
Abstract
The need for well-controlled clinical trials is fundamental to advancing medicine. Care should be taken to maintain high standards in trial design and conduct even during emergency medical events such as an infectious disease outbreak. In 2020, SARS-CoV-2 emerged and rapidly impacted populations around the globe. The need for effective therapeutics was immediately evident, prompting the National Institutes of Health to initiate the Adaptive COVID-19 Treatment Trial. The Special Pathogens Research Network, made up of 10 Regional Emerging Special Pathogens Treatment Centers, was approached to participate in this trial and readily joined the trial on short notice. By trial closure, the Special Pathogens Research Network sites, making up 19% of all study sites, enrolled 26% of the total participants. The initial resources available and experience in running clinical trials at each treatment center varied from minimal experience and few staff to extensive experience and a large staff. Based on experiences during the first phase of this trial, the Special Pathogens Research Network members provided feedback regarding operational lessons learned and recommendations for conducting future studies during a pandemic. Communication, collaboration, information technology, regulatory processes, and access to resources were identified as important topics to address. Key stakeholders including institutions, institutional review boards, and study personnel must maintain routine communication to efficiently and effectively activate when future research needs arise. Regular and standardized training for new personnel will aid in transitions and project continuity, especially in a rapidly evolving environment. Trainings should include local just-in-time training for new staff and sponsor-designed modules to refresh current staff knowledge. We offer recommendations that can be used by institutions and sponsors to determine goals and needs when preparing to set up this type of trial for critical, short-notice needs.
History and Need for Clinical Trials During an Epidemic
New and neglected pathogens with outbreak or pandemic potential continue to emerge due to several factors, including increased global expansion and world travel over the past several decades.1,2 The most notable recent example is the ongoing COVID-19 pandemic caused by SARS-CoV-2. Within a few months after it first emerged in late 2019, the virus had spread across the globe, leading the World Health Organization (WHO) director general to officially declare the outbreak as a pandemic on March 11, 2020. 3
This is not the first time a novel virus has emerged and rapidly taken hold in the human population, (eg, HIV). Many attempts failed to find effective treatments until a double-blind, randomized controlled trial (RCT) demonstrated evidence for a possible therapeutic. 4 In 1987, zidovudine (also known as azidothymidine or AZT) became the first therapeutic approved by the US Food and Drug Administration for HIV. Trials addressing HIV faced a set of challenges that have been seen again in more recent trials for emerging infectious diseases. These include differing and evolving standards of care across trial sites, an unknown disease trajectory, patients seeking alternative medications, and stigma that comes with a diagnosis. Experiences from early HIV trials have informed more recent clinical trial design to try to eliminate some of these challenges.
Since 1976, numerous outbreaks of Ebola virus disease (EVD) have caused substantial challenges to conducting clinical trials and communicating the need to perform robust clinical trials. During smaller outbreaks in Uganda, South Sudan, and the Democratic Republic of the Congo, many therapeutics were used, largely at providers' discretion and without rigorous study. Unfortunately, without the use of well-designed clinical trials and data reporting, it is impossible to determine whether any real clinical benefit exists or to appropriately gauge the potential risks of untested therapeutics. 5 The 2014-2016 West African Ebola outbreak offered an unprecedented opportunity to test candidate therapeutics at scale. Many trials were initiated, but only a few were run in a rigorous enough manner to glean actionable data. The JIKI trial tested the antiviral favipiravir as a proof-of-concept for the utility of a multicenter nonrandomized trial. 6 Due to the lack of a control arm, limited conclusions could be drawn regarding the safety and efficacy of the treatment; however, the study shed light on several previously unknown components of the disease course and demonstrated the feasibility of running a clinical trial during an epidemic and in a limited-resource setting. A second trial, the Partnership for Research on Ebola Virus in Liberia II (PREVAIL II), used an RCT design to compare the efficacy of the monoclonal antibody cocktail ZMapp plus standard of care with standard of care alone. 7 Unfortunately, the trial was unable to enroll the desired number of subjects due to the trial initiating too late in the course of the epidemic. Consequently, the trial failed to reach the predetermined statistical cutoff point to indicate superiority of the antibody cocktail over standard of care alone. The treatment was retested in a later outbreak in eastern Democratic Republic of the Congo in 2018, along with 3 other treatments. 8
Quick deployment and testing of 4 therapeutics was made possible by the development of an ethical framework by WHO known as the Monitored Emergency Use of Unregistered Interventions. Using this framework, a panel of experts compiled a high-priority list of therapeutics that had the potential to be efficacious against the Ebola virus based on in vitro, animal model, and clinical data. High-priority lists such as this need to be established for pathogens with outbreak potential to rapidly determine lead candidates for clinical trials. In the case of the COVID-19 pandemic, the International Society for Influenza and other Respiratory Virus Diseases discussed at a conference in 2018 potential therapeutics against respiratory viruses, including the use of remdesivir as a broad-spectrum antiviral for treatment of Middle East respiratory syndrome coronavirus and severe acute respiratory syndrome. 9 The available in vitro data against related coronaviruses, along with safety data produced during EVD outbreaks, showed the potential utility of remdesivir in treating a new coronavirus.
Performing a high-quality clinical trial is a substantial challenge by itself and running one amid an infectious disease epidemic involves the same robust attention to detail as any other clinical trial, with the added component of time sensitivity and an increased need for emphasis on participant and researcher safety. Several ethical considerations must be addressed, including assurance that response efforts are not hindered, extensive collaboration both locally and internationally, rapid and accurate dissemination of results, equitable recruitment of participants, and use of a placebo arm. These are not new considerations for clinical trials, but they have the potential to be overlooked in an emergency situation. In addition, due to the unknown nature of an emerging infection, the ability to share samples and data can be a vital resource. Trial participants must be made explicitly aware of the possibility for their samples and data to be shared across the research community, presumably with personal identifying information removed, and given a choice to opt out of sample or data banking. WHO has published several documents relating to ethical considerations in performing research during infectious disease outbreaks and public health emergencies.10,11
Some pathogens result in small and/or sporadic outbreaks that do not generate a large enough sample size to draw statistical conclusions. For these pathogens, the efficacy of a given therapeutic can be tested across multiple outbreaks effectively and expeditiously using an adaptive protocol, such as those developed by WHO 11 and those described by Dean et al 12 and Dodd et al. 13 A core protocol can enable a clinical trial to extend across multiple outbreaks, thus enabling accommodation for the changing dynamics. An important factor to consider is a definition for standard of care, especially when a trial is expected to take place across several institutions and even several countries. It is expected that the standard of care may evolve over time, and this must be addressed in both study design and in the statistical methods used to compare multiple sites. Robust engagement from researchers and stakeholders at all levels of healthcare delivery systems is vital in determining the primary research questions, trial endpoints, and major design elements.
Creation of SPRN and ACTT
The Special Pathogens Research Network (SPRN) was established in November 2016 by the National Emerging Special Pathogens Training and Education Center (NETEC), with a mission to create and sustain the infrastructure necessary to conduct multicenter clinical research to improve patient care and outcomes, reduce nosocomial transmission, safely collect critical biospecimens and rich clinical metadata, and expand knowledge of existing and newly emerging special pathogens. SPRN represents a collaboration across 10 Regional Ebola and Other Special Pathogen Treatment Centers (RESPTC), academic institutions, biosafety and research laboratories, and federal partners to develop an infrastructure for rapid deployment and coordination of research studies for high-consequence pathogens in the United States. 14 While not all RESPTC biocontainment units had extensive clinical trials experience, all had executed at least 1 special pathogen expanded access protocol before the COVID-19 pandemic. All clinical enrollment sites are set up under the auspices of large regional patient referral centers and have explicit relationships with large academic medical institutions, which enables them to leverage the clinical trials experience of their broader institutions. In some cases, individual RESPTCs consist of multiple hospitals or healthcare facilities that could potentially serve as additional study sites, expanding the reach of the network.
To facilitate conduct of research across this network, SPRN established a single institutional review board (sIRB) at the University of Nebraska Medical Center (UNMC) with interinstitutional reliance agreements with the other 9 RESPTCs. The proactive establishment of the network and collaboration to ensure all sites would be prepared to act in case of an outbreak enabled a rapid response when the COVID-19 pandemic began.
The first clinical trial protocol SPRN hosted across the network in response to the COVID-19 pandemic was the Adaptive COVID-19 Treatment Trial (ACTT), which was led by the National Institutes of Health. The first phase (ACTT-1) (ClinicalTrials.gov identifier: NCT 04401579) assessed the efficacy and safety of the antiviral drug remdesivir versus placebo to treat COVID-19 illness. 15 Enrollment within SPRN began within 30 days of the first case of COVID-19 diagnosed in the United States. All 10 RESPTCs, some with multiple enrollment sites, joined this international collaborative effort over the short time frame of 32 days (Figure), and the trial was fully enrolled across 60 US and international sites in 58 days (see Table 1 for enrollment numbers for each phase of the ACTT trial). SPRN, which made up 27% of all sites, contributed substantially to the rapid patient accrual, enrolling 29% of the total ACTT-1 study participants. The second phase (ACTT-2) studied the additional impact of adding baricitinib, a Janus kinase inhibitor to remdesivir compared with remdesivir alone. 16 SPRN, which made up 18% of the total sites, contributed an outsized portion, enrolling 28% of the total volunteers, helping the protocol to complete full enrollment rapidly in 53 days. ACTT-3 studied the effectiveness of interferon beta-1a in conjunction with remdesivir compared with remdesivir alone. In ACTT-3, SPRN, making up 19% of all trial sites, also maintained 28% of the total enrollment across all sites (Table 1). 17 In the final stage, ACTT-4, dexamethasone plus remdesivir versus baricitinib plus remdesivir was evaluated for the treatment of patients with severe COVID-19. SPRN sites contributed 19% of the 1,010 subjects enrolled in this study, making up 12% of all sites involved. In all, the preparedness and collaboration of the SPRN network enabled rapid deployment of the ACTT studies: over the course of the 4 stages of ACTT, SPRN made up 19% of the total sites and enrolled 26% of the study population. The ACTT study data resulted in 2 medical countermeasures being recommended for treatment of COVID-19. 18
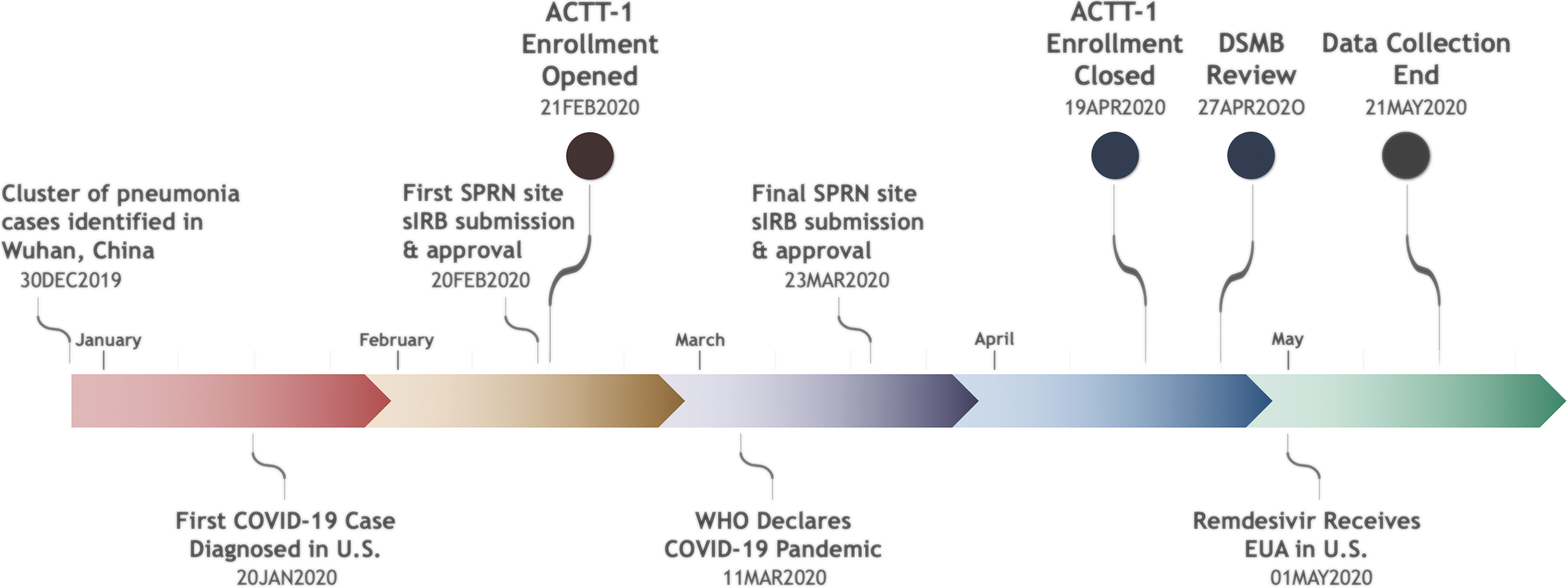
months of the COVID-19 pandemic. Abbreviations: ACTT, Adaptive COVID-19 Treatment Trial; DSMB, Data and Safety Monitoring Board; EUA, emergency use authorization; sIRB, single institutional review board; SPRN, Special Pathogens Research Network; WHO, World Health Organization.
Special Pathogens Research Network Enrollment Contributions
Total subject enrollment across the 10 NETEC RESPTCs that make up the SPRN and percent of total enrollment. Some regional centers included multiple study sites.
Abbreviations: ACTT, Adaptive COVID-19 Treatment Trial; NETEC, National Emerging Special Pathogens Training and Education; RESPTC, Regional Ebola and Other Special Pathogen Treatment Center; SPRN, Special Pathogens Research Network.
Clinical Trial Landscape Across SPRN Sites
The National Institute of Allergy and Infectious Diseases (NIAID), which sponsored the study, hired a contract research organization to manage communications across the entire study network, set up an internet platform for enrollment tracking, manage the database, and collect case report forms and laboratory specimens. The sponsor provided information to the network sites through the contract research organization in between group calls, and directly to study sites on regular leadership calls. SPRN leadership then had internal calls with its study site investigators and teams and sIRB. The IRB communications lead also shared information with the network teams directly. The robust recruitment of participants into the ACTT trials by SPRN shows that it is possible to contribute to a research trial regardless of program size, resources, or experience. In many cases, ACTT-1 was initiated by the special pathogen program at each SPRN site. Where appropriate, the conduct of ACTT-1 was transferred to experienced clinical trials teams elsewhere within the institution. To assess the program readiness and experience across SPRN sites at the start of ACTT-1, a discussion among the 10 SPRN sites of successes and challenges was documented, and a survey was distributed to the 10 site principal investigators to gather data retrospectively. This was determined not to be human subjects research by the UNMC IRB.
Most sites activated for ACTT-1 with only a small core team, half of which had only 1 to 3 dedicated personnel, and in some circumstances, only a single site investigator (Table 2). Access to study personnel was variable across sites and throughout the different trial phases. By the end of ACTT-2, 40% of the sites reported having 10 or more staff supporting the trial. Due to fluctuations in institutional guidelines regarding work assignments and personnel furloughs, turnover of personnel was high between studies. In some cases, workforces were bolstered by individuals reassigned from non-COVID-19 research that had been temporarily halted to support COVID-19 clinical trials. Many sites were able to train personnel who were reassigned from other duties early in the pandemic; however, as institutions resumed normal operations, those individuals were assigned back to their original positions. The need for more personnel was uniform across sites, although the specific staffing needs varied (Table 3).
Clinical Trial Staffing Availability, Across the SPRN Sites
Survey results from the 10 SPRN sites regarding the number of staff available at the start of ACTT-1, during ACTT-1 and ACTT-2, and at the completion of ACTT-2.l. Staff types could include site principal investigators, coordinators, nurses, laboratory technicians, data personnel, or others.
Abbreviations: ACTT, Adaptive COVID-19 Treatment Trial; SPRN, Special Pathogens Research Network.
Clinical Trial Resource Availability, Needs, and Experiences Across the SPRN Sites
Survey results from the 10 SPRN sites when asked about resources available at the start of ACTT-1. Team readiness was assessed through survey questions regarding familiarity with study procedures and the need for laboratory modifications.
Abbreviations: ACTT, Adaptive COVID-19 Treatment Trial; DMID, National Institute of Allergy and Infectious Diseases Division of Microbiology and Infectious Diseases; SPRN, Special Pathogens Research Network.
The majority of sites had the laboratory capabilities in place to handle specimens from a patient diagnosed with COVID-19. Some sites, however, were required to find laboratory space with biosafety capabilities to process these infectious samples; this required either certification of an existing laboratory space and training of laboratory personnel on personal protective equipment requirements or the use of another institutional laboratory space that was certified to handle positive COVID-19 specimens. For programs considering running clinical trials for emerging infectious diseases, a preexisting relationship with their institutional biosafety program and basic science researchers can expedite this process.
Most sites had previous experience running a clinical trial with the team used for ACTT (90%) but less than half had carried out a trial sponsored by the NIAID Division of Microbiology and Infectious Diseases (DMID) (Table 3). Sites with less experience relied heavily on guidance from those with more clinical trial experience (ie, both overall experience and DMID-specific experience). A single sponsor may require similar documentation and processes across various trials; therefore, the expertise from those having conducted DMID-sponsored trials in the past was invaluable. SPRN is in the process of developing tools and resources to assist sites that may not have a preestablished network to rely on for support. Even at sites that employed teams with extensive clinical trial experience, new situations emerged such as the prioritization of multiple ongoing clinical trials. Institutional prioritization strategies were discussed during SPRN calls and these options should be considered at individual institutions onboarding multiple clinical trials for a single disease (Table 4). Six sites reported in-house development of a prioritization schema, with 3 sites prioritizing double-blind RCTs over non-double-blind RCTs and Phase I/II trials as the lowest priority. One site reported trials funded by NIAID receiving priority over trials with other funding sources, and 2 reported all trials being prioritized equally. At sites where multiples studies were being conducted, 5 sites offered patients the opportunity to choose between studies while 3 sites had an oversight group in place to determine which trials patients would be offered. This landscape offers an overview of how the various network sites functioned throughout the various stages of ACTT. The successes and challenges that SPRN sites encountered while implementing and operationalizing the ACTT-1 and ACTT-2 trials are described as follows.
Prioritization of Clinical Trials Across the SPRN Sites
Survey results from the 10 SPRN sites when asked about the prioritization of clinical trials when institution was approached by multiple trial groups. Further questions addressed how potential participants were offered participation in a study if they were eligible for multiple studies. The number of sites providing a “yes” response to each question is indicated.
Abbreviations: DB, double-blind; NIAID, National Institute of Allergy and Infectious Diseases; RCT, randomized controlled trial; SPRN, Special Pathogens Research Network.
Lessons Learned in Clinical Trial Operations
Communication
Rapid and accurate communication during a pandemic is crucial and SPRN sites routinely noted their preference for more communication. Members of SPRN valued regular communication across sites with the sIRB and noted the support given by network leaders. SPRN scheduled twice-weekly conference calls, which included site principal investigators, relevant study personnel, a UNMC sIRB representative, and, when appropriate, sponsor representatives and federal partners. During these meetings, time was set aside for each site to provide an update on their hospital census, study status, and shared successes and challenges with enrollment and study procedures. Enrollment updates and challenges communicated during these calls helped prepare sites that had not yet experienced a surge in cases. These calls enabled sharing of ideas regarding process implementation, purchasing consumables, managing enrollment challenges, and other operational hurdles. As case counts decreased across the country, call frequency was reduced, with additional meetings added on as needed. This integrated team approach was a valued tool that likely enhanced SPRN-related ACTT enrollments. For future trials, such calls are paramount for keeping everyone involved aware of study activity changes. Similar calls could be handled by either telephone or video conferencing. In addition, establishing a listserv for internal notifications or a shared internet chat platform could be considered.
The UNMC sIRB ensured an open line of communication between sponsor and sites through their regular presence on SPRN conference calls. These calls enabled changes to processes, protocols, and other operational procedures to be disseminated quickly across all sites. All 10 SPRN sites were activated by the National Institutes of Health within 32 days of study start and all sites were approved by the IRB the same day, with only 2 exceptions (Figure). The sIRB provided flexibility and support to the sites and assumed responsibility, in conjunction with site principal investigators and regulatory leads, for ensuring all sites had the most up-to-date protocols and consent forms. This was accomplished by increased access to sIRB support staff via email and telephone during the onboarding process. In addition, the UNMC sIRB Research Support System used for IRB submissions and documentation has a messaging feature that enabled communications to be submitted directly in the system for routing to the appropriate personnel. Furthermore, the knowledge, expertise, tools, and checklists provided by the sIRB were noted as particularly helpful across the sites. New resources and checklists are available on the SPRN website. 19
An interesting challenge developed in ACTT due to the deployment of SPRN's sIRB. While most of the ACTT sites in the United States used a private sIRB with whom the DMID had contracted, SPRN sites used their own sIRB, thus creating 2 sIRBs reviewing trial protocols. The reason for using 2 separate sIRBs was a simple matter of timing and onboarding efficiency: due to preestablished contracts between the UNMC sIRB and RESPTCs, the unique flexibility of the UNMC sIRB rapid response protocols, and regular ongoing communication, SPRN was able to immediately begin protocol submissions and approvals. 20 While each sIRB worked effectively within its own network, the use of 2 sIRBs created some communication challenges as each organization had different processes and timelines, leading to delays and miscommunications. In some cases, individual sites received updated information from the sponsor regarding protocol and consent form version changes before the protocol lead was able to update the sIRB submission, leading to reported version control issues between SPRN sites and sIRB. One key implementation finding is the critical nature of the communication channel between all parties involved. SPRN sites noted that care should be taken from the sponsor to set up a consistent channel of communication and ensure the protocol lead and/or network lead are included in all communications.
Sites noted occasional delays in getting pertinent information, which could be improved in future network trials. In addition, a lack of uniform guidance from the 2 sIRBs and sponsors sometimes led to confusion. Furthermore, not all sites were at the same stage of enrollment and the needs for these sites were dependent upon where they were in the process. As the trial continued into other iterations (ie, ACTT-2, ACTT-3, ACTT-4), some of the study teams changed. Occasional delays in communicating this information to both the sponsor and the sIRBs led to outdated distribution lists. These issues are inherent to rapidly evolving study protocols, yet there are clear opportunities to improve processes between the sites, the IRBs, and the sponsor. For example, each site should appoint a single point of contact who receives and distributes all communications from the sponsor and IRBs and ensures completion of required requests. This individual does not need to handle all requests but should be knowledgeable enough in the processes to accurately forward communications to the appropriate party.
Collaboration and Coordination
Collaboration between the sIRBs, sponsors, and SPRN sites was commendable and will serve as a good foundation for moving forward with future clinical trials. The ideal aspect for such collaboration lies in having a network “warm base,” with employees and systems already in place, operational, and working on other protocols routinely. This warm base enables preestablished, battle-tested lines of communication and a network of personnel already familiar with each other in advance of the crisis. For example, before the COVID-19 pandemic, SPRN had exercised a drill in which an investigational therapeutic was requested and delivered, thus providing an opportunity to evaluate strengths and improve weaknesses in this process. At the onset of the COVID-19 pandemic, SPRN has several collaborative activities ongoing, but not to the degree that a pandemic response would require.
As the trial began, sites took an all-hands-on-deck approach, given the urgency of the situation. This approach facilitated camaraderie and a willingness to do whatever it takes to combat the disease. Frequently, the challenges with collaboration occurred when changes happened too quickly for individual team members to react. An abrupt change in processes and procedures without prior communication often derailed the established process. One such example occurred when updates to procedures did not efficiently reach the personnel at the sites, such as splitting samples into primary and secondary inventories for shipping. Also, not all sites were familiar with the rigor of the National Institutes of Health quality assurance processes for clinical trials, which led to an additional unexpected workload for some sites. Sponsors should ensure that details regarding procedures are provided to each site at the start of each trial as they may affect daily procedures, staffing needs, and workflow. SPRN can take an active role in advising future trial sites by compiling a list of common infrastructure needed and procedures required by larger sponsor organizations or creating a list of FAQs that can be asked during onboarding. As the sites became more familiar with the process, they became more comfortable and efficient, leading to improved efficiency in data reporting.
Information Technology
A central location for timely and accurate information sharing helps with compliance across several sites. For ACTT, the dashboard provided by the contract research organization was useful in providing a single location to access all resources related to the protocol. The dashboard included support for online training and information regarding protocols and served as a document tracking tool. Site-specific data were also available on the dashboard. The UNMC sIRB had acquired a new Research Support System just before the initiation of ACTT-1. Some barriers were observed early on as sites worked to onboard all study personnel with the contract research organization dashboard and sIRB Research Support System. Since both systems were new to many personnel, there was a steep learning curve. Frequent changes to the database and conflicting instructions on what to enter in each system also created challenges with data entry. A manual of operations with step-by-step instructions and reporting definitions should be provided to all sites to prevent these challenges in the future. In addition, database changes should be thoroughly vetted and clearly explained before implementation, and a timeline should be provided so that study personnel can anticipate outages and new reporting needs.
Due to changes in visitor polices related to COVID-19, monitors were not allowed to appear in person at many sites. This created a challenge since contracts required sites to enable regular monitoring of their trials. Site monitors were required to go through institutional chains of command before being able to physically visit some sites. Sites that developed or used remote monitoring noted that it was useful, and this should be strongly considered in future trials. Institutions that are considering running clinical trials should coordinate with their information services department to determine the requirements needed to perform remote monitoring.
Regulatory
Even with the newness of the Research Support System, the UNMC sIRB was a powerful tool for ensuring timely review and approval of the protocols across the 10 RESPTC sites. The UNMC sIRB had previously established procedures for rapid review of new protocols in an emergency during the 2014-2016 Ebola outbreak in West Africa. This enabled the sIRB to meet and approve the first ACTT trial in an expeditious manner, which led to the first enrollments in the ACTT trial to occur within SPRN.
Rather than each institution relying on their own local IRB, sites could rely on the UNMC sIRB. This process required sites to include any site-specific language in the consent form and list site-specific points of contact. Sites also had to ensure their local ancillary reviews (eg, institutional biosafety committee, pharmacy and therapeutic, conflict of interest) were completed before full IRB approval. The sponsor produced Spanish-language translations of the consent for sites to use if approved by the local IRB, but sites were responsible for providing translations for other languages if needed. There was variability across sites regarding the local ancillary reviews and the order in which regulatory activities occurred. For many sites, this was their first time working with the UNMC sIRB, and therefore the process for reviews and approvals were new to both study personnel and the local IRBs. While the UNMC sIRB maintained the most up-to-date versions of these documents, sites reported a desire for earlier communication regarding planned changes and amendments. Variations in what needed to be reported to the UNMC sIRB, local IRB, and the sponsor created a final regulatory challenge. In some cases, sites were required to report the same items multiple times; a consistent method of communication across these groups would be useful. This issue could be remedied by determining a clear chain of communication between these entities, understanding that individual sites may need to appoint an IRB representative to assist with studies structured in this way. A list from each entity detailing what documents are needed would also aid in ensuring proper reporting at each stage of the trial.
One unanticipated challenge encountered during the first phase of the study was a limitation on who could receive the study drug through the expanded access/compassionate care route. The medical systems participating in ACTT were often unable to request remdesivir through these routes because of a regulatory limitation that prevented fulfillment of these requests since the drug was available through a clinical trial at these sites. Some of the hospital systems participating in ACTT consisted of several campuses spanning a large geographic area. Within these systems there was usually only 1 campus that conducted the trial. As a result, all other campuses within the hospital system were unable to access the study drug. Sites that ran into this problem noted that it would have been beneficial if they were made aware of these restrictions before agreeing to participate in the trial. This guidance should come from the study sponsor and be provided to sites before initiating a trial. After ACTT-1 was complete and remdesivir was designated as the standard of care, access was no longer hindered. Patients could consent to participating in ACTT-2 or receive remdesivir as part of their care. A second access challenge was the inability to enroll vulnerable populations such as prisoners. Several of the sites serve a large prison population that was inundated with COVID-19 cases. Although the efficacy of remdesivir was unknown at the beginning of the trial, remdesivir was 1 of very few potential therapeutics being tested. While it is understood that there are ethical concerns enrolling vulnerable populations, the previously mentioned inability to acquire remdesivir via alternative routes left these groups with fewer options available to them. As we prepare for future pandemics and epidemics, it will be critical to consider ethical ways to include prisoners and other vulnerable populations, including children and pregnant women. This topic needs to be discussed further, with a particular focus on trials conducted during times of emergency, such as a pandemic. Addressing this topic and how it might be handled in the future needs to occur before the next crisis.
Financial and Personnel Resources
The resources needed to initiate and maintain a large-scale multisite clinical trial were not readily available at all SPRN sites, and many site investigators did not have a full understanding of the magnitude of resources that would be required to maintain this trial. The rapid growth of cases across the country and unpredictability of the disease course meant that resource needs far exceeded what was previously necessary to run a clinical trial, especially personnel (Table 3). Research coordinators and data entry personnel were consistently documented as the biggest needs across sites. Other sites reported needing laboratory technicians, screening personnel, and investigators. Due to the scale of this study and the urgent need to find a suitable therapeutic, many sites did not have an adequate staffing pool at the start. To address this shortage, staff and graduate students from research groups whose work was paused due to the pandemic, as well as medical students and residents, were trained to assist; however, the additional staff were not always trained in clinical research. This required on-the-job training in research processes and ethics and strong oversight to ensure duties were carried out correctly. Research efforts across the board began to surge as the pandemic continued and staff who were knowledgeable and trained were quickly pulled back to their original duties to run other trials or restart trials that had been suspended. This shift in personnel resources seemed to follow the ebb and flow of the pandemic. A core staff was still needed to continue enrollment and sampling, especially for follow-up visits. This points to a clear need to have a core team of staff that can shift to lead a trial such as ACTT when the need arises. This core team can be volunteers from other departments who are interested in performing clinical trial research in an emergency situation. This staffing scenario is similar to the current model of biocontainment unit staffing across the RESPTCs where staff are expected to remain updated on trainings in addition to their daily responsibilities. Should the need arise, these staff are reassigned as available to the biocontainment unit. It would be beneficial to have a document detailing staffing needs for a trial of this size or an approximation of how many staff would be needed for a specific number of enrollees. In addition, for institutions that may deal with special pathogens threats, cross-training of medical staff on research procedures could increase this resource pool.
An impactful challenge with running trials for COVID-19 was identifying personnel willing to enter the designated COVID-19 wards and interact with SARS-CoV-2 patients. As mentioned previously, study staff with limited exposure to working in the biocontainment environment were often pulled from other areas and required extensive just-in-time training to work in this environment. Trainings for donning and doffing of personal protective equipment were provided by each institution based on their guidelines, but resources were available within the NETEC resource repository. 21 As the staff members of each RESPTC's biocontainment unit team were already familiar with various forms of personal protective equipment, these staff often ran the trainings. For activities where patient interaction was required, such as drawing blood samples, obtaining nasal swabs, or administering the study drug, investigators reported that it was important to build a good working relationship and open lines of communication with the floor staff and the primary team. To help facilitate this process, it was necessary to determine when floor staff would be available to assist with clinical study activities so as not to detract from their normal duties.
Recommendations
Several valuable lessons were learned while carrying out this multisite trial and have been described throughout the text. These are summarized as 9 recommendations that can be applied to similar trials in the future (Table 5). Based on the discussions between the 10 SPRN site personnel, these recommendations have been categorized by the operational concept and the responsible group(s) (eg, institution, sponsor, IRB/sIRB).
Recommendations for Establishing Future Clinical Trials During an Emergency Event
Definitions of responsible groups: institution: hospital or university participating in conducting the clinical trial; IRB: local institutional review board; sIRB: single institutional review board; sponsor: organization overseeing the trial and performing data collection and analysis.
To prepare for a similar situation in the future, several tasks can be completed ahead of time. From an institutional standpoint, this includes creating and documenting a plan for rapid implementation of contracts, routing of funding, and redistributing staff. It would be beneficial for institutions to maintain a core group of staff, both clinical and nonclinical, who are trained on clinical research processes and could be transitioned to work on a trial in an emergency. Institutions should also consider creating clinical research learning modules specific to the personnel role, whether it is for a site coordinator, data entry personnel, or laboratory personnel. Sponsors should prepare documents with information that will apply across any study, such as sponsor-specific processes (eg, quality assurance, specimen shipment, site monitoring). Finally, IRBs, whether local or a single IRB, should have training modules for their specific software and procedures, and a list of documents that are necessary to submit throughout the study process, from the beginning through completion.
As a new study is organized, additional resources and processes, if implemented, enable increased communication and an understanding of expectations across all stakeholders. The sponsor should provide each institution with a list of expectations including the number of personnel expected to complete a study and a list of possible restrictions (eg, off-protocol access to study medication). To improve communication and collaboration, sponsors should consider creating a central dashboard that contains study-specific website links, forms, FAQs, and contact information. This dashboard could also be used to inform study personnel of recent and upcoming changes to the protocol, study forms, or study processes. Any changes should also be distributed via a listserv, which should include at least 1 representative from all stakeholders (ie, IRB, sIRB, site principal investigator or coordinator, network lead). Although not necessary for conducting the trial, an open discussion forum would enable idea sharing, which could result in secondary publications, and discussions regarding operational hurdles such as obtaining supplies and retainment of staff. At the study start, any study-specific training required by the institution and the sponsor should be developed and distributed to all personnel. Trainings should be updated as necessary and should include both just-in-time training for newly onboarded personnel and refresher courses for existing personnel. Finally, regularly scheduled conference calls will ensure that all parties are aware of study progress and changes and act as a forum for openly discussing operational hurdles. These calls can occur at multiple levels (eg, across all sites, within a specific research network, within an institution), and each level may require a different frequency.
Conclusions
The amount of data and information regarding treatment options for a new or rare disease can be overwhelming and difficult to sift through, especially when facilities are managing a rapid influx of patients. Regardless of the situation, high-quality RCTs are the most robust way to prove the efficacy of a given therapeutic in a timely manner. Given the sporadic and impromptu nature of emerging infectious diseases, adaptability is the most important quality for success of an RCT during an outbreak. As the diseases that cause epidemics are often not well characterized, there is an added component of learning about the disease pathogenesis and clinical outcomes that goes along with testing therapeutic efficacy. The standard of care may change during the course of an epidemic and this needs to be factored in when analyzing results. In addition, outbreaks can wax and wane in size and personnel may need to shift location or roles.
Participation in the ACTT trials by SPRN shows that programs of any size can participate and contribute. Communication and support are key, due to the rapid nature of the outbreak and the continuous changes in available information. Members of SPRN, even those with extensive clinical trial experience, acknowledged that they substantially underestimated the effort needed to support and conduct this emergency clinical trial to scale for a pandemic; most institutions experienced strain on operational resources due to the sheer number of patients being enrolled, including rapidly onboarding personnel. Preparations before the COVID-19 pandemic focused heavily on a containment approach to a special pathogens threat, a situation vastly different from responding to a growing pandemic; nevertheless, SPRN rose to the challenge and was among the first to activate research trials in the United States. SPRN's experiences in the phases of ACTT serve as a lesson for future emergency clinical trials to ensure that adequate support infrastructure is in place or ready to provide logistical support for clinical trials and to rapidly mobilize additional resources. SPRN will continue to act as a leader in building research infrastructure and producing resources so that clinical trials against emerging pathogens can be rapidly formed across the United States. It is expected that SPRN can offer support and guidance to institutions that may not be engaged with a larger network and provide feedback to sponsors regarding best practices, such as how to obtain informed consent in a biocontainment facility. Ideally, this support and guidance will lead to enhanced response for the next critical need.
Footnotes
Acknowledgments
We would like to thank the members of SPRN for providing their observations and recommendations for conducting future trials, and all those involved in carrying out the ACTT protocols. NETEC is funded by the US Department of Health and Human Services Office of the Assistant Secretary for Preparedness and Response and the US Centers for Disease Control and Prevention, CFDA #93.825. The views expressed are those of the authors and do not necessarily represent the official policies of NIAID, the National Institutes of Health, the US Centers for Disease Control and Prevention, or the Office of the Assistant Secretary for Preparedness and Response.