
Abstract
In cellular immunotherapies, natural killer (NK) cells often demonstrate potent antitumor effects in high-risk cancer patients. But Good Manufacturing Practice (GMP)-compliant manufacturing of clinical-grade NK cells in high numbers for patient treatment is still a challenge. Therefore, new protocols for isolation and expansion of NK cells are required. In order to attack resistant tumor entities, NK cell killing can be improved by genetic engineering using alpharetroviral vectors that encode for chimeric antigen receptors (CARs). The aim of this work was to demonstrate GMP-grade manufacturing of NK cells using the CliniMACS® Prodigy device (Prodigy) with implemented applicable quality controls. Additionally, the study aimed to define the best time point to transduce expanding NK cells with alpharetroviral CAR vectors. Manufacturing and clinical-scale expansion of primary human NK cells were performed with the Prodigy starting with 8-15.0 × 109 leukocytes (including 1.1–2.3 × 109 NK cells) collected by small-scale lymphapheresis (
Introduction
Natural killer (NK) cells connect the innate and adaptive immune system as the first line of defense against virally infected and malignantly transformed cells. NK cells eliminate target cells without previous activation, and cytotoxic activity is regulated by balanced signals from inhibitory and activating surface receptors.1–4 Mechanisms for early killing of target cells are based on release of immune regulatory cytokines such as interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α), 5 death receptor ligand binding to induce caspase-dependent apoptosis,6,7 or secretion of cytotoxic granules composed of granzymes A and B, granulysin, or perforin.1,2
The main NK cell population is defined as CD56+CD3– lymphoid cells, which are divided in two subpopulations: CD56highCD16– regulatory cells and CD56dimCD16+ cytotoxic cells. Because of the advantage of preventing graft-versus-host disease (GvHD) in allogeneic settings, NK cells seem to be ideal for immunotherapies. 8 However, it is still a challenge to expand high numbers of Good Manufacturing Practice (GMP)-grade NK cells for patient treatments, since several infusions of at least 1 × 107 NK cells/kg are necessary.8,9
In the past, GMP-compliant procedures relying on the CliniMACS Plus® (Plus) device were developed successfully for two-step-processes by combination of immunomagnetic CD3 T-cell depletion and CD56+ NK cell separation. However, a fully automated GMP-compliant separation and expansion of clinical-grade NK cells at sufficiently high effector cell numbers remains a great challenge.10–12 Recently, a multiple-step manufacturing process was realized by the CliniMACS Prodigy® (Prodigy) instrument for clinical grade NK cells using an automated T-cell receptor (TCR)-α/β-CD19 depletion, CD56 enrichment, followed by an automated feeder cell-based expansion period, which resulted in a mean 850-fold expansion after day 14. 13
Use of NK cells as an anticancer adoptive immunotherapeutic approach is hampered by various mechanisms that tumor or leukemia cells employ to circumvent the effector cell–based immunosurveillance.14,15 To overcome these tumor defense strategies—so-called tumor immune escape mechanisms—and to enhance the specific targeting to resistant/masking malignant cells, NK cells can be transduced by engineered viral vectors to express chimeric antigen receptors (CARs). These artificial surface receptors consist of an extracellular single-chain variable fragment (scFv) domain to detect specific target molecules expressed on cancer cells. For signal transduction, the CAR molecule contains intracellular signaling domains fused to the scFv. 16 Shortly after binding to CAR target molecules, activation of the CAR NK cells results in strong IFN-γ release and/or secretion of cytotoxic granules (degranulation). 1
CD123 corresponds to the interleukin (IL)-3 receptor α chain that is frequently reported to be highly expressed on acute myeloid leukemia (AML) cells. Therefore, this cell surface molecule constitutes a suitable marker for AML cell detection.17,18
The objective of this study was to establish a fully integrated manufacturing platform and the appropriate quality controls (QC) for clinical-scale production of human primary NK cells. Therefore, the study connected the GMP-compliant NK cell separation by immunomagnetic CD3 depletion and CD56 separation with a NK cell expansion for 14 days in the automated closed Prodigy system. Glucose concentration and pH were monitored as parameters for NK cell growth during the whole expansion period. In-process control (IPC) samples were analyzed for a period of 14 days to assess the NK cell-based cytotoxicity against K562 target cells, levels of surface marker expression, cytokines, cytolytic granule proteins, and degranulation marker CD107a involved in NK cell expansion. NK cell viability was evaluated before cryopreservation and after thawing. The second aim was to implement optimized vector technology in the manufacturing runs during time of expansion. In order to define the best time for optimal successful alpharetroviral vector transduction of expanded NK cells with CAR vectors, IPC samples harvested at different time points in the expansion process in regard to transduction rates were analyzed. The redirected effector-mediated killing activity against the AML cell line KG1a was analyzed by flow cytometry–based cytotoxicity assays and time-lapse tracking experiments, which demonstrated improved retargeted killing that was dependent upon activation levels from expanded NK cells.
Methods
Source of cell material
Peripheral blood leukapheresis (LA) products from healthy donors were obtained from the Institute for Transfusion Medicine of Hannover Medical School following written informed consent, as approved by the ethics committee (#2159-2014) of Hannover Medical School.
Cell lines
The human cell line K562 (chronic myelogenous leukemia) was obtained from the German Collection of Microorganisms and Cell Cultures and was cultivated in Roswell Park Memorial Institute medium 1640 (Biochrom) supplemented with 10% fetal bovine serum (FBS; Biochrom). This CD123-negative cell line was used to determine basal cytotoxicities of freshly isolated, expanded, or thawed primary NK cells. The CD123-positive human cell line KG1a (acute myelogenous leukemia) was purchased from the American Type Culture Collection and was grown in Iscove's modified Dulbecco's medium (Biochrom) supplemented with 20% FBS. The KG1a cells were used in cytotoxic assays to determine the impact of the CAR on the killing capacity of CAR NK cells.
CD3 depletion, selection, and expansion of NK cells using Prodigy
Prodigy (Miltenyi Biotec) presented a closed automated GMP-compliant cell processing system that was used for enrichment of primary human NK cells out of LA, starting with a two-step procedure that consists of CD3 depletion and CD56 selection without changing the tubing set TS 310. All reagents used for manufacturing were GMP-compliant, except AB serum. For clinical manufacturing, clinical grade AB serum will be mandatory. Depletion and selection were based on CD3 CE and CD56 CE reagents (Miltenyi Biotec), respectively. The complete procedure was performed according to the manufacturer's instructions and was based on modified T-cell transduction process software v1.2.1 (Miltenyi Biotec). Different cell fractions—such as LA, positive fraction (PF) after immunoselection, IPCs from expanded cell culture, and end product (EP)—were collected and analyzed. As starting material, LA products with a median of 14 × 109 viable white blood cells (WBC; range 8–15.0 × 109 WBC) were used.
As part of the preassembled tubing set, the CentriCult® unit (Miltenyi Biotec) allowed a maximum volume of 250 mL for cell cultivation. The starting concentration of primary human NK cells was 2–4 × 106 NK cells/mL while expanding the cells for 14 days at 37°C and 5% CO2 in NK MACS® expansion medium supplemented with 5% human type AB serum (Biochrom), IL-21 (1 IU/mL; initial; Miltenyi Biotec), IL-2 (500 IU/mL; Proleukin S; Novartis Pharma GmbH), and IL-15 (140 IU/mL; Miltenyi Biotec), and contained 2% NK MACS® supplement (Miltenyi Biotec). Growth medium was exchanged every 2–3 days, as shown in Fig. 1. During the first 6 days, NK cells grew without shaking, while a gentle shaking mode (0.17–0.3
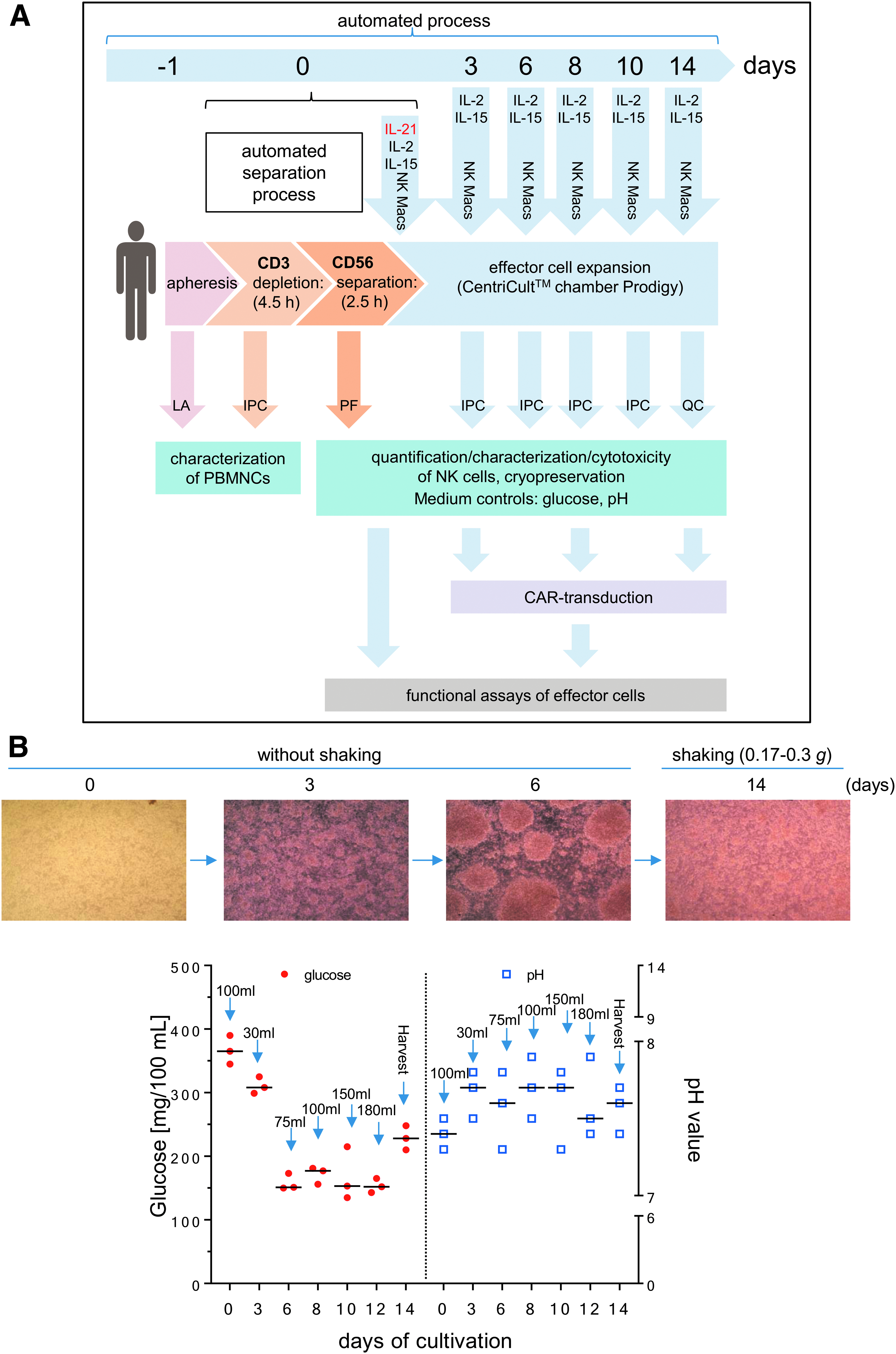
Manufacturing of Good Manufacturing Practice (GMP) grade natural killer (NK) cells.
Alpharetroviral vectors and transduction
Alpharetroviral self-inactivating (SIN) vectors harbored large deletions of transcriptional elements in the U3 of the long terminal repeat regions and included the strong myeloproliferative sarcoma virus promoter as an internal promoter to direct expression of the third-generation CAR transgene (anti-CD123 CAR) followed by an internal ribosomal entry site for enhanced green fluorescent protein (eGFP) expression (Fig. 5A).19,20 The anti-CD123 CAR construct has been described previously 21 and contained a signal peptide derived from granulocyte–macrophage colony-stimulating factor receptor α-chain, anti-CD123 scFv, 22 CD28, CD137 (4-1BB), and CD3ζ. A vector that solely expressed eGFP was used as a control.
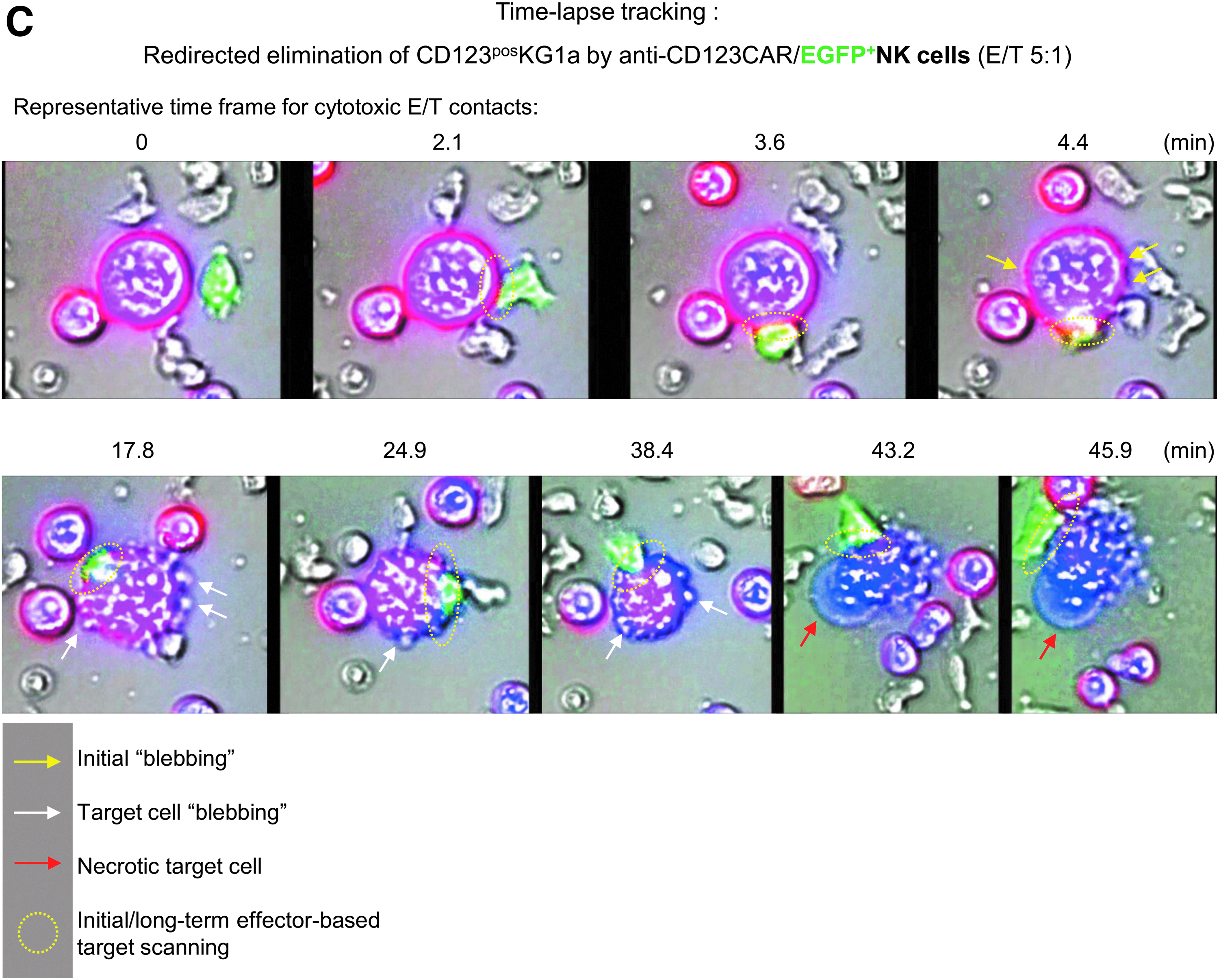
Transduction efficacy and functional characterization of anti-CD123 CAR NK cells. In order to find the appropriate time point for genetic engineering of cultured NK cells, several IPCs were collected during the expansion period (2–3, 8, and 14 days in Prodigy). NK cells were modified with RD114/TR-pseudotyped alpharetroviral vector encoding anti-CD123 CAR/enhanced green fluorescent protein (eGFP) or only eGFP at a multiplicity of infection (MOI) of 1 or 3
HT1080 fibroblasts were used to estimate titers of alpharetroviral vector supernatants using standard transduction protocols. Briefly, 5 × 104 HT1080 cells were seeded in each well of 12-well plates (Sarstedt) the day before transduction. To transduce HT1080 cells, culture medium was replaced with 500 μL of fresh culture medium containing protamine sulfate (4 μg/mL; Sigma–Aldrich). Various volumes of supernatants containing alpharetroviral vector particles were then added to different wells, and the cells were centrifuged for 1 h at 400
A detailed description of the alpharetroviral vector system (pAlpha.SIN.noTATA) and production of alpharetroviral vector supernatants has been published previously.19,20,24 Briefly, 5 × 106 293T cells were seeded onto 10 cm culture dishes the day before transfection. The next day, 293T cells were transfected with a mixture of the appropriate alpharetroviral vector (5 μg), a codon-optimized alpharetroviral gag/pol helper plasmid (2.5 μg; GenBank accession no. HM130053) and RD114-TR envelope plasmid 25 (2 μg) using the calcium phosphate method. After approximately 6 h, the medium was exchanged for 9 mL fresh DMEM containing 10 mM HEPES. To harvest viral particles, supernatants were collected 24 and 48 h after transfection, filtered through 0.22 μm pore-size filters (Millipore), pooled, concentrated by ultracentrifugation, and stored at −80°C until further use.
Transduction of NK cells with alpharetroviral vectors (at a multiplicity of infection [MOI] of 1 or 3) was accomplished with a RetroNectin-based method on 48-well plates, as essentially described by Suerth
Definition of appropriate time periods for successful CAR transduction in expanded NK cells
NK cell samples were collected at different days of expansion (day 2 or 3, day 8, and day 14) for RetroNectin-based transduction on 48-well-plates.19,20 For transduction, alpharetroviral SIN vectors included an expression cassette containing only eGFP, or a combination of an anti-CD123 CAR and eGFP were used. After transduction at a MOI of 1 or 3, NK cells were expanded for 6 days on 48-well-plates containing NK MACS® basal medium supplemented with 5% human type AB serum, IL-2 (500 IU/mL), IL-15 (140 IU/mL), and 2% NK MACS® supplement. Transduction frequency, cytotoxicity, and NK cell degranulation (after 4 h co-culture with KG1a at effector-to-target [E/T] cell ratios of 1:1 and 5:1) were determined by flow cytometry.
Monitoring of glucose and pH
Determinations of glucose concentration and pH value of the cell-free supernatant were performed in parallel every 2–3 days using blood glucose meter Accu Check Aviva and pH indicator strips for a pH range between 6.5 and 10 (MColorpHast™; Merck).
Flow cytometric analysis of surface markers
Purified NK cells and other peripheral blood cells collected in LA, PF, IPC samples, and EP were analyzed for specific surface markers on a Navios flow cytometer (Beckman Coulter) in a no-wash single-platform procedure,26,27 adding Flow-Count fluorospheres™ (Beckman Coulter) to calculate cell concentrations. Cell staining was done using monoclonal antibodies (mABs) such as NK group 2 member D (NKG2D; CD314)-phycoerythrin (PE), NKp30 (CD337)-PE, NKp44 (CD336)-PE, NKp46 (CD335)-phycoerythrin-cyanin-7 (PC7), CD19-fluorescein isothiocyanate (FITC) or allophycocyanin-Alexa Fluor 750 (APC750), CD14-ECD (phycoerythrin-TexasRed®-X), CD69-ECD, CD16-allophycocyanin (APC) or -pacific blue (PB), CD56-PC7, CD3-PB or -allophycocyanin-Alexa Fluor 700 (APC700), and CD45-krome orange (KO) purchased from Beckman Coulter. Additional antibodies (CD137-APC, CD178-APC, and CD253-APC) were manufactured by BD Biosciences. The proportion of living and dead cells in the samples was quantified by 7-AAD (Beckman Coulter) staining. The quality control strategy followed ISHAGE guidelines 27 and is illustrated in Fig. 2.
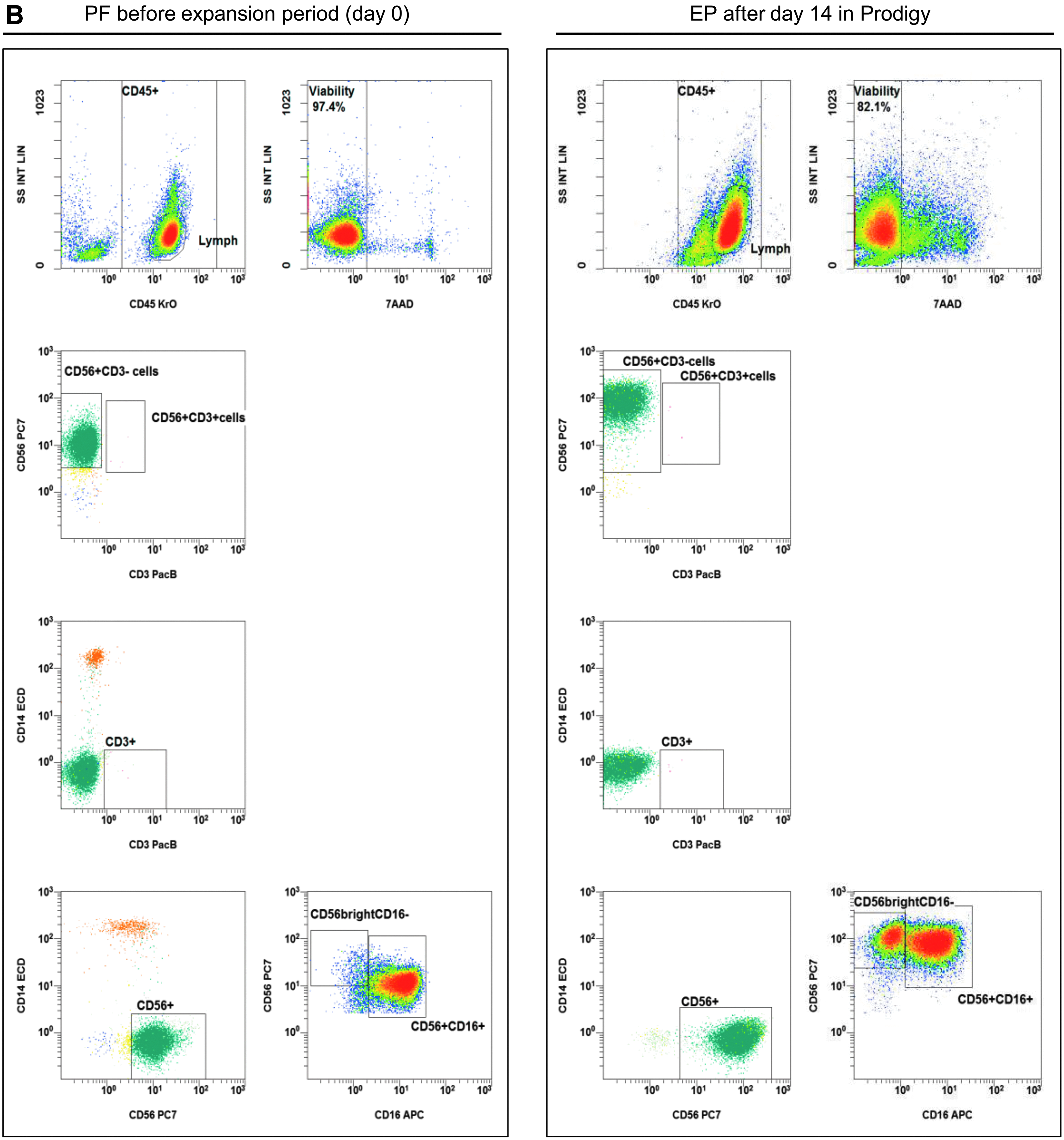
Gating strategy: flow cytometric quality controls for the quantification of CD3–CD56+ NK cells. IPCs from different process fractions (LA, PF, and EP) were stained by monoclonal antibodies (mABs) to visualize CD3–CD56+ NK cells and analyzed by a no-wash, single-platform method based on an eight-color flow cytometric panel.
Flow cytometry–based cytotoxicity assay and CD107a degranulation assay
To assess the cytotoxicity of the NK cells, effector cells were co-cultured with target cell lines at various E/T ratios in NK MACS containing 5% HSA (Octapharma). NK cells were incubated with the K562 cell line for 4 h at E/T ratios of 0.5:1, 1:1, and 5:1 to measure basal NK cytotoxicity, while co-cultivation of CAR-modified NK cells and CD123-positive KG1a cells was accomplished for 24 h at E/T ratios of 1:1 and 5:1 to detect specific anti-CD123 CAR-dependent killing. For detection of viable cells, samples were stained with 7-AAD, and specific mABs (Beckman Coulter) against surface markers were used (NK cells: CD16-APC or -PB, CD56-PC7 or -APC, CD45-KO; K562: CD15-FITC; KG1a: CD34-PC7) in no-wash, single-platform flow cytometric measurements (Navios; Beckman Coulter), as illustrated in Supplementary Fig. S1.
To evaluate NK cell-mediated cytolytic activity based on decline of viable target cells, the following equation was used:
Cytotoxicity (%) = [1 – concentration (co-cultured target cells/μL)/concentration (control target cells/μL] × 100%.
Degranulation of NK cells was detected in the presence of mAB CD107a-PE (Beckman Coulter) after 1 h of co-incubation with target cells at E/T ratios described previously for cytotoxicity assessments. After this 1 h staining with CD107a, Monensin and Brefeldin were added (each diluted 1:1,000; eBioscience), and cells were incubated for an additional 3 h to determine basal NK degranulation using CD123-negative K562 cells or for an additional 4 h to determine CAR-specific NK cell degranulation using CD123-positive KG1a cells. Cells were then washed with PBS (Biochrom). Cell staining and flow cytometric analyses were accomplished as described above for cytotoxic assays.
CAR detection
Anti-CD123 CAR-modified NK cells were incubated with the recombinant and polyhistidine-labeled protein CD123 (1 μg/mL final concentration; Sino Biological, Inc.) for 30 min at 4°C. Unbound CD123 was removed by washing with PBS before transduced NK cells were stained with 7-AAD, CD45-KO, and anti-HIS-APC (R&D Systems) and analyzed by flow cytometric analysis.
Cytokine analysis
Measurements of soluble cytokines, pro-apoptotic markers (TNF-α and IFN-γ), and cytotoxic granule proteins (perforin, granzyme A, granzyme B, and granulysin) were performed in supernatants of cytotoxicity assays using a bead-based immunoassay (LEGENDplex™, human CD8 panel, 13-plex, BioLegend®) according to the manufacturer's instructions.
Cryopreservation and thawing
IPC samples were collected between days 0 and 14 of the NK expansion period (Fig. 1A) in a CentriCult® unit (Prodigy). To freeze the unmodified NK cells, cells were re-suspended in Composol PS® (Fresenius Kabi) containing 2.86% HSA (20% Octalbin; Octapharma). Cells were then mixed with a Composol–dimethyl sulfoxide (DMSO; Sigma–Aldrich) solution (20% DMSO) for cell protection. Freezing was performed in an automated cryo freezer Biofreeze BV50 (Consarctic) before cells were stored in the vapor phase of liquid nitrogen. After 3 months, cells were quickly thawed at 37°C and re-suspended in fresh pre-warmed NK MACS expansion medium (5% AB serum, 2% supplement, 500 IU/mL IL-2, 140 IU/mL IL-15, initial 1 IU/mL IL-21). Cell concentrations were initially set to 2 × 106 NK cells/mL. Samples were measured by flow cytometry after 24, 48, and 72 h to determine recovery, expansion, and surface markers (NKG2D, NKp30, CD178, and CD253).
Time-lapse microscopy
Cell–cell interactions between transduced primary human NK cells and target cells (KG1a) were monitored by fluorescence tracking for 8 h under standard incubation conditions (37°C, 5% CO2, 95% relative humidity) using the IX81 microscope (Olympus). All transduced NK cells expressed eGFP, while KG1a cells were labeled with the intracellular dye eFluor™ 450 (Affymetrix eBioscience™), and extracellular staining was performed with mAB CD34-PE (Beckman Coulter). For co-incubation, effector and target cells were mixed at an E/T ratio of 5:1 on chamber slides. In time-lapse recordings, images of selected slide sections were generated every 30 s with Olympus scanR data software.
Statistical analyses
For statistical analysis and generation of graphs, GraphPad Prism v6.05 (GraphPad Software, Inc.) was used to analyze data sets from three Prodigy runs individually for NK cell fractions (LA, PF, EP, and IPC samples), NK cell expansion rates, recovery, viability, effector cell–based properties, and transduction frequencies. Data are indicated as the median with range (minimum to maximum) within the individual text sections.
Results
Automated separation and expansion of primary NK cells
Three different NK cell manufacturing runs on the Prodigy device were performed by starting the immunomagnetic separations via CD3 depletion followed by CD56 enrichment (Fig. 1A). After removal of CD3+ cells, CD56+ NK cell separation was immediately performed by positive selection using CliniMACS CD56 CE reagent and the same tubing set (TS310). CD3+ cells were depleted in a median frequency of log −3.58 (range log −3.6 to log −3.24) after the depletion step. Following CD56 selection, the overall separation process was increased to a median log −3.66 (range log −3.68 to log −3.6; Table 1). Medians of purity and viability of selected NK cells prior to expansion were >90% (median purity 93.2%, range 78.3–94.9%; median viability 96.5%, range 96.2–97.4%). The median of total NK cell recovery was 65.9% (range 63.2–67.5%), with an absolute number of 818.4 × 103 contaminating CD3+ cells (median, range 574.0–1035.0 × 103) and 543.4 × 103 CD3+ CD56+ cells (median, range 201.6–575.0 × 103; Table 1).
CD3–CD56+ NK cells were finally collected from three independent manufacturing runs in cell collection bags 2 (CCB2). For process evaluation, purity (%), recovery (%), NK subset ratios and cell viability (%) were determined, as well as CD3+ cell depletion and CD56+ cell expansion. Cell viability was assessed by 7-AAD staining and subsequent flow cytometric analyses.
NK, natural killer; LA, lymphapheresis; IL, interleukin.
Subsequent NK cell cultivation steps within the same manufacturing process (Prodigy) were performed by effector cell activation with repeated addition of IL-2 and IL-15 (every 2–3 days), and initial IL-21 (day 0) to reach median expansion rates of 5.85-fold (range 4.18–8.45) after 14 days. However, the often-described loss of freshly separated NK cells between expansion day 0 and 328–30 was detected for all three manufacturing runs and resulted in a lower increase in expansion rates between days 0 and 14. Consequently, it would be more useful to determine NK expansion rates after the initial loss of NK cells, for example between expansion days 3 and 14, as indicated in Table 1 where a median expansion rate of 12.46-fold (range 11.31–17.94) was observed. The NK cell purity (median 99.1%, range 98.8–99.2%) increased with expansion, and viability (median 86.9%, range 86.0–89.2%) slightly decreased. After 14 days of expansion, the T-cell contamination decreased to a median of 140.0 × 103 CD3+ cells (range 135.0–635.3 × 103) and 156.3 × 103 CD3+/CD56+ cells in total (range 97.5–231.0 × 103; Table 1). The ratio of NK cell subpopulations (CD56bright/CD16– cells vs. CD56dim/CD16+ cells) was also examined and displayed a wide range between 0.2 and 2.3 (median 0.9; Table 1).
During the first 6 days of initial NK cell expansion, the cells were grown without any shaking of the cultivation chamber to promote effector cell contact and cell clustering. After 6 days, activated NK cells showed strongly increased cell numbers and much larger cluster formations (Fig. 1B). Due to higher glucose consumption and increased pH, the nutrient/medium supply for effector cells were increased (every 2 days), and the suspension was shaken slightly (shake type 1: every 30 s unidirectional shaking for 2 s) to ensure better mixing of the medium and cells. During the expansion phase, glucose concentrations varied (range 150–390 mg/100 mL), while pH values ranged between 7.3 and 7.9 (Fig. 1B).
IPCs were evaluated to quantify cellular compositions of LA prior purification, purified NK cell product (PF, day 0), and subsequent expanded NK cells after 14 days (EP, Fig. 2A and B). Prior to NK cell separation, LA contained populations of CD14+ monocytes, CD19+ B cells, and CD3+ T cells (including CD3+/CD56+ NKT cells), as well as CD3–/CD56+ NK cells. After depletion of CD3+ cells and separation of CD56+ cells, the population of CD3–/CD56+ NK cells was highly purified, demonstrating nearly no residual CD3+/CD56+ NKT cells and a minimal population of CD14+ monocytes (Fig. 2B, day 0). After 14 days, the residual CD14+ cells and CD3+ T cells (including CD3+/CD56+ NKT cells) were largely removed due to specific cell culture conditions (Fig. 2B, day 14), as the NK MACS® basal medium was specially optimized for NK cell growth. The depletion of monocytes and T cells prevents the development of GvHD, since both immune cell populations are able to produce inflammatory cytokines (
Comparison of surface markers, cytotoxicity, and degranulation before and after expansion of NK cells
The surface expression of NCRs of freshly isolated NK cells was analyzed and compared to the NCR levels detected after 14 days of expansion (Fig. 3A). Total NK cells expressed initially low to moderate levels of NKG2D (median 23%, range 3.3–53.4%), NKp30 (median 0.5%, range 0.1–1.82%), NKp44 (median 1.0%, range 0.3–4.5%), and NKp46 (median 43.3%, range 7.1–44.9%). The surface markers increased during the 14-day expansion in the NK MACS medium supplemented with cytokines IL-21, IL-2, and IL15 (NKG2D: median 97.3%, range 95.1–99.2%; NKp30: median 72.8%, range 36.0–93.1%; NKp44: median 95.5%, range 69.4–95.8%; NKp46: median 71.4%, range 50.8–89.6%). Death receptors (CD178 and CD253) and activation markers (CD69 and CD137) were also expressed at higher levels after cell expansion compared to freshly separated NK cells (Fig. 3B). In particular, the expression of the early activation marker CD69 increased from 2.21% (range 0.2–17.1%) to 95.1% (range 80.8–90.4%). The expression of the apoptotic marker CD178 (FasL) displayed a similar rate of increase starting at 24.8% (range 0.4–39.7%) and reached 89.7% (range 62.2–99.4%) on day 14. The expression of CD137 (day 0: median 0.2%, range 0.2–1.1%; day: 14 median 3.5%, range 1.7–4.23%) and CD253 (day 0: median 33.4%, range 12.2–47.9%; day 14: median 82.9%, range 81.6–100%) increased as well.
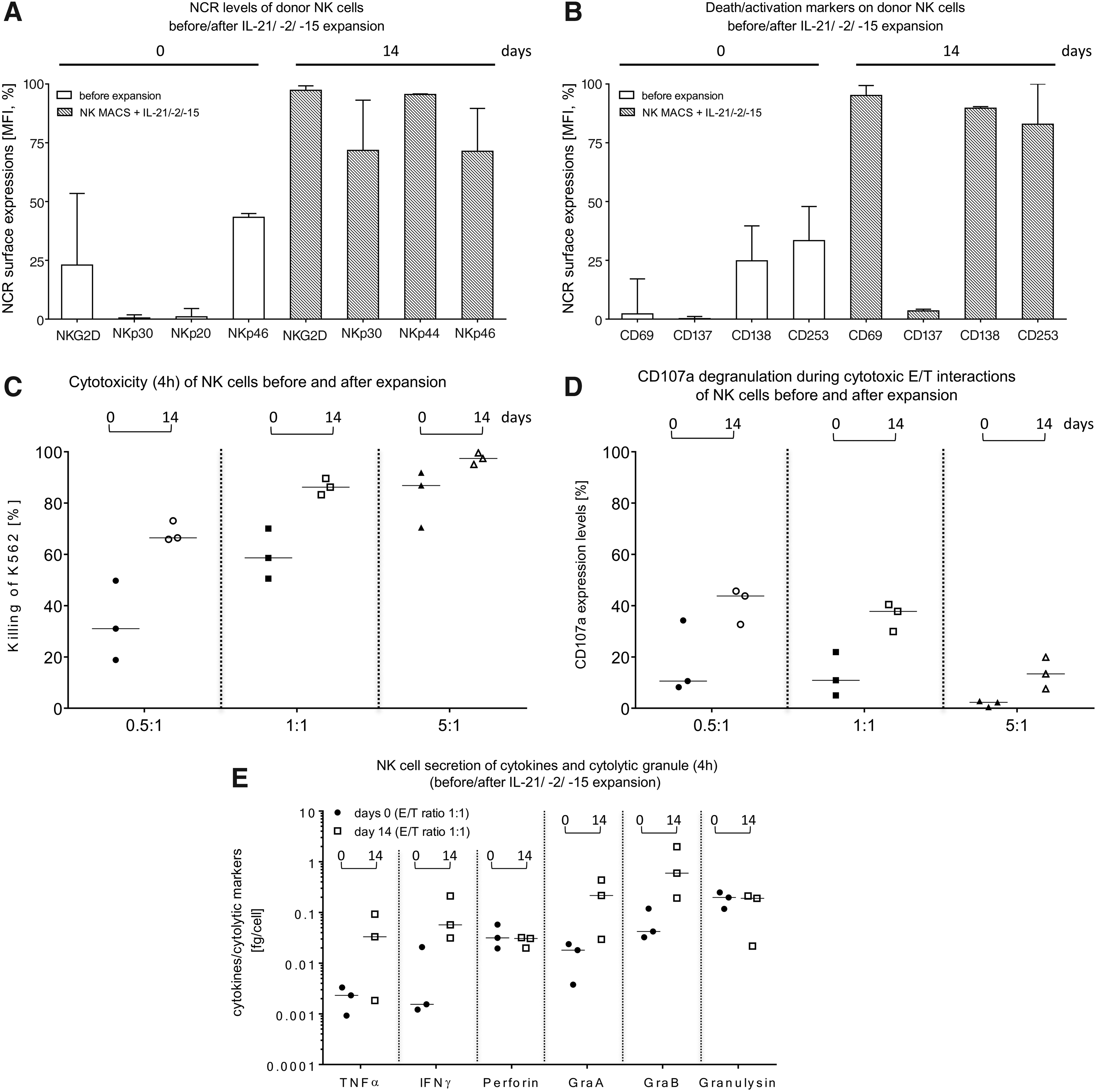
Comparison of cytotoxic properties and activation markers on primary NK cells before and after the expansion process in Prodigy. IPCs (approximately 2.0–2.5 mL) were collected as indicated in Fig. 1A for analysis of NK cell-dependent cytotoxicity, CD107a degranulation, surface marker expression levels, and cytokine secretion. NK cell phenotyping of
Killing activities of non-expanded, freshly separated NK cells were compared to activated NK cells after 14 days using K562 target cells at the indicated E/T ratios (Fig. 3C). In general, the NK cell cytotoxicity increased after expansion and highest killing rates (%) derived at an E/T ratio of 5:1 (Fig. 3C). The strongest increase in NK cell cytotoxicity was observed at an E/T ratio of 0.5:1, with an initial median cytotoxicity of 31.0% (range 18.8–49.8%, day 0), which increased to a median cytotoxicity of 66.5% (range 65.9–73.1%) on day 14. Degranulation assays detected CD107a as a marker for stimulation-induced granule exocytosis. Analyses demonstrated enhanced CD107a expression for expanded (day 14) NK cells compared to non-activated NK cells (day 0; Fig. 3D). It is notable that an E/T ratio of 0.5:1 showed the highest degranulation effect (freshly isolated NK cells: median 10.6%, range 8.2–34.2% vs. expanded NK cells: median 43.8%, range 32.7–45.7%) compared to an E/T ratio of 1:1 (non-activated NK cells: median 10.9%, range 5.0–21.9% vs. expanded NK cells: median 37.7%, range 29.9–40.4%) and 5:1 (freshly isolated NK cells: median 2.3%, range 0.5–2.8% vs. expanded NK cells: median 13.4%, range 7.6–19.9%). Supernatants of cytotoxic experiments with NK cells before and after 14 days of expansion were collected, and pro-inflammatory cytokines (TNF-α and IFN-γ) and cytotoxic granule proteins (perforin, granzyme A, granzyme B, and granulysin) were analyzed (Fig. 3E). Concentrations of TNF-α, IFN-γ, granzyme A, and granzyme B increased during expansion (TNF-α: day 0 median 0.002 fg/cell, range 0.0009–0.003 vs. day 14 median 0.033 fg/cell, range 0.0018–0.092; IFN-γ: day 0 median 0.002 fg/cell, range 0.001–0.02 vs. day 14 median 0.057 fg/cell, range 0.03–0.2; granzyme A: day 0 median 0.018 fg/cell, range 0.004–0.02 vs. day 14 median 0.216 fg/cell, range 0.03–0.43; granzyme B: day 0 median 0.042 fg/cell, range 0.032–0.12 vs. day 14 median 0.598 fg/cell, range 0.19–1.98), while no changes were detected for perforin (freshly isolated NK cells: day 0 median 0.031 fg/cell, range 0.019–0.058 fg/cell vs. expanded NK cells: day 14 median 0.031 fg/cell, range 0.02–0.031 fg/cell) or granulysin (freshly isolated NK cells: day 0 median 0.178 fg/cell, range 0.118–0.248 fg/cell vs. expanded NK cells: day 14 median 0.190 fg/cell, range 0.022–0.212 fg/cell).
Expansion of NK cells after cryopreservation
To assess the stability of expanded NK cells after cryopreservation, IPC samples were frozen directly after separation (day 0) or on day 3, 6, 8, 10, or 14 after isolation during expansion. After 3 months, cryopreserved NK cells were thawed and expanded for 24, 48, and 72 h to determine killing activity and surface marker expression (NKG2D, NKp30, CD178, and CD253; Fig. 4). Due to cell loss on day 3 prior to the cell expansion phase, all NK cells harvested on day 3 were used solely for functional assays (Fig. 4G). NK cells stored in liquid nitrogen on day 10 after separation showed the best ability to expand, especially 72 h after re-cultivation (72 h median 1.1-fold, range 1.1–5.7-fold), while moderate cell revival ability was found on cells harvested on day 6 (72 h median 0.9-fold, range 0.88–1.7-fold), day 8 (72 h median 1.7-fold, range 1.7–3.9-fold), and day 14 (72 h median 2.2-fold, range 1.6–3.36-fold). The amount of NK cells that were frozen immediately after isolation (day 0) decreased during the next 72 h (median 0.4-fold, range 0.4–0.5-fold) due to the well-known loss of NK cells between days 0 and day 6 of expansion (Fig. 4A).28-30 After 24 h of re-cultivation, NK cells demonstrated the lowest viability (medians <80%) for all collected IPC samples and increased during the expansion period after 72 h (medians >80%; Fig. 4B). Activating NKp30 was highly expressed (median >80%; range 95.5–100%) in all NK cells harvested between day 0 and day 14 and re-cultivated after thawing between 24 and 72 h (Fig. 4D). Activating cell surface receptor NKG2D showed similar expression, but NK cells harvested directly after isolation (day 0) demonstrated a lower expression level during expansion for 24 and 48 h (Fig. 4C), and only after 72 h of expansion did approximately 80% (median, range 77.0–99.9%) of total NK cells stain positive for NKG2D. The death receptor ligands FasL (CD178) and TRAIL (CD253) were highly expressed on NK cells harvested between days 6 and 14 (CD178: median >60%, range 46.3–99.5%; CD253: median >70%, range 57.8–99.8%) and cultivated after thawing for 24–72 h (Fig. 4E and F), while CD178 and CD253 were expressed to a lower degree on NK cells harvested immediately after separation (day 0) in all re-cultivation experiments. There was a slight loss of CD178-positive and CD253-positive NK cells after 48 h of expansion, which was overcome after 72 h of expansion.
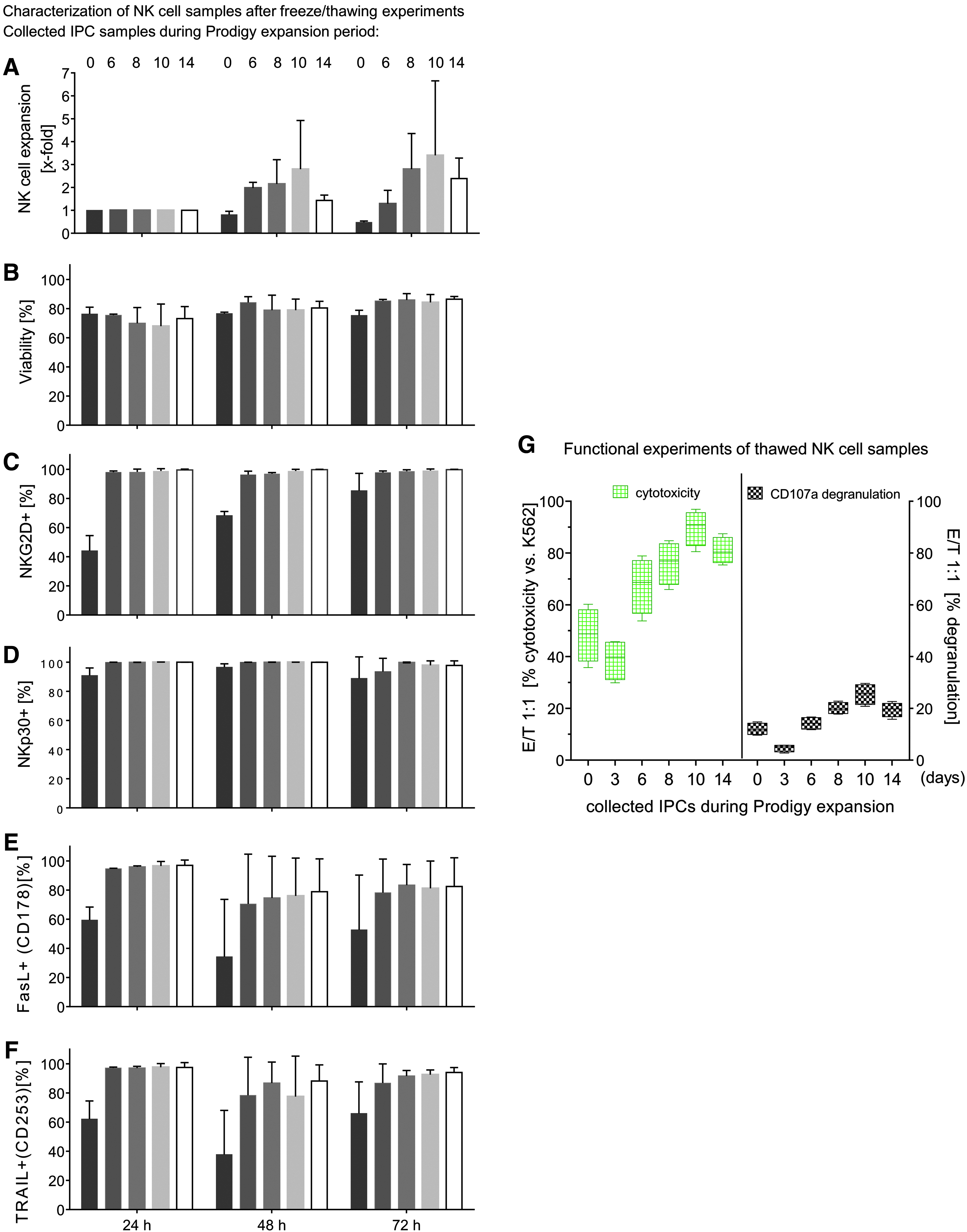
Freeze and thawing experiments from collected IPCs before, during, and after NK expansion in Prodigy. IPCs of isolated and expanded NK cells were harvested/collected directly after immunomagnetic separation (Prodigy) and during NK cell cultivation at different time points (3, 6, 8, 10, and 14 days) and cryopreserved in liquid nitrogen for 3 months before thawing and re-cultivation was started for 24, 48, and 72 h. NK cell expansion properties
Functional assays after cryopreservation
NK cells harvested on various days during the expansion period were analyzed for killing activity and their ability to release cytotoxic granules after 3 months of freezing. Therefore, NK cells from different IPC samples (0–14 days) were re-cultured for 72 h and subsequently co-cultured with K562 target cells (E/T ratio 1:1) to examine their cytotoxicity and degranulation potential. NK cells harvested on day 10 displayed the highest lytic activity (median 90%, range 80.5–96.9%), while the same NK cells collected on day 3 exhibited the lowest cytotoxicity (median 39%, range 29.9–45.8 %). Basal killing activity peaked for NK cells harvested after day 10 (median 90%, range 80.5–96.9%) and declined after day 10 (day 14 median 81%, range 75.4–87.4%; Fig. 4G, left side).
NK cell samples that were assessed in cytotoxic assays were also analyzed for CD107a expression to estimate degranulation. In accordance with the cytotoxic data, the lowest degranulation was detected for cells harvested on day 3 (median 4.7%, range 2.8–5.8%), while peak expression of CD107a was found for NK cells harvested on day 10 (median 25.5%, range 20.8–29.7%). For NK cells harvested on day 14, CD107a levels decreased (median 19.8%, range 15.8–22.7%; Fig. 4G, right side).
Retargeted anti-CD123 CAR NK cells and their ability for cytotoxic killing and degranulation
NK cells were transduced at several time points during the expansion period in order to determine the best time point for transduction efficiency. Alpharetroviral vectors that encoded either an anti-CD123 CAR combined with eGFP (Fig. 5A) or solely eGFP as a control were used to transduce NK cells. Transduction frequency of CAR-engineered NK cells was quantified by flow cytometry to measure the number of eGFP-positive cells. Surface expression of CAR molecules was detected with a polyhistidine-labeled CD123 antigen and anti-HIS mABs conjugated with APC. As shown in Fig. 5A, eGFP signals were higher (21.7%) than signals for CD123 antigen binding (11.2%), which implies a different expression of eGFP and functional CARs, although both genes were part of the same vector (Fig. 5A). The highest transduction efficiencies of alpharetroviral CAR constructs were achieved with NK cells harvested on day 14 (median 5.2%, range 3.2–11.2%), while the lowest transduction rate was determined for NK cells that were modified 2–3 days after NK cell separation (median 0.5%, range 0.2–1.3%). Control experiments with the eGFP vector confirmed these results (day 2–3: median 2.3%, range 1.3–3.5%; day 14: median 9.9%, range 6.9–13.5%), implying that NK cells in expansion are more receptive for alpharetroviral vectors at later time points (Fig. 5A).
Retargeted killing activity of CAR-engineered NK cells was examined with CD123-positive KG1a cells in potency assays for cytotoxicity and CD107a degranulation using different E/T ratios (1:1 and 5:1). After 24 h of co-culturing, a decrease of the KG1a target cell population was determined by single-platform flow cytometric assays. Anti-CD123 CAR/eGFP-modified NK cells were compared to eGFP-positive NK cells (without CAR expression), which served as controls for redirected elimination of KG1a. Anti-CD123/eGFP transduced NK cells showed a higher killing activity against KG1a cells compared to eGFP-modified NK cells at both examined E/T ratios (Fig. 5B). The highest cytotoxic rates were determined from anti-CD123 CAR/eGFP NK cells transduced on days 8 and 14 of expansion, suggesting that this interval is a suitable time point for gene modification of expanded NK cells. Comparison of killed target cells for an E/T ratio of 5:1 (day 8: anti-CD123 CAR/eGFP: median 65.4%, range 38.6–81.4% vs. eGFP: median 31.3%, range 23.7–39.3%) and for an E/T ratio of 1:1 (day 8: anti-CD123/eGFP: median 44.3%, range 31.9–74.1%; eGFP: median 21.5%, range 14.1–32.8%) showed a higher killing rate for an E/T ratio of 5:1.
In general, anti-CD123 CAR/eGFP NK cells exhibited higher CD107a surface levels compared to eGFP-expressing NK cells (Fig. 5B). As also observed for NK cell cytotoxicity, degranulation was enhanced for NK cells modified on days 8 and 14 of expansion, while NK cells transduced on days 2–3 of expansion showed lower CD107a surface levels for both E/T ratios. NK cells modified on day 8 or 14 and subsequently co-cultured with KG1a at an E/T ratio of 1:1 exhibited the highest CD107a degranulation (day 8: anti-CD123/eGFP: median 29.7%, range 18.7–32.8%; eGFP: median: 16.8%, range 12.7–21.7%). In contrast, an E/T ratio of 5:1 displayed lower extracellular CD107a release (day 8: anti-CD123/eGFP: median 25.7%, range 21.9–28.0%; eGFP: median: 13.7%, range 8.8–16.2%). In all examined NK cell samples, an E/T ratio of 1:1 showed higher 107a degranulation rates compared to an E/T ratio of 5:1.
In order to track specific retargeting of E/T cell contacts, anti-CD123/eGFP-transduced NK cells were co-incubated for 8 h with labeled CD123-positive KG1a cells (E/T ratio of 5:1). The scanR automated image and data analysis software (Olympus) was used for time-lapse imaging of redirected cell contacts of CAR NK and KG1a cells that resulted in specific, long-term E/T interactions. Shortly after making contact with target cells, anti-CD123 CAR NK cells initiated membrane protrusions (“blebbing” of target cells) on the KG1a that resulted in apoptotic and/or necrotic cell reactions, while effector cells stayed vital, as indicated in the time-lapse photographs shown in Fig. 5C and Supplementary Video S1. Imaging analysis showed that anti-CD123 CAR-expressing NK cells acted specifically against CD123-positive target cells.
Discussion
In the last decade, several clinical studies (NCT 00995137, NCT 01974479, NCT 03056339, NCT 03415100, NCT 03579927, NCT 03692767, NCT 03690310, NCT03692663, NCT 03692637, and NCT 03824964) have demonstrated the growing interest in CAR-modified primary NK cells as a gene therapy medicinal product against different tumor entities. However, GMP-compliant manufacturing of cells on a clinical scale still has to overcome various hurdles before the CAR NK cell strategy can be made broadly available. This work examined the potential of the closed Prodigy system to separate highly purified primary NK cells in combination with clinical-scale expansion. Although only three Prodigy runs are reported here, all three runs demonstrated consistent results regarding cellular composition, viability, purity, expansion rates, and functionality of the produced NK cells. This implies that a robust standardized manufacturing process can be implemented. However, it will also be important to perform the transduction procedure on Prodigy to validate the optimal transduction time point that was determined in experiments outside of the Prodigy system. Since the NK cell content of the peripheral blood shows individual variations and comprises up to 10–20% of all lymphocytes,31,32 optimal donor NK cell concentrations should be high. For clinical treatment in a Phase II study, Stern
The depletion of CD3+ cells and the separation of CD56+ cells from LA were done in consecutive steps within the same tubing set. The purity of NK cells is critical because of the T cells characteristic to trigger GvHD. It has been demonstrated that patients developed an acute GvHD ≥grade II when receiving ≥0.5 × 105 T cells/kg body weight, while NK cell doses are very well tolerated when T-cell contamination is 0.03 × 105 T cells/kg body weight. 8 The depletion median rate of log −3.6 and a median purity of 93.2% in the final product were very similar to former published data (CD3+ depletion: log −3.5; CD56+ purity: 95.4%) 11 using the Prodigy instrument. The median total recovery (65.9%) and the median viability (96.5%) of the CD56+ cells were also comparable to previously collected data (total recovery: 60.4%; viability: 96.3%), suggesting the robustness of these protocols. 11
Moreover, the translation of the optimized
This study demonstrated increasing levels of NCRs (NKG2D, NKp30, NKp44, and NKp46) and death/activation markers (CD69, CD137, CD178, CD253), as well as a strong increase in cytokines (TNF-α and IFN-γ), cytolytic proteins (granzymes A and B), and enhanced CD107a expression levels on NK cells that were expanded 14 days. The upregulation of the surface markers indicated that NK cells were highly activated after expansion, which was also reflected by functional assays (cytotoxicity, CD107a degranulation) and confirmed by other publications.11,13 The current degranulation experiments showed higher CD107a expression levels for low E/T ratios such as 0.5:1 or 1:1 compared to lower surface CD107a on NK cells at an E/T ratio of 5:1, indicating different predominant killing mechanisms. Factors such as secretory lysosome contents, adhesion promoting structures on target cells, or sensitivity of triggering death pathways may mediate this enhanced NK cell cytotoxicity. 40 The observation of high CD107a degranulation for low E/T ratios was also reported by others.41,42
Monitoring glucose consumption and pH reflect NK cell density and metabolism rates during expansion in the CentriCult® chamber. High NK cell metabolism was associated with increased glucose reduction in the growth medium. To ensure an optimal nutrition supply, NK cells were fed every 2 days during the linear growth phase, which stabilized the glucose concentration in the range of 150–228 mg/mL between day 6 and day 14 of NK cell expansion. During the first 6 days, NK cells are fragile, as demonstrated by the decline of the cell concentration, and accompanied by a low glucose consumption and little exchange of growth medium in the first 3–4 days. For this reason, gentle cell shaking was not applied until day 7. Gentle cell mixing started on day 7 allowed high cell densities. 13 Medium exchange every 2 days limited the pH fluctuation to within a physiologically tolerable range between pH 7.3 and 7.9.
In order to assure NK cell quality after cell cryopreservation, IPC samples were harvested at various time points during the cultivation process for control experiments followed by storing in vapor phase of liquid nitrogen for 3 months. After thawing, NK cells were expanded for an additional 3 days to restore NK cell functions. The highest revitalization was found for NK cells collected on day 10 of expansion, while less growth only occurred for NK cells collected immediately after the separation process. This was also reflected by the lowest expression of NKG2D, NKp30, CD178, and CD253, indicating that NK cells collected directly after separation demonstrated low killing activity and degranulation. Similar results were shown by Childs and Berg
43
who then recommended re-culturing thawed NK cells for several days before administration to patients. Indeed, this study observed increased killing activity of NK cells and degranulation by NK cells collected up to day 10, while day 14-collected EP demonstrated slightly lowered cytotoxicity and degranulation (compared to day 10 NK cells) after re-cultivation. This may be partially explained by the finding that chronic stimulation with IL-2 and IL-15 induced NK cell exhaustion.44,45 On the other hand, cytokines are necessary for
Customized immune therapies with a particular focus on CAR-modified immune cells have emerged in recent years because of the advantage of attacking cancer cells even after development of resistance to conventional agents. CAR NK cells seem to be a potential source for “off-the-shelf” therapies, 46 since they do not cause GvHD while mediating antitumor and antileukemic reactions.47,48 Thus, clinical-grade manufacturing of CAR NK cells has to be developed and optimized. In order to detect adequate time periods for optimal transduction rates by alpharetroviral vector constructs encoding either anti-CD123/eGFP or only eGFP, IPC samples were collected on days 2–3, 8, and 14 after selection. Transduction rates (measured on day 6 after transduction) rose continuously until day 14 after NK selection. However, functional assays against KG1a demonstrated similar killing activity and CD107a degranulation for NK cells transduced on day 8 or day 14 of expansion, suggesting that cytolytic properties of CAR-transduced NK cells might be optimized on day 8 without any opportunity for substantial increase. This implies that NK cells should be transduced on day 8 (with subsequent expansion for 6 days) without any disadvantages in generating highly activated NK cells. Determination of the optimal time period to transduce primary NK cells is required to integrate the transduction procedure into future Prodigy processes. The described RetroNectin-based transduction method seems to be inappropriate for routine Prodigy processes, since the 48-well-plate procedure is an open system and therefore it is not GMP-compliant. In addition, it is difficult to combine the RetroNectin-based method with the closed Prodigy system. An alternative method might be transduction based on vectofusin-1, a synthetic nontoxic histidine-rich peptide that promotes T-cell transduction with lentiviral vectors. 49 This soluble GMP-compliant enhancer could be easily added in advanced Prodigy settings for NK cell transduction. Time-lapse tracking experiments performed by fluorescent microscopy confirmed the results of expanded CAR NK cell cytotoxicity and demonstrated redirected E/T cell contacts of anti-CD123 CAR NK cells toward CD123-positive AML cells and subsequent apoptotic and necrotic reactions in the attacked AML cells.
In the present experiments, the used vectors were SIN vectors that are replication incompetent, demonstrate a singular infectiousness, and decrease the risk of insertional mutagenesis after transduction.21,24 Nevertheless, the freedom of infectious particles in the final product should be achieved. Usually this is accomplished via manifold washing of the cell suspension during the post-transduction cultivation.50,51 Such washing procedures can be integrated into the activity matrix of the Prodigy device. In this context, methods should be implemented to check residual vector concentration in supernatants (Jang
In addition to CAR NK cells, CAR T cells present an additional immunotherapy option to treat AML. The development of T cells expressing CD123-specific CARs demonstrated high cytolytic activities in
Footnotes
Acknowledgments
The authors thank D. Lenz for excellent technical support. This project was funded by grants of the DFG (Collaborative Research Center 738, CRC738, and Cluster of Excellence REBIRTH), the Integrated Research and Treatment Center Transplantation (IFB-Tx, Ref. No. 01EO0802 and 01EO1302), and the European Union (Horizon 2020 program SCIDNET).
Author Disclosure
J.K. and V.H. were employees of Miltenyi Biotec GmbH, although V.H. left Miltenyi Biotec shortly after the experimental phase. A.S. is co-inventor on a patent application describing alpharetroviral SIN vectors. The remaining authors declare no financial or commercial conflicts of interest.