
Abstract
Lentiviral vectors are used in laboratories around the world for
Introduction
The third-generation lentiviral vector design and original protocol for high-titer virus production was developed in the Naldini and Trono laboratories.1–3
In the 20 years since, lentiviral vectors have become commonly used for gene delivery for preclinical research applications and are now becoming the vector of choice for
This gene therapy delivery system is well-characterized, effectively delivers genetic material into dividing and nondividing cells, stably integrates into the host cell genome to maintain long-term expression in target cells, and delivers larger amounts of genetic material than other vector systems. Furthermore, third-generation lentiviruses are thought to be nonpathogenic and non-immunogenic, although long-term investigations are still underway. Guidance on broaching this and related subjects with regulatory authorities in the course of developing a clinical trial plan is available. 10
The third-generation lentiviral vectors described here are considered to be replication-incompetent and self-inactivating vectors, due to the number of essential genes that have been deleted from the third-generation lentiviral packaging system.
11
These third-generation lentivector systems have a number of additional safety features over the second-generation: the viral
Preclinical gene therapies made with substantially similar lentiviral vectors to the clinical material can be used for efficacy testing in various model systems,
The goals of this report are to share the Standard Operating Protocol (SOP) for preclinical lentivirus production and the process development considerations for clinical-grade lentivirus manufacture, where the quality of the product, consistent production, and GMP compliance must be met, as well as the requirements set in the relevant manufacturing and clinical trial authorizations. A broad overview of processes related to the manufacture of GMP lentiviral vector-based gene therapies is shown in Fig. 1; specific considerations for converting preclinical viral manufacturing protocols and platforms to scaled-up GMP manufacture are provided after the protocol itself and the troubleshooting section.
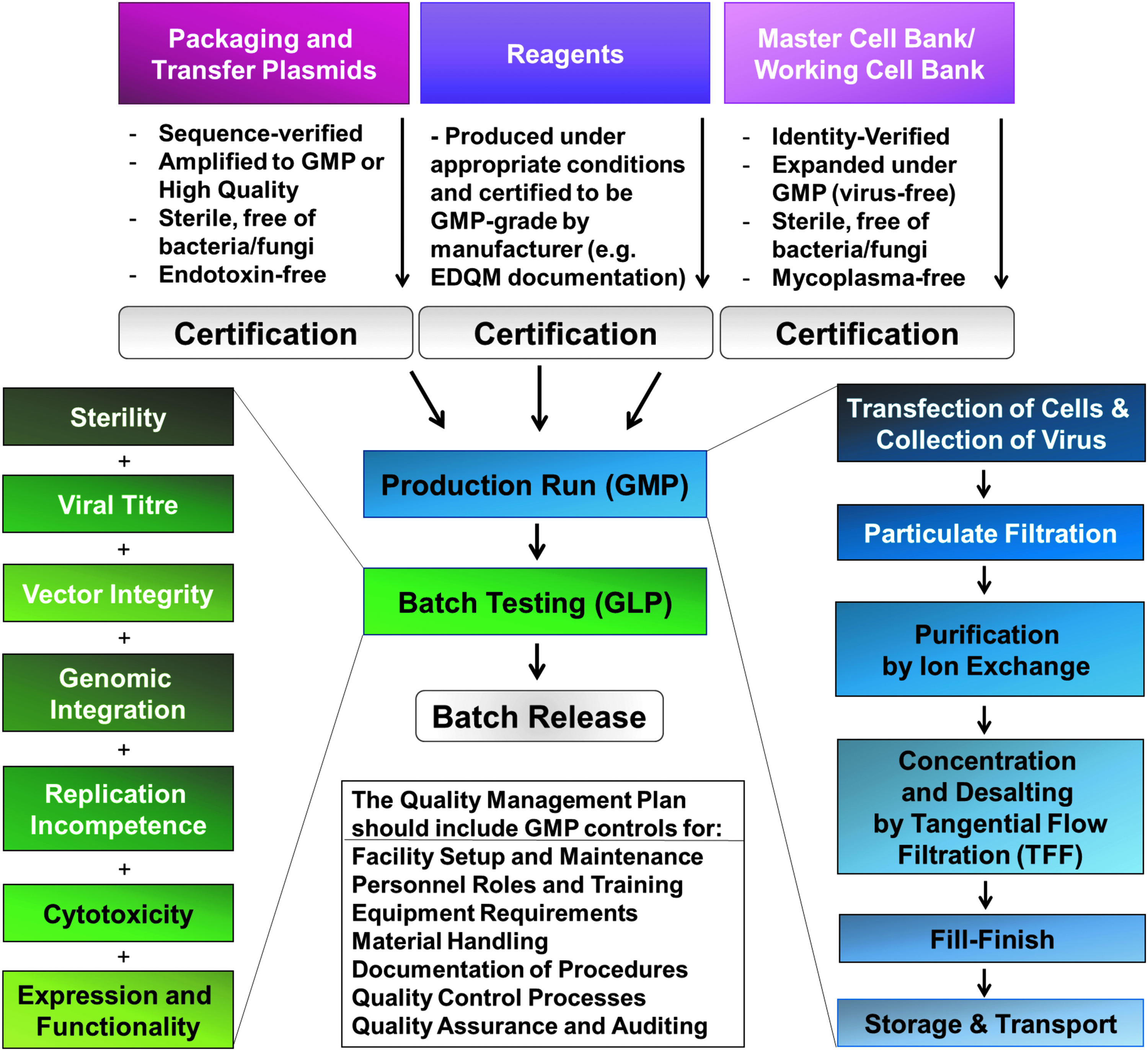
Process for lentiviral production under Good Manufacturing Practice.
Materials
HEK293T producer cell line
HEK 293 cells were generated in 1973 by isolation and subsequent adenoviral-mediated transformation of human embryonic kidney cell cultures in Alex van der Eb's laboratory in Leiden, The Netherlands. The 293T cells are derived from HEK 293 cells but stably express the SV40 large T antigen, which can bind to SV40 enhancers-of-expression vectors to increase protein production. This cell line is easy to grow, transfects readily, ectopically expresses genes, and has been widely used in research laboratories for many years. HEK293T are also used as cell factories to generate lentiviral vectors 23 (as described here in “Experimental Procedures”).
Plasmids encoding the genetic payload and viral packaging components
The transfer vector should contain the desired transgene(s) and other components required for viral integration and expression in the host genome. The full sequence justification of pMUH, a versatile third-generation lentivector, 10 is given in Table 1. This and similar third-generation lentivector transfer plasmids contain multiple cloning sites for insertion of target genes, as well as a fluorescent reporter or selection marker for preclinical use. Using this vector plasmid, the promoter driving gene expression, and up to three genes, can easily be swapped to achieve flexibility for preclinical viral vector development and to achieve the leanest lentivector construct sequence for producing a targeted gene therapy.
RSV, Rous sarcoma virus; LTR, long terminal repeat; HIV-1, human immunodeficiency virus type 1; RRE, Rev response element; cPPT/CTS, central polypurine tract and central termination sequence; UbC, human ubiquitin C promoter; MCS, multiple cloning site; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element; SV40 poly(A) signal, SV40 polyadenylation signal; SV40 ori, Simian virus 40 (SV40) origin of replication; f1 ori, f1 bacteriophage origin of replication.
It is inadvisable for fluorescent reporter proteins to be included in viral vectors used for gene therapies in humans, since these proteins are thought to be immunogenic. 24 However, efficacy tests in cell cultures or animal models can make use of them. Furthermore, European regulatory guidance urges careful and well-justified use of antibiotics during plasmid preparation and viral production in order to avoid the cultivation of antibiotic-resistant strains of bacteria. Such considerations should be taken into account when designing the specific characteristics of a transfer vector.
Packaging plasmids
The packaging genes used for preclinical third-generation lentivirus manufacture are contained on three separate plasmids, which are co-transfected into the producer cell line along with the transfer vector. These three plasmids are described in Tables 2–4. The four plasmids are shared among academic laboratories, but would have to be specifically licensed for use in clinical-grade lentivirus manufacture.
β-globin poly(A), human β-globin polyadenylation signal; CMV, cytomegalovirus; VSV-G, vesicular stomatitis virus G glycoprotein.
pRSV-Rev (Addgene #12253, from the laboratory of Prof. Didier Trono)
The sequence of the pRSV-Rev plasmid is described in Table 2. This is a cDNA expressing plasmid in which the joined second and third exons of HIV-1
pMD2.G (Addgene #12259, from the laboratory of Prof. Didier Trono)
The sequence of the pMD2.G plasmid encoding Vesicular stomatitis virus glycoprotein is described in Table 3. This cDNA expressing plasmid encodes vesicular stomatitis virus glycoprotein (
pMDLg/pRRE (Addgene #12251, from the laboratory of Prof. Didier Trono)
The sequence of the pMDLg/pRRE plasmid encoding viral core proteins is described in Table 4. This plasmid encodes
Reagents and consumables
Reagents used in preclinical manufacture are shown in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/hgtb). Many of these products are made or sterilized under high-quality conditions, traced from origin, and certified for GMP use.
Consumables, such as conical tubes and tissue culture flasks, used in preclinical virus manufacture, which are made or sterilized under high-quality conditions, traced from origin, and certified for GMP use, are suitable for use in GMP manufacture as well as for research purposes.
Instrumentation and equipment
1. Small incubator 37°C
2. Large orbital shaker/incubator
3. Bunsen burner
4. Microwave
5. Balance
6. pH meter
7. Gel electrophoresis and imaging system
8. Blue light transilluminator
9. Vortex mixer
10. DNA spectrophotometer
11. Biological safety cabinet
12. CO2 system
12.1. Tanks
12.2. Regulators
12.3. Hoses
12.4. Changeover unit
13. Incubator 37°C, 5% CO2
14. Benchtop centrifuges
14.1. Mini (pulse spin, 1.5 mL tubes)
14.2. Micro (1.5 mL tubes)
14.3. Small (50 mL tubes)
15. Ultracentrifuge
16. Refrigerator (4°C)
17. Freezers (−20°C and −80°C)
18. N2 storage tank
19. Light microscope
20. Bead or water bath
21. Vacuum pump
22. Pipettes
Experimental Procedure
Making reagents for cell culture and transfection
Cell culture medium (containing 10% fetal bovine serum)
In 440 mL of Dulbecco's modified Eagle's medium containing high glucose, add 50 mL of fetal bovine serum (FBS; for a final concentration of 10%), 5 mL of 200 mM L-glutamine (for a final concentration of 2 mM), 5 mL of 100 mM sodium pyruvate (for a final concentration of 1 mM), and 5 mL of 10,000 IU/mL penicillin/streptomycin solution (for a final concentration of 100 IU/mL). Store at 4°C.
Calcium chloride transfection reagent (CaCl2 2.5 M)
Dissolve 36.75 g of calcium chloride dehydrate in 100 mL MilliQ/PureLab Flex2 water. Sterilize by filtration with a 0.45 μm filter (Millipore). Store 1.5 mL aliquots for up to 6 months at −20°C. Working concentrations of CaCl2 are obtained by diluting the stock concentration in deionized tissue-culture-grade water (
Borate-buffered saline transfection reagent (BBS 2 × )
Dissolve 4.09 g of NaCl, 2.6625 g of BES, and 0.0525 g of Na2HPO4; add deionized tissue-culture-grade water to reach a volume of 250 mL. Adjust pH to 6.95. Sterilize by filtration with a 0.45 μm filter (Millipore). Store 14 mL aliquots for up to 6 months at −20°C.
Expanding HEK293T cell line for viral production by reanimation and passaging
1. Remove a vial of HEK293T expanded master cell line stored at −20°C.
2. Thaw at 37°C for approximately 2 min.
3. Add 1 mL of cells to 10 mL of cell culture medium in a 50 mL conical tube.
4. Centrifuge two balanced conical tubes at 250 g for 4 min.
5. Re-suspend cells in 10 mL of cell culture medium containing 10% FBS.
6. Count cells with a hemocytometer (for approximately 105 cells/mL).
7. Plate 10 mL of fresh cell culture medium–containing cells into a 10 cm dish or equivalent flask.
8. Place cells in the incubator at 37°C and 5% CO2 overnight.
9. When cells are 70% confluent, after 1–4 days, they are ready for passage.
10. Remove the serum-containing medium and rinse with phosphate-buffered saline (PBS).
11. Place trypsin-EDTA diluted in dPBS (1:10 or 0.05%) onto the cells and watch for signs of dissociation.
12. Once cells have dissociated, they can be collected and spun down.
13. The cell pellet should be re-suspended well in approximately 2 mL of cell culture medium.
14. The cells should then be further diluted in an appropriate volume of cell culture medium to plate the cells at 104–105 cells/mL.
Optimizing the transfection
Alternative transfection methods can be used for larger lentiviral vector production volumes. 26
1. Plate HEK293T cells in four 10 cm tissue culture dishes.
2. Cells are ready for transfection at 70% confluence, about 24 h after plating.
3. Thaw 2 × BBS and 2.5 M of CaCl2 solutions at room temperature.
4. Prepare different concentrations of CaCl2 (0.20 M, 0.25 M, 0.30 M, 0.35 M) in cell culture-grade dH2O, by diluting 80 μL of CaCl2 in 920 μL of dH2O and so on.
5. Place 10 μg DNA (GFP control plasmid) into each of four conical centrifuge tubes.
6. Add 500 μL of diluted CaCl2 per tube.
7. Pipette gently up and down.
8. Add dropwise 500 μL 2 × BBS per tube.
9. Pipette gently up and down.
10. Let the reaction stand for 30 min at room temperature in the hood.
11. Add the mixture drop by drop over each dish. Cover the entire area of the dishes.
12. Place cells in the incubator at 37°C and 3% CO2 overnight.
13. Check the fluorescence under the microscope after 24 h.
14. Determine which concentration gives the best transfection efficiency; >95% of cells should be green to achieve high levels of viral production.
Plating HEK293T for production of third-generation lentiviral vectors
Do not allow cells to grow over-confluent prior to viral production; it will lower the titer.
1. To coat plates, prepare 20 cm dishes or equivalent flasks.
2. Dilute 20 mL of poly-L-lysine in 80 mL of dPBS.
3. Pipette 10 mL of this 1:5 PLL:dPBS solution in each plate (10 × 20 cm).
4. Let plates stand for >15 min at room temperature in the hood.
5. Aspirate. The plates are now coated. Plate cells immediately after coating.
6. To plate cells, collect HEK293T from confluent plates.
7. Remove media and wash 4 × 20 cm confluent plates with 6 mL of dPBS.
8. Add 4 mL of Trypsin-EDTA (0.5%) to each plate, along with 6 mL of dPBS.
9. Incubate at room temperature for 2 min or until cells start to detach.
10. Add 10 mL culture medium and collect all cells.
11. Centrifuge two balanced tubes of cells at 250 g for 4 min.
12. Re-suspend cells in 160 mL of culture medium.
13. Count cells with a hemocytometer (the target is approximately 5 × 105 cells/mL).
14. Plate 16 mL of fresh cell culture medium containing cells into each dish. Place cells in the incubator at 37°C and 5% CO2 overnight.
15. Cells are ready for transfection at 70% confluence, about 24 h after plating.
Transfecting HEK293T to produce third-generation lentiviral vectors
Transfection efficiency is a major factor in lentiviral vector yields. Plasmid DNA concentration and quality, cell culture density, and pH variations of BBS are some of the factors that can affect transfection efficiency. If low transfection efficiency (<60%) persists, addition of sodium butyrate within 24 h post transfection can potentially enhance protein expression levels.
1. Thaw 2 × BBS and 2.5 M of CaCl2 solution at room temperature.
2. Prepare proper concentration of CaCl2, as previously determined for this batch of reagent.
3. Mix plasmids together, as described in Table 5.
4. Add 11 mL CaCl2 to the tube (for 10 × 15 cm plates).
5. Pipette gently up and down.
6. Add dropwise 11 mL of 2 × BBS to the tube (for 10 × 15 cm plates).
7. Pipette gently up and down.
8. Let the reaction stand for 30 min at room temperature in the hood.
9. Add 2.25 mL of the mixture into each dish. Cover the entire area of the dishes.
10. Place cells in the incubator at 37°C and 3% CO2 overnight.
The transfer vector length will vary according to the transgene size. When using different plasmid lengths, the DNA copy number should be always maintained as specified above, which can be achieved by increasing or decreasing the DNA mass.
Collecting virus-rich supernatants
• All waste that is possibly contaminated with virus should be inactivated by incubation with disinfectant for 30 min, and all of this waste should be collected in dedicated bags. These bags should be transferred to an autoclave using closed containers and inactivated by autoclaving at 121°C for 20 min.
• All autoclaved waste should be incinerated using the appropriate clinical waste route using an approved contractor.
• In the event of spillage of possibly viral vector-contaminated material, the area will be disinfected using appropriate disinfectant. The used paper towels and any other disposable material should be collected in dedicated bags and autoclaved as above. All work should be performed in a contained area. The wider environment is unlikely to become contaminated. However, a plan should be in place for both a contained spill and uncontained spill (
1. After 18 h, replace the culture media with 16 mL of fresh cell culture medium.
2. Do not collect this transfection media (remove and dispose in appropriate containment).
3. Put cells back in the incubator at 37°C and 3% CO2.
4. After 24 h, remove cell culture medium and place it into four 50 mL conical tubes. This is the first collection of media containing viral vectors.
5. Add 16 mL of fresh cell culture medium to each plate.
6. Put cells back in the incubator at 37°C and 3% CO2.
7. Centrifuge two balanced tubes of supernatants at 250 g for 4 min to remove cell debris.
8. Repeat with two more balanced tubes of supernatants (containing approximately 40 mL each).
9. Sterile filter the debris-free supernatants using a 0.45 μm filter (approximately 160 mL of supernatant in total).
10. Label this bottle. Wrap the cap with parafilm. Keep at 4°C overnight.
11. Repeat the following day (second collection of media containing viral vectors).
Serial ultracentrifugation for viral vector concentration
• When following the protocol described here in its entirety, yields of concentrated lentiviral vector reach physical titers between 1011 and 1012 particles/mL and functional titers between 107 and 1010 particles/mL. Note that some transgenes may produce cytotoxicity during viral production, resulting in lower viral titers. To overcome this problem, use of an inducible expression system 27 to control transgene expression during virus production is recommended.
• GMP manufacturing protocol requires non-ultracentrifuge-based concentration methods and more extensive filtration (Table 8) such as commercially available tangential flow filtration systems, which could be used to process the GMP viral material.
1. Place conical adaptors into SW28 buckets and SW28 tubes into conical adaptors.
2. Use 30 mL ultracentrifuge tubes (Beckman).
3. Place 24 mL of collected cell culture supernatant in each tube (from day 1 collection).
4. Centrifuge in SW28 at 67,684
5. Discard supernatant and replace tubes into buckets.
6. Place 24 mL of collected cell culture supernatant in each tube (from day 2 collection).
7. Centrifuge in SW28 at 67,684
8. Discard supernatant and keep tubes inverted.
9. Aspirate media off the edges of tubes.
10. Re-suspend the pellet with 4 mL of Hank's Balanced Salt Solution (HBSS):
a. Add 2 mL in the first tube and wash the five subsequent tubes.
b. Add 2 mL in the first tube and again wash the five subsequent tubes.
11. Load 4 mL of HBSS on top of 2 mL of 20% sucrose in dPBS.
12. Use 6 mL quick-seal tubes (Beckman). Add HBSS to complete fill.
13. Centrifuge in SW41 at 75,418
14. Discard supernatant and aspirate any remaining media at edge of tube.
15. Re-suspend the pellet gently in 100 μL of HBSS. Avoid formation of bubbles.
16. Place viral suspension into a low-bind Eppendorf tube.
17. Re-suspend remaining pellet gently in 100 μL of HBSS. Avoid formation of bubbles.
18. Add viral suspension into the same Eppendorf tube (200 μL total).
19. Seal tube with parafilm.
20. Shake tube on vortex at low speed for 30 min at room temperature.
21. Spin at 4000 g for 10 s to collect the virus-containing liquid.
22. Make twenty 10 μL aliquots of the virus and store at −80°C.
Troubleshooting
Guidance on troubleshooting is given in Table 6.
BBS, borate-buffered saline.
GMP considerations involving manufacture of lentiviral vectors for clinical use
Premises
• Premises should be clean, in good repair, for dedicated use, and secure. Conditions such as lighting, temperature, and humidity should be controlled within appropriate parameters so as not to affect the medicinal products or starting materials.
• Premises must be designed and controlled to ensure an aseptic environment for manufacturing of ATMPs. GMP procedures and practices must be in place for ATMPs, as sterilization of the finished product is not an option.
• Positive pressure is used to process sterile products for aseptic manufacturing. Pressure cascades should be clearly defined and monitored with alarms.
• Clean areas should be supplied with air which has passed through filters of an appropriate efficiency. Clean air devices should be classified in accordance with ISO 14644-1.
• The Quality Management System of the GMP manufacturing facility must include an emergency plan dealing with accidental release of viable organisms. The plan should describe procedures for containment, ensure the protection of personnel, and describe means of decontamination.
• Offices, restrooms, storage space, and laboratory animal laboratories should be separate from the manufacturing and testing areas. Manufacturing and testing areas should be separate from each other as well, with these procedures carried out by separate personnel.
• Quality control (testing) laboratories must be Good Laboratory Practice (GLP)-approved laboratories for assessing titer, sterility, replication incompetence, and other validation testing. Efficacy tests such as protein expression and functionality after gene delivery in relevant cells or tissues does not have to occur at GLP grade and can occur in standard academic research labs.
HEK293T producer cells
• GMP-grade producer cell lines for viral vector manufacture should be sourced from a suitable facility such as the American Type Culture Collection (ATCC) and maintained as a Master Cell Bank (MCB). The cell line can then be expanded and tested to be used as a certified Working Cell Bank (WCB). Documentation should be provided for receipt and storage of these starting materials.
• Establishment of seed lots and cell banks, including master and working generations, should be performed under appropriate conditions. This should include an appropriately controlled environment to protect the seed lot itself, the cell bank being produced, and the personnel handling these materials.
• During the establishment of the seed lot and cell bank, no other living or infectious material (
• Once a HEK293T cell bank is licensed from ATCC or sub-licensed from a contract manufacturing organization, this resource must be maintained to GMP standards, and must be certified free of mycoplasma and adventitious agents such as bacteria and fungi. Further, this cell line must be validated, including genotyping for identity verification and testing for the presence of human viral contaminants.
• Specific considerations for safety during viral manufacture include the risk of biological agents within the HEK293T human-derived cell line used for production, including blood-borne viruses: HIV, hepatitis B virus (HBV), hepatitis C virus (HCV), and hepatitis D virus (HDV). These are all dangerous pathogens, which can cause serious or fatal infectious diseases and cancers. HIV causes acquired immune deficiency syndrome (AIDS), while HBV, HCV, and HDV can cause hepatitis, liver cirrhosis, and cancer. While the HEK293T producer cell line has been used for decades with no known pathogens, the MCB acquired from ATCC and any WCBs derived from it may need further testing to validate the absence of pathogens. These cells can be safely disposed after use, upon exposure to a detergent such as TriGene, which destroys bacteria, fungi, and viruses.
• Documentation should be logged and made available to support traceability, including the number of generations (
• Cell banks should be stored and recovered in such a way as to minimize the risks of contamination (
Transfer and packaging plasmids
• The transfer vector plasmid contains the gene of interest (the transgene) with expression driven by a promoter sequence. Advice should be requested from the relevant regulatory authority regarding the subtype of lentivector transfer plasmid used. The type of vector used for preclinical studies and clinical-grade manufacture should be the same. The promoter and transgene sequence should be as close as possible to the final version.
• The lentivector transfer plasmid used for preclinical studies may also contain an ampicillin resistance gene or other selection marker for amplification in competent bacteria and other elements necessary for effective infection and gene delivery. Advice should be requested from the relevant regulatory authority regarding any restrictions on using a selection marker in the clinical-grade transfer plasmid. Guideline documents from the European Medicines Agency indicate that all beta-lactam antibiotics, such as ampicillin, and streptomycin should be avoided during virus production, as they are known to provoke sensitivity in certain individuals.
• The transfer plasmid often includes a fluorescent reporter gene, separated from the target gene by a cleavage site or internal ribosomal entry site, to identify cells that have been targeted in order to verify the extent of viral transduction. It may be useful to have this reporter tool for preclinical testing, but it is advisable to remove this gene for the purposes of viral manufacture for clinical applications because these proteins may be immunogenic. 24
• The transfer vector plasmid and the packaging plasmids used for clinical-grade viral vector manufacture should be amplified under GMP conditions. “High-quality” amplification may be suitable for early-phase (
• Documentation should be logged and made available to support traceability, including the source of the plasmids, any testing conducted on the plasmids, and the storage conditions. These should be consistent with specifications in the marketing authorization/clinical trial authorization, and any deviations should be recorded.
• The third-generation lentiviral vector system has a major safety advantage compared to previous systems, but this comes with a cost. Lower titers are generally obtained in third-generation compared to second-generation lentivirus manufacture system due to the higher number of separate plasmids (four instead of three, respectively) that must be successfully co-transfected to produce functional particles. Stable inducible packaging cell lines for lentiviral vectors have been proposed to overcome this problem.28,29 These cell lines are capable of producing higher amounts of vectors by expressing all lentiviral components at high levels within the cells. This method would also reduce the costs for scaled-up lentiviral manufacture, offering an additional benefit for clinical applications.
• When changing the components or manufacturing process during a transition to GMP, safety issues must be considered for the altered vector system. It is recommended that such changes occur early during transition to clinical-grade production of the gene therapies, as these can impact costs and regulatory considerations.
Reagents and consumables
• Many of the consumables (
• A GMP-grade reagent sourcing form should be created and used to order items and to record lots received. This process will ensure that raw materials can be traced to facilitate recall of products if necessary.
• Starting materials should be confirmed for release by quality control personnel before use in GMP manufacture. All incoming materials should be checked to ensure the lot corresponds to the order. All materials received must be logged.
• All starting materials should be stored under appropriate conditions and in an orderly fashion to permit batch segregation and stock rotation. Starting materials should be appropriately labeled.
• Labels applied to containers, equipment or premises should be clear and unambiguous. The format of labeling should be consistent throughout the facility and should include status (
• GMP manufacturing procedures must protect the product and the starting materials from the risk of contamination, within the accepted bioburden level described in the marketing authorization or clinical trial authorization.
• Where sterilization of starting materials is required, it should be carried out by heat if possible. Alternatively, other appropriate methods such as irradiation and filtration can be used for inactivation of biological materials.
• If plasmid and/or host-cell DNA persists in the medium, virus-rich supernatants can be treated with bacterially derived DNase or alternatively with Pulmozyme, a human DNase approved for human use by the Food and Drug Administration. 30 Pulmozyme treatment would provide a DNase with human rather than animal origin, but could significantly increase the cost of viral vector production and could potentially require an additional step during downstream processing to remove the enzyme.
• Ensure all reagents have a certificate of analysis.
• Ensure all reagents have an EDQM designation (if applicable).
• Ensure all reagents are traceable from point of origin.
• If a reagent has an animal or human origin, additional scrutiny may apply. For example, use of fetal calf serum in cell culture is common, especially for supporting the maintenance of HEK293T cells used for lentiviral and AAV production. However, such human- or animal-derived products must be justified for use in GMP manufacture; suitable origin of materials and traceability standards will apply according to relevant regulatory advice (
• As a substitute for small-scale tissue culture flasks or plates, bioreactors can be packed with macroporous microcarriers (
Instrumentation and equipment
• Many preclinical protocols utilize open-system, small-scale manufacturing. Open-system manufacturing may be sufficient for producing high-titer lentivirus for a first-in-human clinical trial. However, it is suggested that high-throughput closed-system manufacturing are generally used for GMP manufacture.
• The equipment (
• Manufacturing equipment must be adequately maintained. Repair and maintenance operations should not present any hazard to manufacturing processes or products themselves. Parts of production equipment that come into contact with the product must not react with the product in any way that would affect the quality of the product.
• All equipment must be cleaned to prevent contamination, and cleanings should be documented. Single-use disposable material should be used wherever possible. Sterilization of multi-use equipment in contact with the product must be validated.
• Equipment should be calibrated and/or inspected by appropriate methods at defined intervals to ensure proper performance, and these checks should be documented.
• Defective equipment should, if possible, be removed from production and quality control areas, or at least be clearly labeled as defective.
• Many scaled-up cell culture systems are disposable, single-use bioreactors to achieve closed-system processing. While this presents a significant advantage over flasks (which are limited in scale) or stainless steel containers (which require cleaning), single-use bioreactors have greater cost and environmental impact. In addition, the materials used in any disposables must be validated, including: the potential for leaks, the presence of leachables and extractables, the contact time with biological material, and any changes in risk due to temperature.
• It may be better to make the switch to closed-system manufacturing early in the translational process, so that process development concerns can be addressed during early phases in the project, and batch testing data can be used to specify the parameters to be met by future batches intended for authorized clinical use.
• Table 7 describes several options for scaled-up closed-system manufacturing using bioreactors for the growth of cells and collection of supernatant.
• Table 8 describes several options for tangential flow filtration as a scaled-up GMP-compatible method of purifying and concentrating viral particles.
GMP, Good Manufacturing Practice.
Manufacturing runs
• Prior to beginning a production run, it should be ensured that premises, equipment, and cell lines are in suitable conditions for GMP manufacturing.
• At every stage of processing, products and materials should be protected from microbial and other contamination.
• Generally ATMPs cannot be terminally sterilized. Therefore, manufacturing of the active substance and the finished product is required to be conducted in appropriate conditions to ensure aseptic manufacturing. For non-sterile raw or starting materials, additional steps may need to be taken to ensure subsequent aseptic manufacturing (
• Measures to prevent cross-contamination appropriate to the risks identified should be put in place. The manufacture of viral vectors and gene therapy medicinal products should be separated spatially and temporally from other products.
• When seeking a contract manufacturing organization as a partner for GMP manufacture of the clinical-grade product, it may be useful to query not only the platform technologies and manufacturing processes used, but also whether any other products are being concurrently manufactured in the same space. These other products may cause concern regarding multi-product manufacture within the same facility.
• Procedures for maintaining facility infrastructure, logging resources, maintaining equipment, documenting procedures, training staff, practicing waste disposal, reporting to regulatory bodies, and carrying out internal and external audits must be in place prior to production, as well as procedures for quality control and quality assurance.
Virus production
• Before any processing operation is started, steps should be taken to ensure that the work area and equipment are clean and free from any starting materials, products, product residues, or documents not required for the current operation.
• Centrifugation of products can lead to aerosol formation and containment of such activities to minimize cross-contamination is necessary.
• The growth-promoting properties of culture media should be demonstrated to be suitable for its intended use. If possible, media should be sterilized
• Addition of materials or cultures to fermenters and other vessels and sampling should be carried out under carefully controlled conditions to prevent contamination. Care should be taken to ensure that vessels are correctly connected when addition or sampling takes place.
• The gene therapy itself and its clinical application should be described in the Investigational Brochure. Details on manufacturing and testing of the viral gene therapy and the raw materials that go into it (
• Many of the reagents used in preclinical lentivirus manufacture are made or sterilized under high-quality conditions, traced from origin, and certified for GMP use. Any human- or animal-derived products (
• For both preclinical and GMP manufacture, HEK293T producer cells can be transiently transfected to allow packaging of the viral vector containing genetic payload. In this protocol, the affordable and efficient method of calcium phosphate co-precipitation is used, although other proprietary methods can be used. Stable cell lines used for production must be specified to the regulatory authorities, including any viral vectors used and the raw materials used in their production.
• Ultracentrifugation is commonly used to concentrate lentiviral particles for preclinical applications in gene delivery particularly for
• The final formulation in which the viral particles are re-suspended may be a buffered salt solution or suitable alternatives. Any use of growth media, supplements, transfection reagents, and resuspension media during manufacture must be documented and justified in the IMPD.
Documentation of procedures and materials
Documentation allows the recording of quality controls, aids quality assurance, and provides evidence to support batch release. The goal of documentation is to establish, control, monitor, and record all materials and activities which directly or indirectly may affect the quality of medicinal products.
• All calibration and cleaning of equipment and instruments must be documented.
• All handling of materials and products, such as receipt and quarantine, sampling, storage, labeling, dispensing, processing, packaging, and distribution, must be documented.
• All reagents, producer cells, plasmids, and viral vectors must be documented (see Table 9).
• Documentation for manufacturing and testing may be paper-based or electronic, and a data management plan should be in place. Shadow copies allow data to be retrieved if accidentally deleted. Facility management must ensure that any servers containing documentation are housed in temperature-controlled room with restricted access and with redundant power supply. Data should be backed up regularly to a mirrored server in a separate building.
• Batch documentation should be kept for 1 year after expiry of the batch to which it relates or at least 5 years after certification of the batch by the QP, whichever is longest. For any investigational medicinal products, the batch documentation must be kept for at least 5 years after the completion or formal discontinuation of the last clinical trial in which the batch was used.
Footnotes
Acknowledgments
We thank Professor Didier Trono for providing the plasmids pRSV-Rev, pMD2.G, and pMDLg/pRRE. This work was supported by the Wellcome Trust and Engineering and Physical Sciences Research Council through an Innovative Engineering for Health grant (CANDO). The authors are grateful to the ATMP Manufacturing Community for linking a network of professionals with expertise in this area.
Author Disclosure
No competing financial interests exist.