
Abstract
Muscle-derived progenitor cell (myoblast) therapy has promise for the treatment of denervated, weakened, and fibrotic muscle. The best methods for injecting myoblasts to promote fusion and retention have yet to be determined, however. Mesenchymal stem/stromal cells have also been reported to have beneficial effects in restoring damaged tissue, through increasing vascularization and reducing inflammation. The interactions between human primary skeletal myoblasts and bone marrow-derived mesenchymal stem/stromal cells were examined using time-lapse images put into video format. Of interest, there is a high degree of cell-to-cell interaction with microparticles transferring between both cell types, and formation of nanotubules to bridge cytoplasmic contents between the two types of cell. This model provides an in vitro platform for examining mechanisms for cell-to-cell interaction preceding myoblast fusion.
Introduction
Autologous muscle-derived progenitor cell (myoblast) therapy has promise for the treatment of denervated, weakened, and fibrotic muscle. Although promising, there is room for improvement and the best methods for injecting these progenitor cells have yet to be determined. Key goals in the field are to enhance understanding and to improve mechanisms to enhance fusion of donor myoblast nuclei into muscle fibers to promote the sustained retention needed for myoblast therapy of skeletal muscle tissue.
Mesenchymal stem/stromal cells (MSCs) have also been reported to have beneficial effects in restoring damaged tissue, through increasing vascularization, producing beneficial factors, and reducing inflammation while temporarily modulating the immune system (reviewed in refs.1–4 ). The mechanisms that could contribute to improved myoblast and MSC therapy, alone and in combination, are under investigation by our group and others. 5 MSCs can promote an increase in MMP-2/9 expression in myoblasts and stimulate their mobilization, differentiation, and fusion. 6
For donor myoblast fusion into the recipient fiber to occur, the fusion partners must adhere their membranes, open up fusion pores to allow cytoplasmic material exchange, and then merge into one cell. 7 The study of muscle development in Drosophila has paved the way for understanding this process, and knowledge is constantly updated.7,8 Proteins that could contribute to the process, previously implicated in the mammalian myoblast fusion process, are nephrin, 9 Kirrel, 10 GRAF1, 11 or others currently under study. 7 Increasing levels of fusion of implanted myoblast nuclei into existing, damaged muscle fibers is a significant goal.
We have studied the interactions between primary human skeletal myoblasts and human bone marrow-derived MSCs using time-lapse images (Figure 1) put into video format. The integration of lentiviral vectors expressing enhanced green fluorescent protein and tomato red in the two cell types, respectively, allowed tracking of each cell in relation to neighboring cells over time in the culture, without photo-bleaching. The methods for videomicroscopy to examine MSC and myoblast interactions over days in culture, as described in the current report, represent an advance over current methods where fluorescent antibody-based dyes rapidly blanch or leach out of the respective cell type.
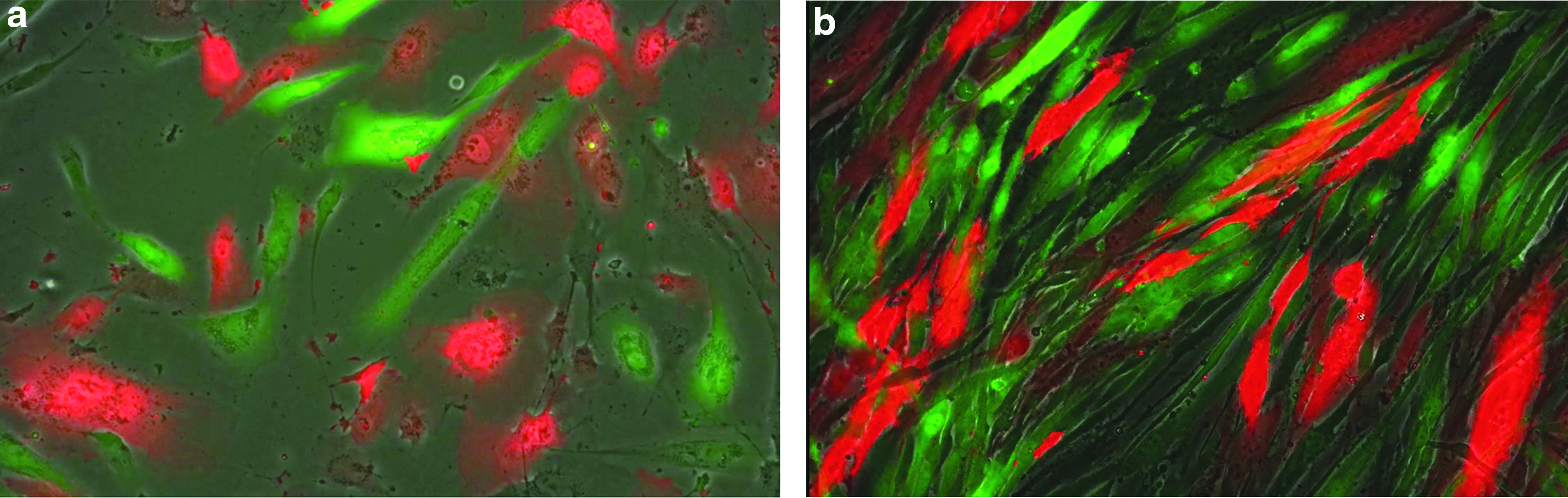
Single image captured from video showing interaction between human myoblasts labeled with a lentiviral vector carrying the eGFP gene and human bone marrow-derived MSCs labeled with a lentiviral vector carrying the gene for Tomato Red. Microparticles shed from both cell types and extensive cell-to-cell communication can be observed at
In the current studies there was a high degree of cell-to-cell interaction with production of visible microparticles by both cell types, as shown by parcels of membrane-bound eGFP and tomato red deposited behind the migrating cells, and formation of nanotubules that could bridge communication and potentially allow exchange of cytoplasmic material between the two types of cell. When the microparticles bind and release contents into a new target cell, as observed in the Supplementary Video S1 (Supplementary Data are available online at www.liebertpub.com/hgtb), there is not a yellow coloration as when red and green are overlaid, but rather the fluorescent molecules are rapidly assimilated into the target cell without a visible color change.
We have not seen obvious fusion of the two cell types in vitro during the times studied (up to 96 hr of co-culture), although this data does not preclude fusion in vivo. The model provides a platform for examining factors that could promote better myoblast fusion. Fusion proteins of the factor under study and the fluorescent molecules described here (such as Kirrel-tomato red and nephrin-eGFP) could be created to allow a direct observation of the protein localization during interaction between the two cell types.
We have previously described an ovine model for tongue regeneration using autologous myoblasts. 5 Our future goal is to co-administer autologous myoblasts with MSCs into the tongues of human patients with severe dysphagia, to potentially enhance the size and function of their muscle tissue. The co-administration of MSCs could enhance survival, vascularization, and potentially fusion of injected myoblasts. The type of durable cell labeling followed by time-lapse imaging over a period of days that we describe in this report is useful to understand the cell-to-cell interactions that occur between different types of cells that are under consideration for co-administration to sites of tissue damage in “next-generation” combination cell therapy products.
Methods
Lentivirus
To produce live, replication-incompetent lentivirus, Lenti-X 293 T-cells (Clontech) were transfected with 25 μg of the lentiviral transfer vectors, 25 μg of pCMV-Δ8.9 (packaging plasmid expressing gag and pol), and 5 μg of pMDG-VSVG (vesicular stomatitis virus glycoprotein-pseudotyping envelope) using TransIT Reagent (Mirus Bio LLC), following manufacturer's instructions. One day posttransfection, medium was changed to Ultraculture Serum Free Media (Lonza). The third day posttransfection (or two days after media change), vector supernatants were collected and concentrated by ultrafiltration using Centricon Plus 70 filter units (Millipore) and then filtered through 0.45 μm Spin-X Centrifuge filter tubes (Costar). The vectors were aliquoted and stored at −80°C. The viral titer was calculated by performing serial dilutions of virus on target cells and assaying for fluorescent reporter expression after 3 days.
Lentiviral transductions
Human myoblasts were transduced by the lentiviral vector pCCLc-MNDU3-LUC-PGK-EGFP-WPRE, and MSCs by lentiviral vector pCCLc-MNDU3-tomato-WPRE. For each cell type used, transduction efficiency as a function of MOI was measured using a GFP-expressing vector in order to adjust for differences in susceptibility to lentivirus between the cell types.
Transductions were performed by plating cells at 6667 cells/cm2. The following day, lentivirus was added to culture medium at an MOI of 40 in the presence of 10 μg/ml protamine sulfate (APP Pharmaceuticals), used to increase nonspecific viral uptake by cells. 12 The next day the medium was replaced and cells were cryopreserved four days posttransduction. Transduction efficiency was determined by flow cytometry for vectors containing fluorescent markers. The MSCs labeled with the tomato vector were 87% red, as assessed using flow cytometry.
Isolation and culture of bone marrow mesenchymal stem cells
Whole bone marrow was purchased commercially (Lonza) from normal donors. To isolate human MSCs, mononuclear cells were recovered from 25 ml bone marrow aspirates with a Ficoll-Paque density gradient (GE Healthcare) and resuspended in complete culture medium (CCM), composed of modified Eagle's medium (MEMα) supplemented with 10% Premium Select Fetal Bovine Serum (Atlanta Biologicals), which was lot selected to achieve maximum growth, with additives of 1% penicillin/streptomycin and 1% L-glutamine. After 24 hr, nonadherent cells were discarded, and adherent cells were washed twice with PBS and then incubated in fresh CCM.13,14 Cells were grown in 37°C and 5% CO2. When MSCs reached approximately 60% confluency, cultures were lifted with trypsin treatment and reseeded at 1000 cells/cm2. The medium was changed every third day.
Culture of human myoblast human skeletal muscle cells
Purchased human myoblasts (Gibco) were thawed and plated on collagen-coated flasks (Corning) with myoblast media (MBM) and placed in a 37°C 1% O2 incubator. MBM is composed of Ham's F-10 (Hyclone) with L-glutamine media supplemented with 20% Premium Select Fetal Bovine Serum (Atlanta Biologicals), recombinant human fibroblast growth factor (rhFGF; Gibco), and dexamethasone (Sigma-Aldrich). 6 The medium was changed every 2–3 days and cultures were trypsinized when cells reached 60% confluency. 6
Cocultures
MSCs cultured to 80% confluency and myoblasts cultured to 60% confluency were washed with PBS and treated with TrypLE (Life Technologies) and plated in a 1:1 ratio on collagen type I-coated dishes (Corning). 7 Cultures were maintained in MEMα/Ham's F-10 with L-glutamine combination media supplemented with 20% FBS with rhFGF and dexamethasone. All cocultures were maintained at 37°C in 20% O2 and 5% CO2. Time-lapse images were acquired using the Nikon Biostation IM (Nikon Inc.) and of randomly selected locations within a single culture dish over a 96 hr period using phase-contrast and filters for TRITC and FITC at both the 10× and 20× objectives. The images for each point were put together to create time-lapse videos capturing the cell-to-cell interactions occurring within the coculture over time.
Footnotes
Acknowledgments
This work was funded by NIH Transformative Grant R01GM099688 (J.A.N.). A.B.B. is funded through a generous donation from the National Foundation of Swallowing Disorders (NFOSD). J.A.N. is funded by the California Institute for Regenerative Medicine, NIH, and the University of California–Davis Stem Cell Program. P.C.B. and M.A.K. are funded by the University of California–Davis, Department of Otolaryngology/Head and Neck Surgery. S.P. is funded by the California Umbilical Cord Blood Collection Program (UCBCP). H.D., K.P., and C.N. are funded through the UC Davis Stem Cell Program MSC, Vector, and Karyotyping Cores, respectively.
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.