
Abstract
The β-hemoglobinopathies sickle cell anemia and β-thalassemia are the focus of many gene-therapy studies. A key disease parameter is the abundance of globin chains because it indicates the level of anemia, likely toxicity of excess or aberrant globins, and therapeutic potential of induced or exogenous β-like globins. Reversed-phase high-performance liquid chromatography (HPLC) allows versatile and inexpensive globin quantification, but commonly applied protocols suffer from long run times, high sample requirements, or inability to separate murine from human β-globin chains. The latter point is problematic for in vivo studies with gene-addition vectors in murine disease models and mouse/human chimeras. This study demonstrates HPLC-based measurements of globin expression (1) after differentiation of the commonly applied human umbilical cord blood–derived erythroid progenitor-2 cell line, (2) in erythroid progeny of CD34+ cells for the analysis of clustered regularly interspaced short palindromic repeats/Cas9-mediated disruption of the globin regulator BCL11A, and (3) of transgenic mice holding the human β-globin locus. At run times of 8 min for separation of murine and human β-globin chains as well as of human γ-globin chains, and with routine measurement of globin-chain ratios for 12 nL of blood (tested for down to 0.75 nL) or of 300,000 in vitro differentiated cells, the methods presented here and any variant-specific adaptations thereof will greatly facilitate evaluation of novel therapy applications for β-hemoglobinopathies.
Introduction
The major hemoglobinopathies sickle cell anemia (SCA) and thalassemia as a group represent the commonest monogenic disorders. The most severe and potentially lethal forms affect the components of the main adult hemoglobin α2β2 tetramer (HbA). Whereas SCA is caused by a pathologic β-globin HBBE6V variant, 1 α- and β-thalassemia are caused by loss or depletion of the eponymous globin chain and consequential toxic excess of its counterpart.2,3 Lifelong management with blood transfusions and medication, such as iron chelation to avoid iron overload and hydroxyl urea to reduce transfusion requirements in SCA patients, reduces suffering by anemia, hemolysis, and disease-related severe pleiotropic effects. Such palliative treatment is prohibitively expensive for standard care in low- and middle-income countries, and poses the persistent threat of blood contamination and long-term damage to vital organs.4–10 Curative treatment by allogeneic transplantation of hematopoietic stem cells, however, is only available to the fraction of patients for whom suitable donors can be identified and, despite ongoing improvements, carries a significant risk of treatment-related mortality. 11
The severity of hemoglobinopathies in combination with a high global disease burden, estimated annually at >330,000 affected births (17:3 SCA:thalassemias), 12 has prompted many laboratories and companies to work toward therapies and cures. For β-hemoglobinopathies in particular, these have long included small-molecule drugs, but lately and more successfully also include gene therapy treatment of hematopoietic stem and progenitor cells (HSPCs) either by gene addition, up to the level of clinical trials, or by diverse approaches based on genome editing.13,14 The latter field has seen tremendous growth and scientific progress, especially based on double-strand breaks introduced by designer clustered regularly interspaced short palindromic repeats (CRISPR)-guided endonucleases and more recently based on CRISPR-directed base editing. 15 In the case of designer nucleases, target genomic DNA sequences can be disrupted by imprecise repair based on non-homologous end joining (NHEJ), or can be modified in a targeted fashion in the presence of suitable repair templates based on homology-directed repair.
The cornerstone of drug-based and many gene therapy strategies is that endogenous γ-globin, which is encoded by the HBG1 (Aγ-globin) and HBG2 (Gγ-globin) genes on the β-globin locus, may be induced as a therapeutic substitute for β-globin by transcriptional reprogramming of HSPCs. Moreover, γ-globin and specific exogenous β-globin designer variants have anti-sickling properties and thus improved efficacy in the treatment of SCA compared to wild-type β-globin. Gene addition vectors for the treatment of β-hemoglobinopathies thus provide exogenous β-globin or, for higher efficacy in SCA, γ-globin or anti-sickling β-globin variants. 16 These and other gene addition approaches, such as the activation of γ-globin by provision of artificial transcription factors 17 or by shRNA-mediated knockdown of γ-globin repressors, 18 conceptually depend on permanent and usually lentiviral delivery of the therapeutic transgene. For clinical application of therapies based on genome editing, delivery of designer nucleases or base editors, and possibly of specific repair templates, will instead be transient. Therapeutic strategies currently under investigation are the recreation of deletions associated with high γ-globin levels in the β-globin locus, 19 the deletion of specific control elements required for maintenance of low γ-globin levels, 20 or the repair of the specific causative mutation and thus reinstatement of normal β-globin expression.15,21–23 Whatever additional approaches may be explored in this fast-moving field, quantitative assessment of therapeutic efficiency is and will remain critical.
A key parameter for the assessment of therapeutic efficiency is the ratio of β-like to α-like globin chains, which in normal adults is 1:1. This should give a ratio slightly <1 for β- to α-globin, owing to their stoichiometric 1:1 contribution to HbA (>95% of total hemoglobin) and the additional presence of normally minor quantities of α2δ2 (HbA2, <3.5%) and α2γ2 (residual fetal hemoglobin, HbF, <1%) heterotetramers in adult blood. To this end, immunoblots are a frequently applied research method for protein quantification, but are characterized by high sample requirements, high reagent cost, and low reproducibility of measurements across experiments because of the requirement of multiple blots for internal controls and calibrators. 24 Moreover, the relative purity of globin chains in blood and erythroid samples renders antibody recognition unnecessary for the specific purpose of globin-chain detection, as hemoglobin makes up 99% of erythroid dry mass. 25 This knowledge is exploited by routine diagnostic methods widely applied for population screening of hemoglobinopathies, such as cation-exchange high-performance liquid chromatography (HPLC), 26 isoelectric focusing, 27 or cellulose acetate electrophoresis, 28 which rely on the detection of hemoglobin tetramers and, particularly if applied in combination, 29 allow sensitive detection of different hemoglobin variants. However, those methods give limited information on the specific quantities of constituting chains, in particular for the γ-globin chains, Aγ and Gγ, which differ by a single amino acid. In partially differentiated cell culture samples or chimeric models, these limitations are exacerbated by the presence of additional proteins or hybrid hemoglobin species. Advanced proteomics approaches achieve high levels of specificity and sensitivity, 30 but their application is limited to few specialized laboratories. Widely accessible, robust detection of individual globin chains may instead be achieved by reversed-phase HPLC (RP-HPLC), which separates proteins based on their hydrophobicity. 31
RP-HPLC data are commonly used in cell and gene therapy publications, but frequently source data are not shown at all32–34 or indicate technical shortcomings (such as sloping baselines). 35 Protocol optimization for RP-HPLC based on C4 column resin has resulted in sensitive detection of down to 0.1 μg hemoglobin 36 and baseline separation of all human globin species, 37 but at runtimes of 70 min and 100 min, respectively, that are too long for routine application. The most significant speed improvement in RP-HPLC application to hemoglobinopathies has come about by a change of column materials (C18 instead of C4 resin) in two studies, but without data for cell culture or animal samples.38,39 Moreover, both advanced protocols employed high-pressure gradient systems at speeds of 3.5 mL/min 38 or 2.0 mL/min, 39 at which the low-pressure gradient systems commonly used in research settings cannot achieve satisfactory mixing of the mobile phases used. In both cases, change of column material reduced peak widths and thus reduced run times down to 16 min, but both reports utilized liberal (0.48 μL of blood) 39 to apparently excessive (hemoglobin equivalent to 1 mL of blood) 38 sample material per injection. In the former study, this would already be equivalent to >2 × 106 erythrocytes per single readout. Existing sensitive research protocols therefore suffer from long run times, whereas diagnostic protocols are not optimized for minimal sample utilization. Accordingly, the overall framework parameters of existing RP-HPLC protocols are not suitable for gene therapy work, where sample material is always limited, numerous samples call for short run times, γ-globin chains are routinely of great interest, and close to clinical application, measurement of human globin chains in the background of transgenic or chimeric murine models is required. This study therefore sought to accommodate short run times, measurement at far-UV wavelengths, common low-pressure gradient technology, and measurement of murine and human globin chains usually encountered in gene-therapy work in a clearly defined set of RP-HPLC parameters for the authors' own experimentation and as a common reference in the field.
Materials and Methods
Study subjects and human blood collection
All experimentation on human peripheral blood (PB) and PB-derived cells was based on approval by the Cyprus National Bioethics Committee (Applications EEBK/EII/2012/02 “Advancing Gene Therapy Vectors for Thalassaemia” and EEBK/EII/2013/23 “ThalaMoSS”) and on written informed consent by all study subjects. Human blood samples were collected in VACUETTE tubes with EDTA-anticoagulation (Greiner Bio-One). Available to this study was donor blood from healthy adult and cord blood controls, from both a carrier and a homozygous patient for the HBBIVSI-110(G>A) mutation, and from a homozygous HBBE6V (SCA) patient. For characterization of the standard separation protocol (Table 1), combination of adult (primarily HbA, α2β2) and cord blood (primarily HbF, with greatest contribution α2Gγ2 and secondarily α2Aγ2) analyses served to determine elution times of human α-, β-, Gγ-, and Aγ-globin chains. Purified HbA2 (H0266; Sigma–Aldrich) served as a standard for δ-globin elution times.
RP-HPLC protocol for human globin chains
Besides the parameters defined here, precise elution times and peak separation will also depend on variation in tube lengths and diameters, column packing, and the degree of deterioration of guard and main column.
Baseline mobile phase 40% B; column temperature 70°C; flow speed 1.0 mL/min; max. backpressure (at injection) 90 bar (9 MPa, 1,305 psi).
RP-HPLC, reversed-phase high-performance liquid chromatography.
Mice and murine blood collection
Transgenic mice carrying the human β-globin locus were gratefully received from Dr. Kenneth R. Peterson (University of Kansas, Lawrence, KS). Other mice were commercial strains obtained from Harlan/Envigo. Murine blood was collected with EDTA as anticoagulation agent (1.5 μL of 0.5 M EDTA, pH 8.0 for up to 250 μL of blood) and stored at 4°C until processing. All handling of animals followed institutional guidelines and regulations. Unless indicated otherwise, mouse samples employed in this study were based on C57BL/6J (C57Bl/6J/OlaHsd) genetic background and the corresponding Hbbs haplotype, resulting in a typical single murine β-globin peak. 40 Analysis of peripheral blood samples for C57BL/6J mice and YAC mice, which show reduced murine Hbbs expression owing to competition with the human β-globin locus, were used to identify elution times for C57BL/6J α- and β-globin chains (Supplementary Fig. S1). Other mouse lines used to characterize the hybrid (murine/human) separation protocol (Table 2) were Balb/c (Balb/cOlaHsd), CBA (CBA/CaOlaHsd), DBA (DBA/2OlaHsd), and CD-1 (Hsd:ICR(CD-1®)).
Hybrid RP-HPLC protocol
The RP-HPLC protocol optimized for low run times and good separation of murine and human β-globin chains at good peak widths. Alternative conditions exist that will give either superior separation or narrower peak shape for the same stationary and mobile phases (see Supplementary Fig. S2).
Besides the parameters defined here, precise elution times and peak separation will also depend on variation in tube lengths and diameters, column packing, and the degree of deterioration of guard and main column.
Baseline mobile phase 36.5% B; column temperature 0–4°C (on ice); flow speed 1.0 mL/min; max. backpressure (at injection) 360 bar (36 MPa, 5,220 psi). Applying a lower flow speed of 0.8 mL/min will reduce backpressure to a maximum of 320 bar, at slight delay and broadening of elution peaks (data not shown).
Lentiviral vector production
Lentiviral (LV) vectors were produced as reported. 41 Briefly, near-confluent HEK293T cells were seeded 1:8 in 100 mm TC-treated culture dishes (Sigma–Aldrich) and 8 mL complete Dulbecco's modified Eagle's medium (cDMEM: DMEM, 10% fetal bovine serum [FBS], 1 × penicillin/streptomycin, and 1% GlutaMAX; Thermo Fisher Scientific). Next day, 100 μg of polyethylenimine (PEI; Polysciences, Inc.) in 500 μL 1 × HBS (140.5 mM of NaCl, 50 mM of HEPES, 0.75 mM of Na2HPO4, pH 7.1–7.14) per dish were mixed with 5 μg of envelope plasmid pMD2_VSVG, 15 μg of packaging plasmid psPAX2, and 20 μg of transfer vector (lentiCRISPRv2 42 constructs expressing start codon-specific [5′-GCTTGCGGCGAGACATGG-3′] and exon1-specific [5′-GCTGGGGTTTGCCTTGCTTG-3′] sgRNAs and Cas9 endonuclease), incubated for 20 min at room temperature and added to HEK293T cultures in fresh 6 mL of DMEM, supplemented with 1% FBS and 1 × penicillin/streptomycin. Medium was replaced with 8 mL of cDMEM at 4 h after transduction and after collection of vector-containing supernatant at 24 and 48 h, with a final collection at 72 h. Supernatant was kept at 4°C throughout, filtered (0.45 μm polyvinylidene fluoride; Merck Millipore), and centrifuged at 20,000 g for 4 h at 4°C. The resulting vector pellet was suspended in 100 μL of StemSpan SFEM II (STEMCELL Technologies) medium (240 × concentration) and stored at −80°C. For the purpose of this study, vector-containing supernatants were used for transduction without vector titration or subsequent assessment of vector copy number.
Expansion and differentiation of human umbilical cord blood–derived erythroid progenitor-2 cells
Human umbilical cord blood–derived erythroid progenitor-2 (HUDEP-2) cells 43 were maintained in SFEM II, supplemented with 100 ng/mL of human stem cell factor (hSCF; PeproTech), 10–6 M dexamethasone (Sigma–Aldrich), 5 μg/mL of doxycycline (Clontech Laboratories), 3 IU/mL erythropoietin (EPO; Binocrit 4,000; Sandoz GmbH), and 2 × penicillin/streptomycin (Thermo Fisher Scientific). Cells were cultured at a density of 0.5–1 × 106/mL in 25 cm2 Nunc flasks (Thermo Fisher Scientific) and incubated at 37°C in 5% CO2 humidified atmosphere. Of note, SFEM II was used as basal medium instead of Iscove's Modified Dulbecco's Medium (IMDM) suggested by the original protocol. 43 Comparison of both identically supplemented media resulted in 24% higher cell death in HUDEP-2 samples cultured with IMDM instead of SFEM II at day 4 of differentiation (data not shown).
Erythroid differentiation was induced by transfer of cells into StemSpan SFEM II containing 2% FBS (qualified, Australian origin; Thermo Fisher Scientific), 3% human serum (Sigma–Aldrich), 3% human plasma (harvested as supernatant of double-centrifuged EDTA-anticoagulated donor blood), 3 IU/mL EPO, 10 μg/mL of insulin, 1,000 μg/mL of holo-transferrin, 3 IU/mL heparin (all from Sigma–Aldrich), 2 × penicillin/streptomycin at a cell density of 1 × 106/mL, and 2.5 μg/mL of doxycycline. Two days after the induction of differentiation, the differentiation medium was topped up to maintain cellular density, and doxycycline-containing cultures were additionally supplemented with 1.25 μg doxycycline per mL of old medium. Differentiated HUDEP-2 cells were harvested for analyses at day 4 of differentiation.
Isolation, expansion, transduction, and differentiation of human CD34+ (hCD34+) cells
PB-derived human CD34+ (hCD34+) cells were isolated according to Protocol C, established by Cosenza et al. 44 Expanded cells at day 6 post-isolation were cultivated in 10 mL of expansion medium at a density of 0.5 × 106/mL, composed of 10 mL of StemSpan SFEM II, 100 μL of StemSpan CC100 Cytokine Cocktail (STEMCELL Technologies), 10–6 M of dexamethasone, 1 × penicillin/streptomycin, and 2 IU/mL of EPO to promote survival and efficient expansion of erythroid cells. Cellular expansion was continued until day 10, resulting in a total number of 21.6 × 106 cells.
CD34+ cells (1.1–1.5 × 106) were seeded in each well of a 12-well plate (Iwaki, Asahi Glass Co.), filled up to 2 mL with expansion medium. After 1 h, 50–75 μL of concentrated LV vector (first transduction step) was added to each well, and the plate was rocked back and forth several times. Twenty-four hours later, cells were gently re-suspended, ensuring homogeneous cellular distribution, and 1 h later, an additional 50–75 μL of LV vector was added in each culture (second transduction step). After an additional 24 h, 500 μL of fresh expansion medium containing 5 μg/mL of puromycin (Santa Cruz Biotechnology) was added in each well, initiating the enrichment of the virally transduced cells at a final concentration of 1 μg/mL of puromycin. For transduction with our HBBT87Q version 45 of the GLOBE vector, 46 a single-hit transduction was performed instead, as described elsewhere, 47 including hourly agitation for 6 h after transduction.
Differentiation of the untransduced controls and transduced hCD34+ cells was induced 24 h after initiating puromycin selection by cultivating cells in differentiation medium composed as follows: 70% α-Minimum Essential Medium (α-MEM; Corning CellGro), 30% FBS defined (HyClone Laboratories, Inc.), 10–5 M 2-mercaptoethanol (Sigma–Aldrich), 10 IU/mL of EPO, 10 ng/mL of hSCF, and 1 × penicillin/streptomycin. Cells were differentiated at a concentration of 0.5–1 × 106/mL for 4 days before their harvest for analyses. Fresh differentiation medium (1.5 mL) was added at day 2 of differentiation.
T7 endonuclease I assay
T7 endonuclease I (T7EI) assay was performed as previously described, 48 with minor changes, and was based on extracted genomic DNA of hCD34+ cells 48 h post-transduction and 24 h after puromycin-mediated selection. T7E1 assays were thus applied at a time point when DNA quality and yield were high, but before the culture endpoint, when DNA yield would be poor but when disruption efficiencies from lentivirally encoded endonucleases would still be higher. As extrapolated from independent transductions with endonucleases, efficiencies at the culture endpoint can be expected to be approximately 3.6 × the efficiency observed here (unpublished data). In brief, a primer pair (Exon1_FW 5′-AAAGCCATGACGGCTCTCCCACAAT-3′; Exon1_RV 5′-CGGCAATGGTTCCAGATGGG-3′), flanking the CRISPR/Cas9 target sites (start codon and exon 1) was used to polymerase chain reaction (PCR) amplify the target region of edited and mock-treated cells, respectively, as template. Subsequently, the purified PCR amplicons were denatured and slowly annealed to create heteroduplexes with loop-out regions at editing events, which were identified by digestion for 20 min at 37°C with 6 units of T7EI (New England Biolabs) and by analysis of products by agarose (2.5%) gel electrophoresis, before densitometric determination of band intensities and calculation of disruption efficiencies. Fold changes that are shown in parentheses in the main text, but no other values in text or figures, show values normalized for initial differential disruption efficiencies of CRISPR/Cas9-treated samples, so that those values give an approximate extrapolation of the γ-globin level that would be expected at 100% disruption efficiency. To this end, the baseline Cas9-only γ-globin level was subtracted from that measured for CRISPR/Cas9 samples, dividing the result by the respective disruption efficiency and adding back to that quotient the Cas9-only baseline γ-globin level.
Sanger sequencing and TIDE analysis
Exon1_FW/Exon1_RV-amplified PCR products spanning the CRISPR/Cas9 target sites were also subjected to Sanger sequencing based on the BigDye Terminator v3.1 Cycle sequencing kit (Applied Biosystems) using Exon1_FW as sequencing primer and 25 cycles of 96°C for 10 s, 50°C for 5 min, and 60°C for 4 min on a Veriti thermocycler (Life Technologies). After purification on Performa DTR Gel Filtration Cartridges (Edge Biosystems), mixed sequencing traces spanning the target site were produced by capillary electrophoresis on a Hitachi 3031xl Genetic Analyzer with Sequence Detection Software v5.2 (Life Technologies). Tracking of Indels by DEcomposition (TIDE; https://tide.nki.nl) was then used to decompose the mixed traces and determine the insertion and deletion pattern typical of the designer nucleases in hand. 49
Immunoblot assay
Cells were washed and re-suspended in 20 μL of radioimmunoprecipitation lysis buffer (20 mM of Trizma-HCl [pH 7.4], 150 mM of NaCl, 1% NP40, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 5 mM of EDTA; all Sigma–Aldrich) per 1 × 106 cells, supplemented with 1 × protease inhibitors (Roche), for analysis of total cellular protein. After centrifugation, the supernatant was mixed 1:1 with sample buffer (0.125 M of Trizma-HCl [pH 6.8], 2% SDS, 21% glycerol, 4% β-mercaptoethanol, 0.05% bromophenol blue). Lysate equivalent to 0.5 × 105 and 2.5 × 105 cells per well, for detection of α-globin and γ-globin, respectively, and corresponding β-actin bands, were loaded in parallel with a molecular weight protein marker (Nippon Genetics) on sodium dodecyl sulfate/polyacrylamide gels, separated by electrophoresis and blotted onto nitrocellulose Parablot NCP membranes (Macherey-Nagel GmbH) using wet electrophoretic transfer. Subsequently, quantitative protein transfer was confirmed by staining of membranes with Ponceau Red solution (Sigma–Aldrich), before blocking and overnight incubation with primary antibodies at 4°C, specifically anti-γ-globin (51-7) mouse monoclonal IgG1 (#sc-21756), anti-α-globin (H-80) rabbit polyclonal IgG (#sc-21005; both Santa Cruz Biotechnology, Inc.; dilution at 1:1,000), and anti-β-actin (AC-15) mouse monoclonal IgG1 (#A-1978; Sigma–Aldrich; dilution 1:10,000). Thereafter, washed membranes were incubated with the corresponding horseradish peroxidase–conjugated secondary antibody, specifically Goat-αMm-IgG(H + L) (#115-035-003) or Goat-αRabbit-IgG(H+L) (#111-035-003; both at 1:8,000; Jackson ImmunoResearch Laboratories). Bands were visualized using chemiluminescence detection (Lumisensor; GenScript), captured using a UVP Biospectrum 810 Imaging system (UVP) with VisionWorks™ LS 7.1 software, and quantified using the ImageJ quantification software. For the α-globin blot shown, linear image adjustment in Photoshop (Levels from 255 down to 109; Adobe) was required to reduce excessive background signal and allow band quantification.
Cytocentrifugation samples
For phenotypic characterization, differentiated hCD34+-derived cells were cytocentrifuged on a Cellspin II with an EASY rotor (Tharmac/Hettich) and sequentially stained in dianisidine (1.5% o-Dianisidine; Sigma–Aldrich; in methanol) for 2 min, in H2O2/ethanol solution (50% ethanol, 0.9% H2O2 in distilled water) for an additional 2 min, in May–Grünwald staining solution (Sigma–Aldrich) for 5 min, and finally in Giemsa staining solution (Fluka Analytica; Sigma–Aldrich) for 10 min. After a rinse in distilled water, sample slides were air-dried and mounted (Entellan®; Merck) with a coverslip. Slides were visualized under an IX73P1F inverted microscope, using LED illumination, a 40 × lens, an XC50 camera, and averaging seven frames per image in CellSens 1.7 (Olympus Corp.).
RP-HPLC analysis
In line with routine procedures for RP-HPLC analysis of thalassemic samples,38,39,50 blood samples were lysed in HPLC-quality distilled water (H2O; 4 μL of PB in 996 μL of H2O), vortexing and incubating at room temperature for 10 min. For cell culture samples, the sample material was centrifuged at 2,000 RCF for 10 min, re-suspended in 15 μL of H2O/300,000 cells, and twice frozen at −80°C and thawed. After 10 min of centrifugation at 21,000 RCF and 4°C, 800 μL of PB lysate were transferred into HPLC vials and 9/10 volume of culture-derived lysate to 100-μL HPLC microvials inside HPLC vials, respectively (all Altmann Analytik). This study did not analyze the resulting cell pellet for residual protein content or globin aggregates. Unless indicated otherwise, 3 μL of blood lysate (equivalent to 12 nL of blood) and 15 μL of HbA2 (Sigma–Aldrich; 200 ng/μL) or culture-derived lysate (equivalent to 300,000 cells), respectively, were injected per HPLC run.
Analyses were performed on a modular Prominence HPLC machine with an SPD-M20A diode array detector, an LC-20AD (low-pressure gradient) binary pump, and an SIL-20AC HT cooled autosampler to facilitate automated overnight runs (all Shimadzu). The stationary phase was an Aeris Widepore 3.6 μm XB-C18 25 cm 4.6 mm column behind a SecurityGuard UHPLC Widepore C18 4.6 mm guard column (all Phenomenex). Mobile phases were Phase A: 0.1% trifluoroacetic acid (TFA; Sigma–Aldrich) and 0.033% (6.4 mM) sodium hydroxide (330 μL per liter of 19.4 M NaOH; 50% NaOH for HPLC; Fluka), resulting in pH 3.0, and Phase B: 0.1% TFA in acetonitrile (E-CHROMASOLV for HPLC; Sigma–Aldrich). Gradients used were as given in Tables 1 and 2. Reagent cost, excluding glassware and column depreciation, is between €0.05 and €0.10 per sample, with acetonitrile (at a current price of €30 per 2.5 L through government tenders in Cyprus) as the main reagent expense.
Results
Separation of human globin chains
Toward lowered backpressure and greatest peak capacity and thus possibly reduced separation times, an HPLC column with core-shell technology was chosen. 51 The specific column, a Phenomenex Aeris Widepore XP-C18, was selected as a replacement of a previous fully porous C18 column,38,39 and has certified stability at pH 1.5–9.0, at backpressures up to 600 bar (60 MPa) and at temperatures up to 90°C. To minimize background noise in the analysis of rare samples at far-UV absorbance measurements, the study additionally drew on acetonitrile supplemented with 0.1% TFA without inclusion of other organic solvents as the organic mobile phase. As aqueous phase, 0.1% TFA was chosen, adjusted from pH 1.9 to 3.0 with 0.033% (6.4 mM) sodium hydroxide for increased column life. Likewise, 70°C was chosen as the highest routine separation temperature to extend column life, although still higher temperatures within the stability range gave even smaller peak widths and baseline separation of relevant peaks (see Supplementary Fig. S3; the effect is most pronounced for heme), potentially allowing a further reduction of runtimes for specific applications. After testing many permutations of gradients, temperatures, and speeds of the mobile phase (see Supplementary Fig. S4 for exemplary separations at 70°C column temperature), the study adopted for sample injection during an initial 1 min gradient for human samples, as it was found that this gave the narrowest peak widths for human α- and β-like globins. The protocol arrived at for rapid baseline separation of all human globins found in normal adults is shown in Table 1 and compares favorably in every aspect with separations on columns previously used in the authors' laboratory (see Supplementary Fig. S5). The sensitivity of the protocol is demonstrated in Fig. 1 for separations of the equivalent of 12 nL commonly encountered human-derived blood samples and the peak height equivalent amount (3 μg) of a commercial HbA2 sample. All samples shown were analyzed after storage of blood lysates at 4°C for >2 months. Although the present method is also suitable for the detection of the therapeutically relevant HBBT87Q variant45,52 and would thus allow post-treatment analyses of corresponding gene addition experiments in the presence of normal HBB (see Supplementary Fig. S6), separation and precise quantification of normal and T87Q β-globin chains would benefit from variant-specific optimization of conditions. Of note, the molar absorption of proteins is dependent on solvent conditions, such as pH, 53 so that changes in the solvent affect relative absorption of globin chains (as illustrated by differences in relative γ-globin peak areas for same-sample separation of cord blood by two different protocols on the same C18 Jupiter column; Supplementary Fig. S5). Consequently, total absorption of β- and α-like globin chains is not necessarily identical for normal samples. Whereas absolute quantification will require absolute external standards, relative quantification is instead performed by normalization to an internal reference peak assumed constant (such as α-globin for β-hemoglobinopathies) followed by calculation of fold-changes compared to a control sample. In this vein and of value for the analyses of rare samples, the present protocol is able to detect β- to α-globin ratios for sample amounts as low as 0.75 nL of PB (equivalent to 113 ng of hemoglobin; see Supplementary Fig. S7). In the following, typical gene and cell therapy applications were chose to demonstrate the utility of the analysis, including application-specific optimization of the method for co-detection of human and murine β-globin chains.
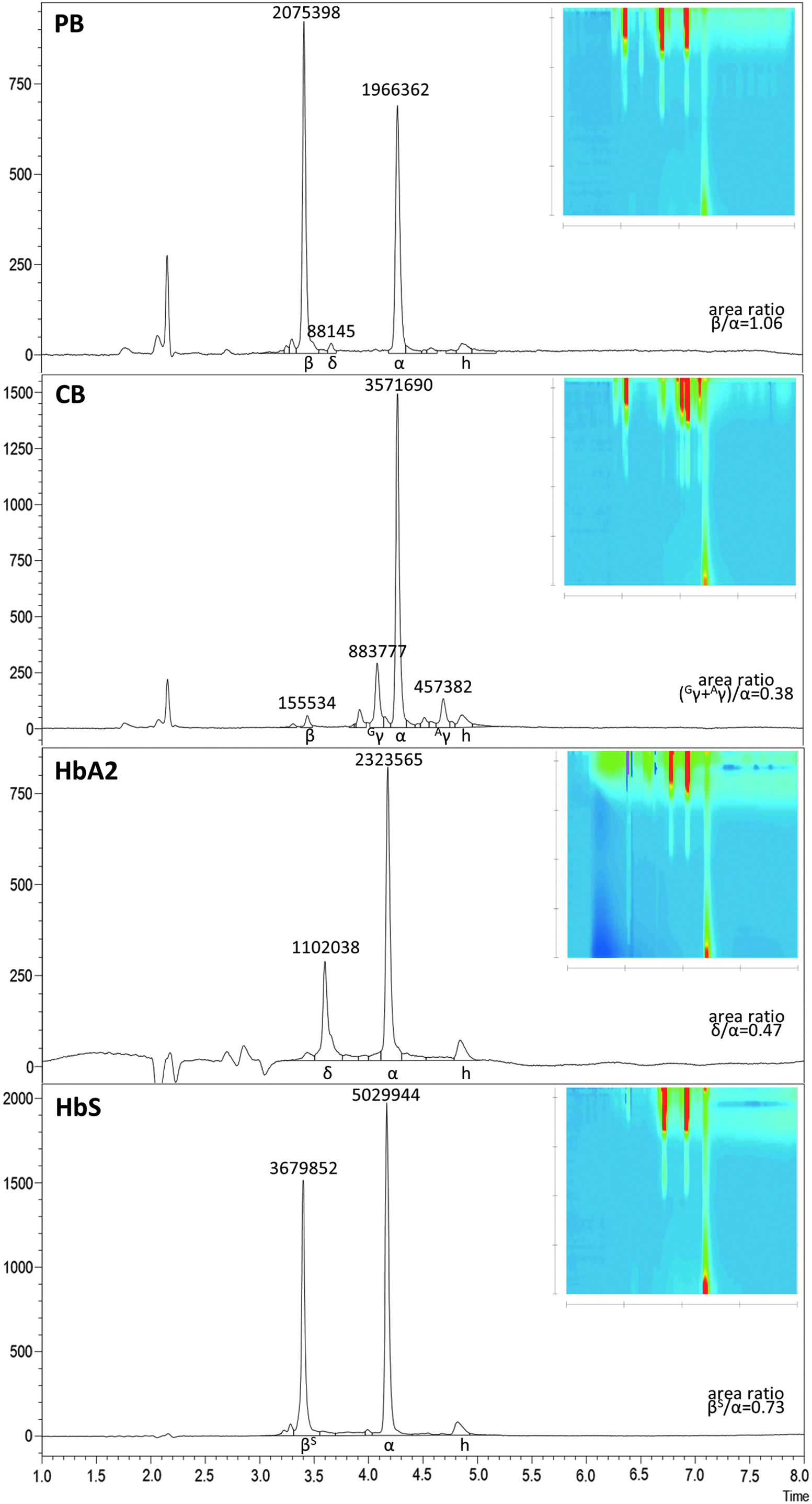
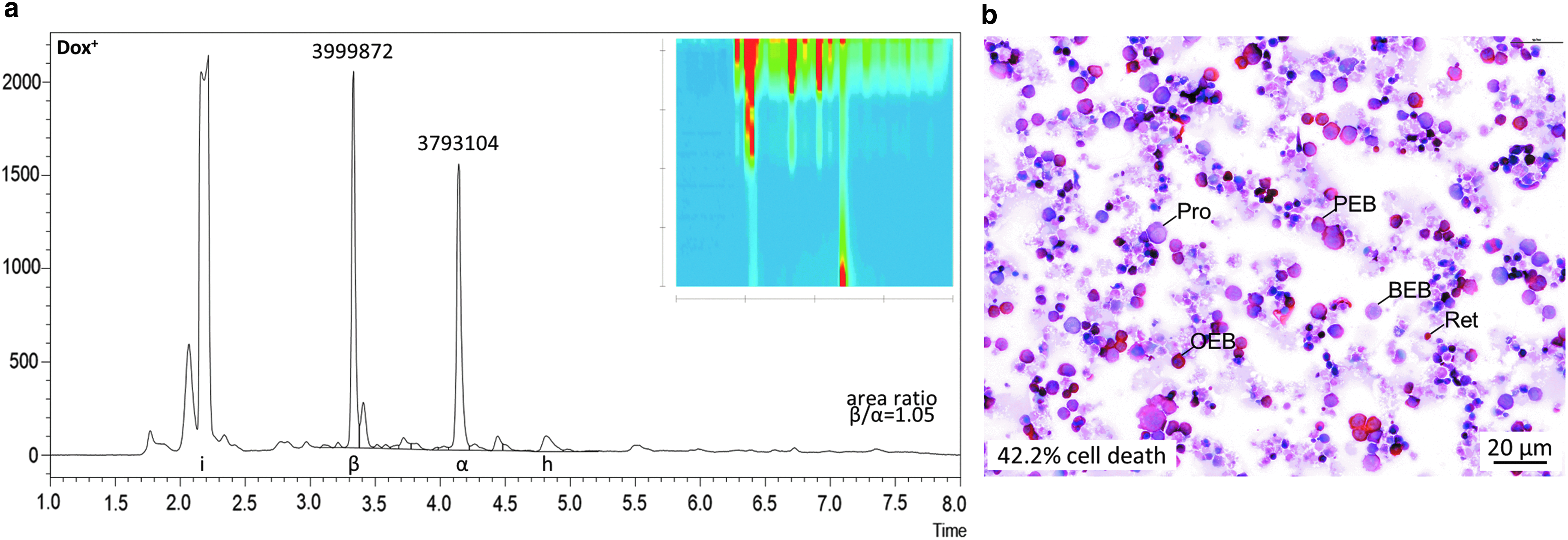
Analyzing differentiation of HUDEP-2 cells
HUDEP-2 cells have been adopted by many groups as a cell line with properties similar to human adult CD34+ basophilic erythroblasts, with ongoing optimization of differentiation protocols.20,43 These cells proliferate and survive owing to doxycycline-inducible transgene expression of human papilloma virus 16 proteins E6/E7, whose downregulation upon doxycycline withdrawal in turn allows erythroid differentiation. Here, RP-HPLC was employed to assess globin chain expression after 4 days of differentiation at a doxycycline concentration of 2.5 μg/mL (Fig. 2a), and the state of differentiation was analyzed with corresponding cytocentrifugation analysis (Fig. 2b), which has lower sample requirements than alternative flow-cytometry analysis of erythroid differentiation.20,54
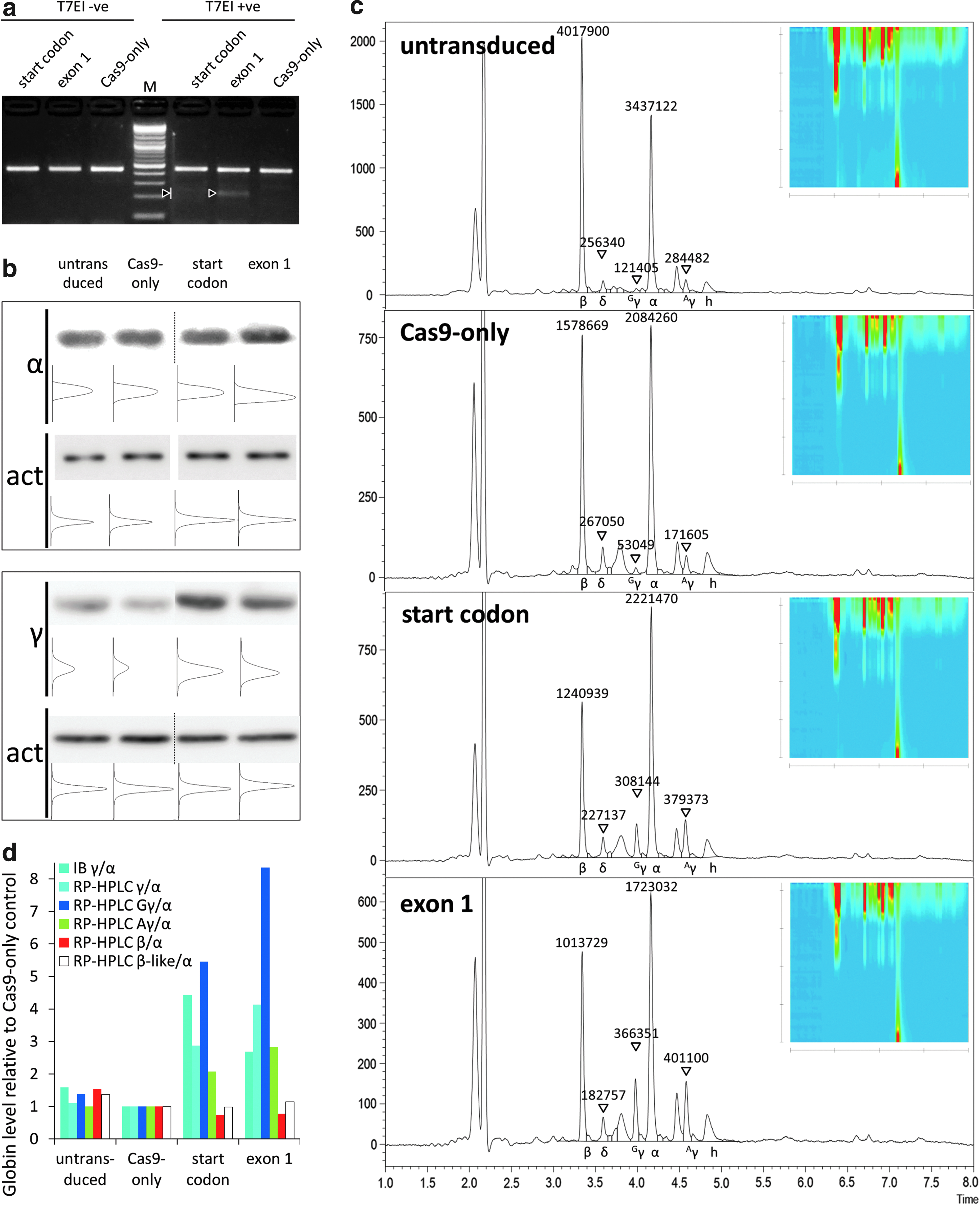
Assessment of γ-globin induction after genome editing of CD34+ cells
With the advent of the versatile CRISPR/Cas9 genome-editing platform, 55 gene disruption by the efficient NHEJ pathway has been adopted by many groups for functional analyses and even for correction of disease.56–60 Disruption of the open reading frame, however, has a fixed percentage of in-frame repair, which might maintain partial function of the target gene. This observation was clearly established for the repetitive, structural dystrophin protein, 61 but might to some extent also hold for NHEJ-mediated disruption of the transcription factor and γ-globin repressor BCL11A, 62 where it would thus lower the efficiency of γ-globin induction. This prompted a comparison of the disruption of BCL11A exon 1 with disruption of its start codon, where most types of insertion or deletion would abolish any production of functional BCL11A protein. Thus, primary CD34+ cells of a carrier for the HBBIVSI-110(G>A) mutation were treated with buffer only, Cas9 only, and dual-promoter CRISRP/Cas9 lentiviral vectors, disrupting either the BCL11A start codon or a 5′-proximal target of its exon 1, respectively. We chose a carrier sample for this analysis, arguing that cellular saturation of β-like globin expression would be reduced compared to a normal sample and that the level of γ-globin background expression would be lower than in a thalassemic sample. 63 Independent analyses for the two designer nucleases in question using TIDE-based decomposition of sequence traces indicated for an independent sample that 70% and 52% of editing events for the start-codon-specific nuclease and the exon-1-targeting nuclease, respectively, introduced a single-nucleotide insertion (Supplementary Fig. S8). For the present sample, start-codon disruption had an overall efficiency of 9.7%, that of exon 1 of 14.6%, as detected by T7E1 assay (Fig. 3a) 24 h after onset of erythroid differentiation. These indicative differences in disruption efficiencies, albeit an underestimation of disruption achieved at the culture end point, could be used to normalize detected changes in γ-globin expression and thus allow functional comparison independent of differential transduction efficiencies, as shown in parentheses below. Immunoblots (Fig. 3b) and same-sample RP-HPLC analysis (Fig. 3c) revealed consistent results of an induction of γ-globin expression with both dual-promoter vectors, with slight deviation in the relative γ-globin induction by start-codon and exon-1 disruption. Measured by RP-HPLC, raw (and efficiency-normalized) fold changes of γ-globin were 3.0 × (22.2 × ) for start codon and 3.4 × (17.5 × ) for exon-1 targeting compared to Cas9-only. Critically, RP-HPLC analysis avoided normalization of same-sample analyses, which is required across membranes for immunoblotting, and in contrast to immunoblots showed greater γ-globin induction by exon-1 targeting compared to start-codon targeting before normalization for disruption efficiencies (Fig. 3d). Additionally, the RP-HPLC analysis allowed differential quantification of induction for each γ-globin chain, showing for this sample preferential induction of Gγ for both BCL11A-specific designer nucleases (with Gγ:Aγ = 0.8 for SC and Gγ:Aγ = 0.9 for Exon 1 in bulk cultures) compared to the Cas9 control (Gγ:Aγ = 0.3), toward a restoration of a fetal γ-globin expression pattern of predominantly Gγ.64,65 Whereas for immunoblots sample material for additional analysis of β-globin expression was insufficient, a single analysis run by RP-HPLC allowed the therapeutically most relevant quantification of the overall balance of β-globin- to α-globin-like chains. Analyses comprising all major globin chains are critical for the assessment of treatment efficiency in β-thalassemia patients and in the present case of a β-thalassemia carrier revealed a concomitant decrease of β-globin expression and no overall increase of β-like globin chains after induction of γ-globin. This was in agreement with observations for normal individuals 66 and with the expectation that erythrocytes from healthy carriers already produce saturating amounts of β-like globin chains.
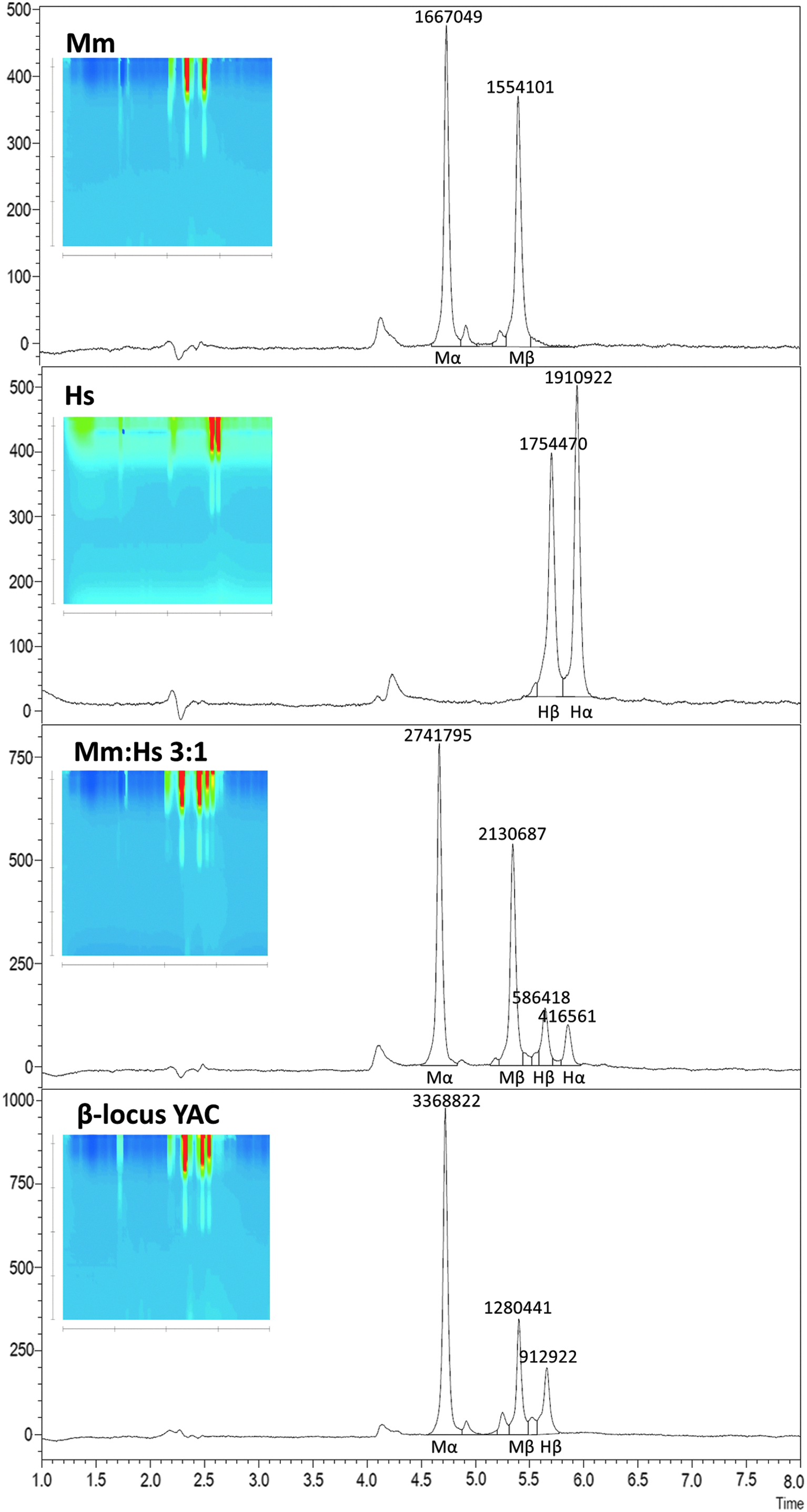
Detection of human β-globin in humanized mouse models
Preclinical assessment of HBB gene addition in animal models, be it in murine HSCs of thalassemic animals or in human HSCs transplanted into immunodeficient mice, usually necessitates the detection of human β-globin chains against a murine globin background. To this end, this study set out to develop a modified protocol for the separation of murine and human β-globin chains. At standard operating temperatures from room temperature to 70°C, use of different organic solvents, such as isopropanol, methanol and 3:1 (v:v) acetonitrile:methanol, against 0.1% TFA did not achieve separation of β-globin chains from both species (data not shown). Variation of temperature for an otherwise identical HPLC gradient (Supplementary Fig. S2) showed that extremes of temperature for standard HPLC equipment (0–4°C or 85°C) allowed separation of murine and human β-globin chains for the original setup of column and solvent combination. Frequent operation at acidic pH and at high temperatures would be detrimental to column life, which prompted separation at 4°C. After trying different combinations of baseline and gradient concentrations, the study arrived at the specific combination of baseline and gradient running phase composition given in Table 2 and illustrated for representative samples in Fig. 4. This is a compromise between alternative conditions that either achieved complete baseline separation of human and murine β-globin chains for broad and shallow peaks, or that showed overlap between both chains at albeit narrower and thus more accurately quantifiable peaks (see Supplementary Fig. S3). The specific hybrid protocol does not resolve Hbbs from Hbbd mouse strains, but detects additional globin peaks for CD-1 and DBA mouse strains (see Supplementary Fig. S1). Most importantly, the protocol allows separation and quantification of murine and human α- and β-globin chains, for example for mixtures of murine and human peripheral blood (Fig. 4c) and for mice transgenic for the human β-globin locus (Fig. 4d), and would thus be suitable for the analysis of human–mouse chimeric blood, for routine screening of HBB-transgenic animals (see Supplementary Data for automatic analysis of 45 mouse samples in 6.5 h), and for follow-up of lentiviral HBB gene-addition in thalassemic mice.
Discussion
This article presents two optimized RP-HPLC analysis protocols for rapid quantification of human globin chains and combined quantification of murine and human globin chains, respectively, and explores their fields of application in gene therapy studies. To showcase the method, this study demonstrated the suitability of core-shell-based RP-HPLC columns for rapid and highly sensitive routine blood globin-chain measurements, assessment of in vitro erythroid differentiation, quantification of γ-globin induction after genome disruption by designer nucleases, detection of HBBT87Q lentiviral transgene expression in normal human background, and detection of human globin chains against murine background in chimeric or transgenic blood samples. Moreover, high-quality readouts from crude blood lysates after prolonged storage at 4°C or based on frozen cell pellets validated the established methodology as a robust tool for analyses of third-party samples in collaborations. In order to demonstrate the utility of the methods described, they were used to investigate different phenomena for the gene therapy of hemoglobinopathies. First, analysis of BCL11A knockout allowed differential quantification of induction for Gγ- and Aγ-globin chains. Second, comparison of the start codon and of a proximal exon-1 region of BCL11A for NHEJ-mediated disruption showed comparable efficiency for both approaches, in line with the finding that the majority of exon-1 targeting events introduced a +1 frameshift. Third, the constraints on protein expression and competition at the β-globin locus were demonstrated for in vitro differentiation of gene-edited primary human HSCs of a β-thalassemia carrier, by showing that, as for carriers in vivo, induction of γ-globin expression does not alter the overall balance of α-globin- to β-globin-like chains but instead leads to concomitant downregulation of β-globin. For additional applications, including baseline separation and accurate quantification of HBBT87Q, for the detection of other therapeutically relevant β-globin variants, such as βE and βC and the highly anti-sickling HBBG16D,E22A,T87Q variant, 67 which were not available to this study, the current protocols may serve as a template for the establishment of dedicated rapid analysis methods.
In conclusion, the RP-HPLC protocols presented here allow single-step assessment of globin expression for precise, simple and inexpensive quantification of human globin chains in human and murine samples. Ease of sample preparation, automation and speed of analyses, and minimal sample requirements make the present core-shell-based RP-HPLC particularly suitable for precious primary and gene therapy–related cell material.
Footnotes
Acknowledgments
We are indebted to our blood donors for sample provision and to Dr. Kenneth R. Peterson for the provision of β-locus YAC mice. 68 We are grateful to our mouse facility staff for their support and advice. This study was co-funded by the European Union's Seventh Framework Program for Research, Technological Development and Demonstration under grant agreement no. 306201 (THALAMOSS), by an Erasmus+ Fellowship, by a Ph.D. Student fellowship from TELETHON Cyprus, and by the Republic of Cyprus through the Research Promotion Foundation under grant agreement YΓEIA/BIOΣ/0311(BE)/20 and through core funding of The Cyprus Institute of Neurology and Genetics.
Author Disclosure
No competing financial interests exist for any of the authors.