogen agnostic and more generalizable. Consequently, it can make better predictions for metagenomic samples and novel DNA sequences. Yet it can also be combined with database approaches to capitalize on available information. However, only classifying superkingdom and phylum is a significant limitation; further taxonomic refinement would make BERTax more useful. Because BERTax requires significant training time and runs slower than other DL methods, one might first use a high-throughput method and pass only unassigned or low confidence sequences to BERTax.
DL, Image-Based Approach to Classify Fungi
DL can be used for image data as well as sequence data. An ML approach to classify microscopic fungi images was developed based on DNNs and bag-of-words, 54 and it is faster and cheaper than traditional techniques that involve human visual inspection and biochemical tests. The method extracts image features using steps from previously trained DNNs, clusters and aggregates the features to reduce dimensionality, and classifies original images into fungal species using a support vector machine or random forest model. 54
To test the DL classifier, 180 preprocessed microscopic fungal scans from the Digital Images of Fungus Species database were used. Data was partitioned by sample preparation, and parameters were optimized through 5-fold cross validation. The Fisher vector representation outperformed bag-of-words, and classification accuracy was approximately 75% to 97% for all but 2 species (accuracy 50% to 60%). The DL classifier struggled with samples displaying high variation in arrangement, appearance, and preparation. 54
By reducing the need for sequencing, image-based DL techniques could decrease sample classification time and cost. This preliminary study shows proof-of-concept for repurposing DNNs and coupling them with flexible feature representations encapsulating varied data, though further developments are required to fully realize these benefits. Poor performance on highly variable samples might foreshadow difficulty translating to biodetection contexts. Expanding the Digital Images of Fungus Species database to include images of higher-resolution, multiple coexistent fungal species and other sample preparation protocols would enhance applicability and performance.
Determining virulence from images could boost relevance, since threat estimation could be more informative than pathogen identity. While the potential gains from an image-based DL method are high, the example classifier is limited by reliance on traditional cell culture. Nevertheless, the current implementation could identify samples requiring sequencing and further analysis.
ML for Spectral Data
Mass spectrometry (MS) analysis of proteomic or metabolomic factors holds promise for phenotype prediction and diagnosis and is explored broadly in clinical settings.55-59 Pathogen detection via MS is well studied for emerging viral diseases 60 but requires intensive curation. Classifying directly from raw MS data using DL models, 61 especially those designed for natural image classification, 62 might overcome these hurdles.
Case Study: MS Analytics for Sample Categorization
In an article published in 2021, 62 researchers transformed MS profiles into images and encoded them into feature vectors, which they used in logistic regression, support vector machine, random forest, and gradient boosted trees for classification (Figure 4A). They used multiple publicly available image models that had been pretrained on “natural images.” 63 Classifiers predicted whether MS profiles from cancer tissue biopsies were derived from a malignant tumor with peak performance of 0.876 area under the receiver operating characteristic curve.
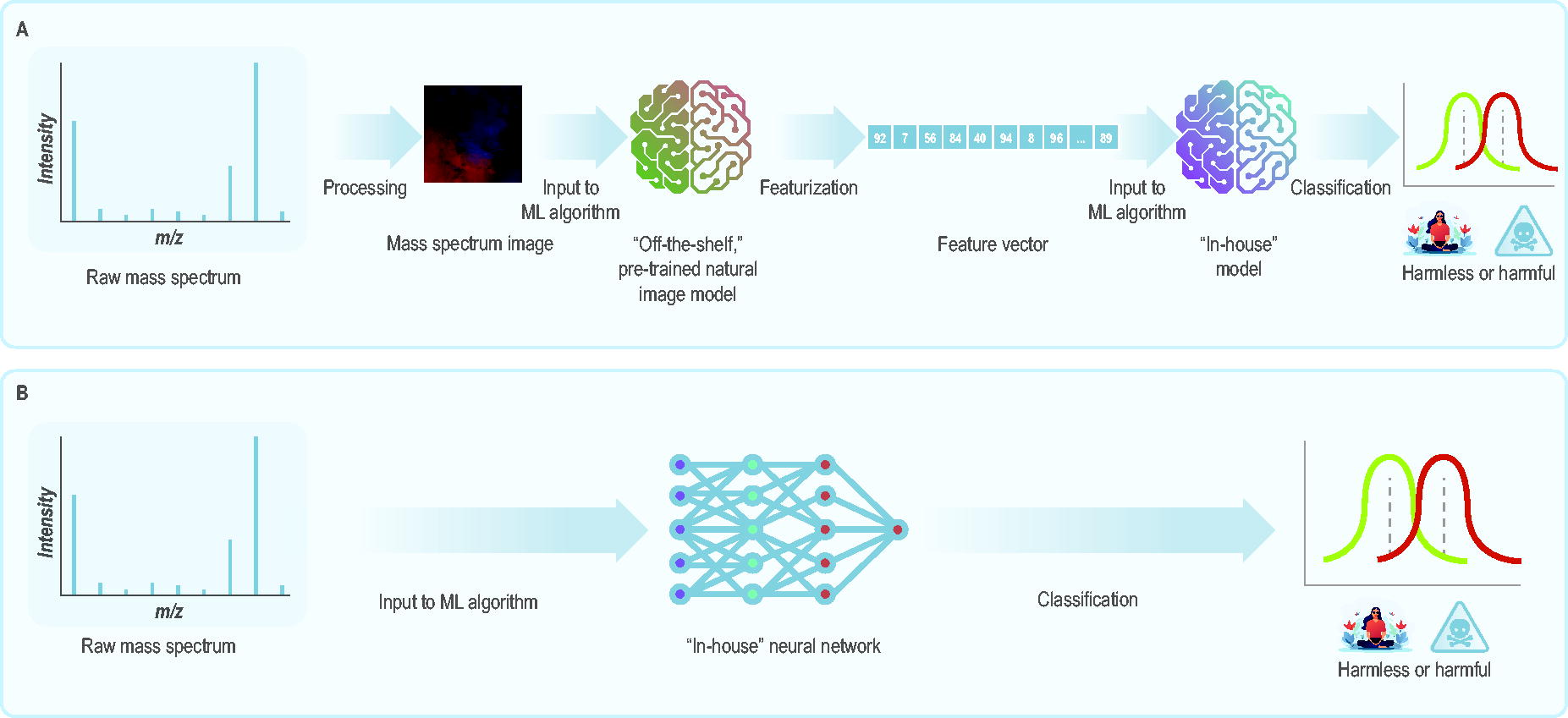
(A) Schematic of the SWATH-MS-based pipeline. 62 The pipeline could be used to distinguish between harmless and harmful samples based on their mass spectra, though those spectra first need to be processed into images before they can be ingested into the machine learning workflow. (B) Schematic of the DLearnMS pipeline. 64 The pipeline consists of a neural network that distinguishes between 2 sets of samples (eg, harmless vs harmful, healthy vs diseased) based directly on their raw mass spectra. Abbreviations: ML, machine learning; SWATH-MS, sequential window acquisition of all theoretical mass spectra.
These analyses show that public pretrained models can generalize to MS image analysis, despite being pretrained on different image classes. Further development of DL models designed and architected for proteomics applications could enhance biodetection by identifying signature pathogen proteins. However, large data storage solutions would be necessary.
Case Study: DL Approach to Detect Biomarkers in LC-MS Proteomics Data
DLearnMS is an NN designed to classify liquid chromatography-mass spectrometry (LC-MS) maps as diseased or healthy with minimal preprocessing (Figure 4B). 64 Lacking experimental data, researchers simulated the training spectra. From UniProt, they randomly selected 20 human proteome peptides for the “healthy class,” then added 9 spiked peptides for the “diseased class.” The DLearnMS algorithm relies on class labels and employs layer-wise relevance propagation for network and feature selection, detecting differentially abundant peaks as biomarkers. Published LC-MS data were used as a benchmark to evaluate performance. DLearnMS recovered 7 of the 9 spiked peptides, detected fewer false positives than other methods, and required less preprocessing.
Because DLearnMS uses raw MS, it avoids information loss and dimensionality reduction, increases interpretability, and minimizes steps between data collection and model prediction. ML techniques developed on synthetic data require quality assurance to ensure transferability to real-world data. Overfitting is a risk since the distinguishing elements in a synthetic dataset are known. Given this study’s limited scope, it is unclear if DLearnMS could be used for high-throughput applications. Finally, because DLearnMS is based on observing differences between 2 data classes, the model might need to be retrained for environmental samples, which would have different background signals than clinical samples.
Case Study: Raman Spectral Processing for Pathogen/Toxin Detection
Image-based DL is also applied to Raman spectra for clinical diagnostics 65 and holds potential for agnostic biosurveillance. Surface-enhanced Raman scattering, for example, has been used for environmental and foodborne pathogen detection 66 and to predict multidrug-resistance profiles in nosocomial pathogens. 67 A CNN model was trained with 2,000 bacterial Raman spectra to distinguish 30 microbial pathogens. 68 Support vector machine evaluation of Raman spectra identified bacterial toxins and their concentrations from spectral data. 69 While most studies combining Raman spectroscopy (RS) and ML (RS+ML) have leveraged ML for dimensionality reduction, classification, validation, regression, and clustering, others applied uncommon or novel methods.70-81
Current approaches require spectral preprocessing to remove background signal. 82 Dimensionality reduction is often performed before model training, resulting in model features that are composite quantities. Full Raman spectra can contain complex patterns, making it difficult to determine individual structural components in a sample, but analyzing minimally preprocessed data with ML increases available information.
RS+ML performs well in biomedical settings. 65 However, most studies used small sample sizes and/or did not validate results. Model performance varied by biological sample and choice of ML method, so RS+ML analyses must first be standardized and optimized if they are to be reliable in biodetection settings. Additional research is needed on samples with complex environmental backgrounds. Indeed, differences in growth conditions and sample preparation affect an organism’s mass and Raman spectra, presenting a major challenge for agnostic biodetection.
Transcriptional Signatures of Infection
The preceding methods were designed to identify and characterize pathogens. Alternative, host-based strategies could provide complementary information and unique advantages. For example, transcriptional signatures in host cells could be used to diagnose infection. 83
A 2022 meta-analysis 83 evaluated published host signatures of infection84-92 against 17,105 Gene Expression Omnibus 93 transcriptional profiles from whole blood cells and peripheral blood mononuclear cells. The profiles included bacterial and viral infections, plus some parasitic infections and noninfectious conditions. To score different signatures, the authors devised a standardized framework that relates gene expression levels for positive and negative genes associated with the signatures. Most signatures were robust at detecting viral or bacterial infection with median area under the receiver operating characteristic curve values greater than 0.7, though some signatures demonstrated cross-reactivity with unintended infection types.
By identifying nascent infections, host transcriptional signatures could enable earlier and broader detection and reduce diagnosis time. While host-based approaches might struggle to identify individual pathogens, this loss of specificity might not be a drawback for pathogen-agnostic detection.
The potential value in host-based techniques is clear, but their feasibility for complex, pooled, or environmental samples is undetermined. This scoring system was designed for biomedical applications but could be extended if the environment is treated as the “host” (Figure 5). There could be nonmicrobial environmental signatures that reflect the presence of a given pathogen or class. The applicability range for such an indirect signature system is unclear, but the approach should be considered, as it is inherently pathogen agnostic.
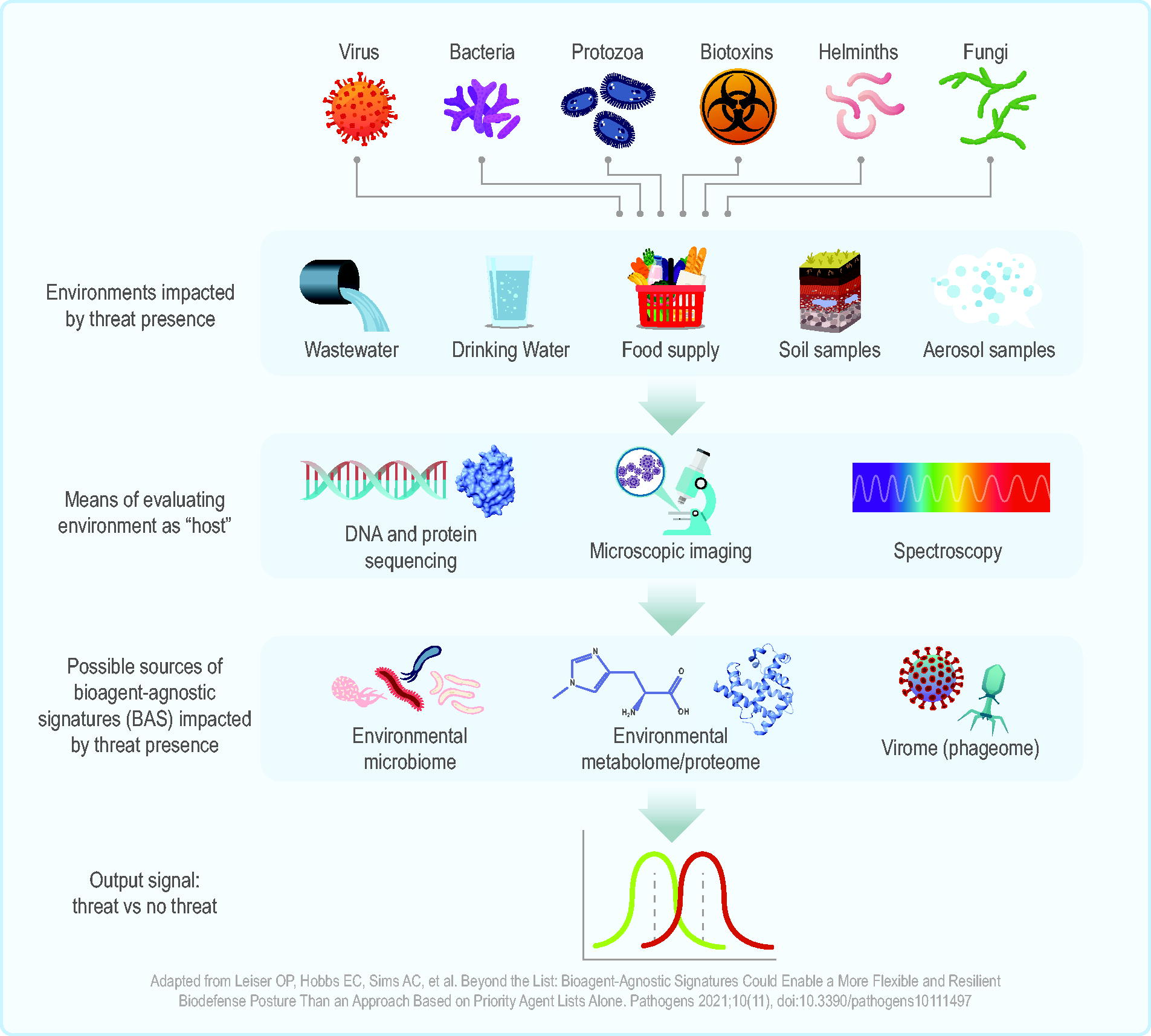
Conceptualization of host-based signatures in an environmental context. A range of potential bioagents could impact various aspects of our environment, from the soil and air to our wastewater and food and water supply. These impacts could be evaluated with analytical techniques such as sequencing, imaging, and spectroscopy. The data from these assessments could be used to identify bioagent-agnostic signatures that differ based on threat absence or presence. Adapted from Leiser et al. 2 Abbreviation: BAS, bioagent-agnostic signatures.
Discussion and Conclusions
Discussions about nontargeted and One Health assays for outbreak preparedness94-96 highlight a clear need for agnostic diagnostics. Although environmental biodetection technologies are less common, ML platforms pretrained for clinical/biomedical use could be adapted for biosurveillance of environmental threats. We examined strengths and weaknesses of ML biodetection approaches with varying input data type and format, ML algorithms, use cases, performance, and maturity level. The unifying theme is identification via raw or minimally preprocessed data, with the aim of achieving untargeted detection or diagnostics readouts.
The systems’ framework depends on models capable of classifying input data and effective methods for learning representations of these datasets. The latter supports reference independence, as flexible representations could capture threat function even if the encoding of that function does not exist in reference knowledgebases.
The DL approach for classifying fungi based on microscopic images 54 avoids cell culture but requires more training data to bolster performance and reliability. Recently developed DL models for bacterial image classification were trained on thousands of images.97,98 Alternatively, if insufficient training images are available, models pretrained on nonmicrobial images could be leveraged. A recent model for classifying bacterial images as Gram-positive or Gram-negative was pretrained on the ImageNet-1k dataset and then retrained on bacterial images. 98
Generalizability hurdles pose another challenge for agent-agnostic biodetection. Given the high resolution of RS and the variability in performance of different combinations of biological samples and ML methods, specific use cases might need a specialized approach. The development cost and delay could render these methods less suitable. Instead, if spectral signatures for threat function could be identified using existing models, focusing on these more generalizable features could aid first-pass threat identification, with further assessments reserved for characterizing potential threats.
FUTUSA 39 and SeqScreen 35 both predict protein function from sequence, though SeqScreen’s approach seems more mature. In contrast to FUTUSA, SeqScreen provides well-defined functional labels, incorporates manual data curation, combines several functional annotation algorithms, and assigns both taxonomic and functional labels. However, FUTUSA, unlike SeqScreen, can predict the functional impact of point mutations.
Reference-independent prediction of unknown protein function is appealing. While initial training requires reference sequences, the resultant embeddings and classification model can assign functions to previously unseen sequences and are more portable than reference databases. Because BERTax 53 operates independently of reference genome databases, it could characterize unknown DNA sequences when other approaches fail.
Host-based strategies are also appealing because they would be inherently pathogen agnostic. Host signatures could be more robust at detecting pathogens, since host response patterns may be “evolutionarily conserved” across “a wide range of pathogens and toxins that elicit disease.” 2 By defining molecular host pathways of disease and assessing how a pathogen would disrupt those pathways, researchers could discover unknown pathogens and functional categories that SeqScreen 35 might not capture.
DL-based tools 99 can already predict protein structure and could also potentially predict function. It could be worthwhile to explore improving performance and generalizability of large language models, like that used in ProteinBERT, 7 given their success in other domains and the language-like nature of protein sequences. As frameworks like large language models become larger and more complex, their capacity to serve as pretrained models that can be fine-tuned for biodetection will be amplified.
Currently, no single algorithm or model addresses all biosurveillance needs. Using complementary platforms together could help overcome limitations and enhance potential (Figure 6). For example, platforms operating on processed 62 and unprocessed 64 mass spectra could be paired. DLearnMS64 could be used to identify detailed and interpretable distinguishing features between healthy and diseased samples, which could then inform high-throughput, multisubject analysis from the SWATH-MS (sequential window acquisition of all theoretical mass spectra)-based platform (Figure 6A). 62
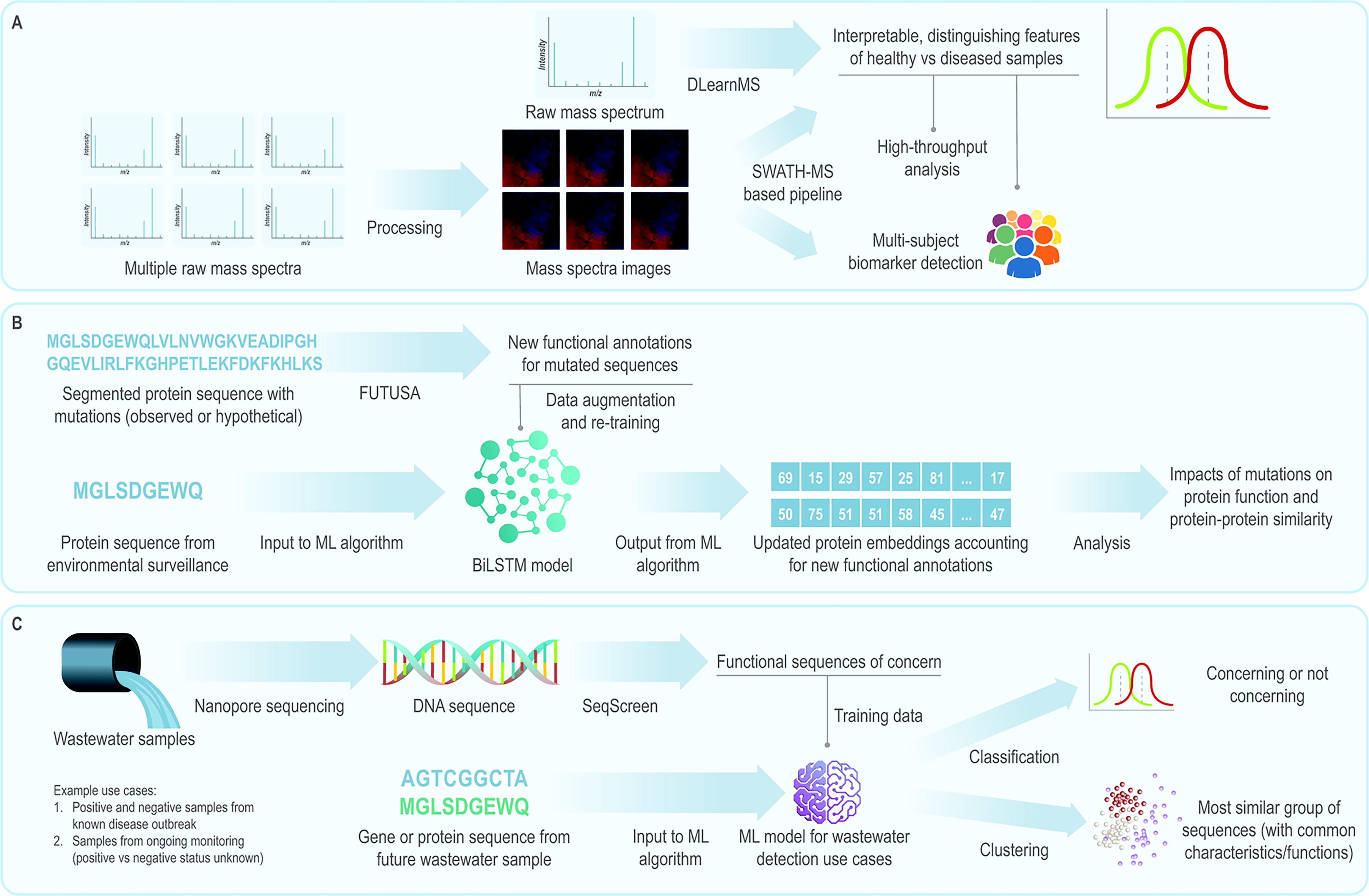
(A) Schematic illustrating how the DLearnMS and SWATH-MS-based pipelines62,64 could be applied in tandem. (B) Schematic illustrating how the FUTUSA 39 and BiLSTM model 49 pipelines could be applied in tandem. (C) Schematic illustrating how nanopore sequencing and SeqScreen 35 could be used in tandem. Abbreviations: BiLSTM, bidirectional long short-term memory; FUTUSA, function teller using sequence alone; ML, machine learning; MS, mass spectrum; SWATH-MS, sequential window acquisition of all theoretical mass spectra.
Coupling FUTUSA 39 and BiLSTM for protein embeddings 49 could be fruitful for probing protein function (Figure 6B). FUTUSA could first be used to create new functional annotations for mutated protein sequences. Those new annotations could then be used to augment the training data set and re-train the BiLSTM model. Updated embeddings could be generated from the re-trained BiLSTM model and used to assess protein sequences from environmental surveillance, potentially giving insight into the effects of mutations on protein function and protein-protein similarity.
Similarly, combining nanopore sequencing and SeqScreen 35 could enhance their value (Figure 6C). Nanopore sequencing could be performed on wastewater samples to generate input sequences for SeqScreen, which could identify FunSoCs. Those identifications could then be used to train a ML model to characterize future wastewater samples or classify them as concerning or not concerning.
Although future biodetection needs are complex and uncertain, the assessments above indicate that ML capabilities are well positioned to address emerging challenges (Figure 7). Efforts to extend and synergize such platforms are still needed to bring their potential utility to practice.
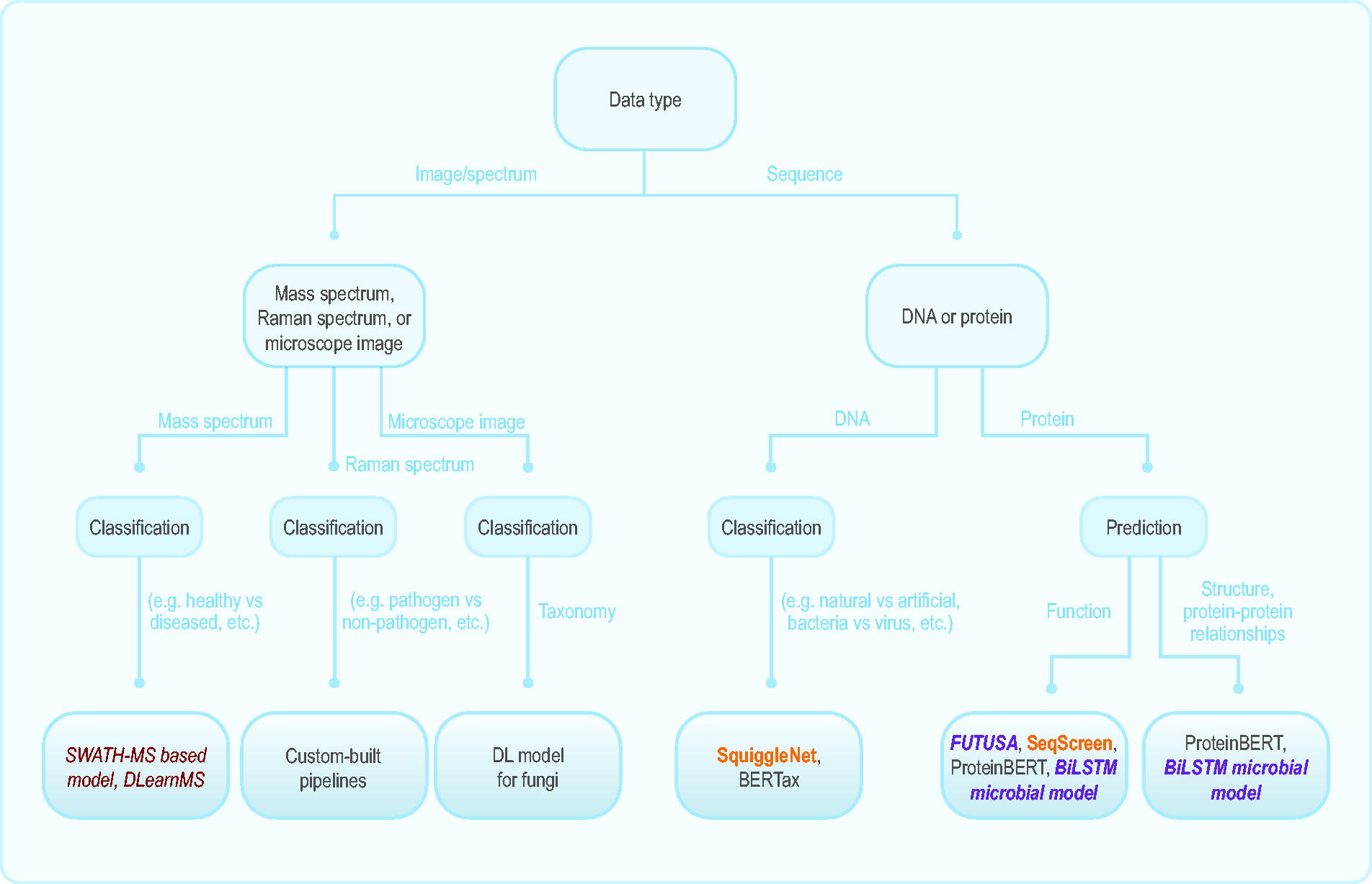
Decision flowchart to aid in identifying the most appropriate pipeline(s) to use based on the available input data and desired task. Pairs of pipelines in orange, red, and purple could be used complementarily, as described in the text and depicted in Figure 6. Abbreviations: BiLSTM, bidirectional long short-term memory; DL, deep learning; FUTUSA, function teller using sequence alone; ML, machine learning; SWATH-MS, sequential window acquisition of all theoretical mass spectra.
Moreover, to bolster confidence in risk evaluation and minimize false positives, evidence from multiple algorithms should be weighed before acting. Thus, an integrated, complementary approach to implementing these technologies will likely best serve the goal of agnostic biosurveillance.
Footnotes
Acknowledgments
We thank Monica Borucki for helpful discussion and feedback, Melanie Mendez for editorial assistance, and Jeremy Turner for graphic design support. We also thank the leadership and team members of the Department of Homeland Security Science and Technology Directorate (DHS S&T), Hazard Awareness and Characterization Technology Center (HAC-TC) for helpful discussion and review. Molecular graphics were created with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the National Institute of Allergy and Infectious Diseases Office of Cyber Infrastructure and Computational Biology. This work was performed under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. This effort was funded by DHS S&T HAC-TC contract number 70RSAT23KPM000036.