
Abstract
During the production of some adeno-associated virus (AAV) serotypes, a large amount of vectors is found in the medium of producing cells. For their purification, previous protocols used tangential flow filtration (TFF) of the medium followed by iodixanol gradient centrifugation. Taking advantage of the higher purity of the medium than the cell-derived material as the source of AAV, we tested a simple method that combines production of large culture medium volumes containing AAV from cell stacks with medium clarification+TFF without further time-consuming and nonscalable centrifugation. To test this, we selected AAV2/8, which is emerging as a favored serotype for transduction of liver, muscle, and retina and abundantly found in the extracellular medium. We show that yields and in vitro infectivity of AAV2/8 vectors produced from the culture medium using this method are higher than those of vectors purified from the same cell lysate using a conventional CsCl2 gradient ultracentrifugation-based method, although purity appears inferior. In addition, we found that the transduction efficiency of AAV2/8 purified from medium was similar to that of AAV2/8 purified from the same cell lysate in the murine liver, muscle, and retina. Considering that the purification protocol from the medium we describe requires 3 hr as opposed to the 63 hr of a conventional two-round CsCl2-gradient ultracentrifugation+desalting, we conclude that TFF of the medium containing AAV2/8 represents a quick and scalable method to purify research-grade vectors for use in animal models.
Introduction
Recombinant vectors based on the adeno-associated virus (AAV) are emerging as leading gene therapy vectors owing to their safety and low immunogenicity. AAV vectors can transduce a broad range of nondividing cells, thus allowing their use as an efficient gene delivery vehicle in the treatment of several chronic diseases (Brunetti-Pierri and Auricchio, 2010). Among the most efficient and safe AAV serotypes for in vivo application, AAV2/8 is emerging as one of the most promising for liver (Nathwani et al., 2011), muscle (Wang et al., 2005), and retina (Colella and Auricchio, 2012). Although in vivo application of AAV vectors has the potential to safely deliver therapeutic transgenes in vivo long-term, one of the major challenges is their versatile and efficient production.
Traditionally, AAV upstream production by triple transfection of human embryonic kidney 293 (HEK293) cells (Grimm et al., 1998; Salvetti et al., 1998) was carried out in plates, cell factories, or roller bottles (Liu et al., 2003).
Downstream AAV purification methods include CsCl2 ultra-high-speed density gradient centrifugation (Zolotukhin et al., 1999) or chromatography, with the former being adopted by many investigators. CsCl2 ultracentrifugation, however, is very time-consuming (Ayuso et al., 2010). Iodixanol ultra-high-speed density gradient centrifugation (Hermens et al., 1999; Zolotukhin et al., 1999) is similar to CsCl2 ultracentrifugation, but with higher vector recovery yields although expensive and relatively time-consuming. The majority of chromatographic purification methods use either ion-exchange (Urabe et al., 2006; Qu et al., 2007; Okada et al., 2009) or affinity-based techniques (Auricchio et al., 2001; Brument et al., 2002; Kaludov et al., 2002; Zolotukhin et al., 2002) to purify AAV. The latter use either antibodies directed to the assembled AAV capsid, which are mostly serotype specific (Anderson et al., 2000), or heparin/sialic acid-based affinities, which are based on the property of some AAV serotypes (i.e., AAV2/2 and AAV2/5), to bind with high-affinity heparin sulfate proteoglycan (Summerford and Samulski, 1998) or sialic acid, respectively (Walters et al., 2001). Although affinity chromatography is less time-consuming and easier to scale-up than ultracentrifugation, its specificity to some AAV serotypes and the purification of empty capsids with full capsids has limited its widespread use. The CsCl2 or iodixanol ultra-high-speed density gradient centrifugation, although time-consuming, remains the most diffused downstream process in most laboratories that produce research-grade AAV lots.
Recently, Vandenberghe and coworkers have reported that vectors based on serotypes other than AAV2/2, including AAV2/8, are efficiently released into the culture medium after transient transfection of HEK293 cells (Lock et al., 2010; Vandenberghe et al., 2010). The harvesting from supernatants rather than cell lysates, which are highly enriched in nonviral proteins, significantly simplifies the downstream processing of AAV vectors for research and clinical applications.
Here, we tested a simple tangential flow filtration (TFF) concentration method, which is not followed by cesium chloride/iodixanol gradient purification, to purify AAV2/8 vectors that are released into the culture medium. The procedure is fast, reproducible, efficient, and amenable for large-scale production of research-grade AAV2/8 vectors. Our studies compared total yields, infectivity, and transduction levels in murine muscle, liver, and retina of AAV2/8 vector purified from the medium to those of vector purified using traditional methods from cellular lysates, and found that they are overall similar.
Materials and Methods
Plasmids
Plasmids used for AAV vector production are (1) the pAd helper plasmid that contains the adenovirus E2A, E4, and VA RNA helper genes (Zhang et al., 2000), and (2) the pAAV2/8 (Gao et al., 2008) packaging plasmid with AAV rep and cap genes, the pAAV cytomegalovirus promoter (CMV)-enhanced green fluorescent protein (eGFP), or pAAV thyroxine-binding globulin promoter (TBG) eGFP (Auricchio et al., 2001) cis-plasmids that contain the eGFP expression cassette for ubiquitous (CMV) or liver-specific (TBG) expression flanked by two identical inverted terminal repeats (ITRs). All plasmids were produced and purified by Puresyn (Malvern, PA).
Cell culture
HEK293 cells (from human embryonic kidney carcinoma) and HEPA 1–6 cells (from mouse hepatoma) (ATCC, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Monza, Italy) with 10% fetal bovine serum (Life Technologies) at 37°C in 5% CO2.
Calcium phosphate transfection and harvesting
For each large vector preparation, a suspension of 2.2×109 low-passage HEK293 cells was triple-transfected by calcium phosphate with 1,000 μg of helper, 520 μg of packaging, and 520 μg of pAAV cis-plasmids and plated in CellSTACK-10 (Corning, Amsterdam, The Netherlands). For each small vector preparation, a suspension of 1.1×109 low-passage HEK293 cells was triple-transfected by calcium phosphate with 500 μg of helper, 260 μg of packaging, and 260 μg of pAAV cis-plasmids and plated in CellSTACK-5 (Corning). The following day, the medium was changed with serum-free DMEM and cells were harvested 72 hr after transfection. HEK293 cells from one CellSTACK were harvested by trypsinization and centrifuged at 2,600×g for 20 min. The supernatant (referred to as “medium”) was transferred to a separate tube for further purification or testing. In Fig. 2a, we show the average yields of seven large and two small preps; in Fig. 2b, we show the average infectivity of three large and two small preps.
CsCl2 purification
Cells were lysed by three rounds of freeze–thaw to release AAV2/8 particles. The lysate was then incubated with both DNase I (8,000 U for large vector preparation and 4,000 U for small vector preparation) and RNase A (200 U for large vector preparation and 100 U for small vector preparation) (Roche Diagnostics, Monza, Italy) for 30 min at 37°C to get rid of nucleic acids and with 10% Octyl-βD-glucopyranoside (Sigma-Aldrich, St. Louis, MO) to complete lysis. AAV vectors were then purified by two sequential CsCl2 gradients. Gradient fractions were measured by refractometry, and those with refractive index ranging between 1.3660 and 1.3740 were pooled. All fractions were desalted in phosphate-buffered saline. Glycerol was added to the concentrated AAV lots to a final concentration of 5% (v/v), and the preparations were aliquoted and stored at −80°C.
TFF purification
One liter of the culture medium from the same single CellSTACK-10 (500 ml of the culture medium from CellSTACK-5) from which AAV2/8 vectors were purified from cell lysate was incubated with the same amount of DNase I and RNase A as described for CsCl2 purification, and clarified through a 0.45 μm pore size filter (SLHA 033 SS; Millipore, Billerica, MA). The clarified feedstock was then concentrated by TFF, using a NovaSep-LS holder with customized 0.25-inch (inner diameter) tubing and ports (TangenX Technology, Shrewsbury, MA) and a 0.02 m2 Sius-LS single-use TFF screen channel cassette with a 100 kDa molecular weight cutoff HyStream membrane (TangenX Technology). A 67-fold concentration to 15 ml was performed according to the manufacturer's recommendations with a transmembrane pressure of 10–12 psi maintained throughout the procedure. The TFF filter was discarded after each run, and the system was sanitized with 0.2 N NaOH between runs.
Glycerol was added to the concentrated AAV lots to a final concentration of 5% (v/v), and the preparations were aliquoted and stored at −80°C.
Vector purity
Purity of viral preparations was assessed by loading 1×1010 genome copies (gc) on sodium dodecyl sulfate 4/12% polyacrylamide gels (SDS-PAGE) run under reducing conditions. Protein was detected by Coomassie staining of SDS-PAGE.
Vector genome titration (quantitative polymerase chain reaction)
To eliminate nonencapsidated DNA, before quantitative polymerase chain reaction (qPCR), samples were incubated with 10,000 U of recombinant DNase I RNase-free (Roche Diagnostics) for 1 hr at 37°C, then boiled at 99°C for 5 min, and put on ice for 5–10 min. Encapsidated viral genomes were quantified using an iCycleriQ real-time PCR (Bio-Rad, Hercules, CA) using primers directed to the AAV2 ITRs. The sequences of the AAV2 ITRs primers are 5′-GGAACCCCTAGTGATGGAGTT-3′ (fwd ITR) and 5′-CGGCCTCAGTGAGCGA-3′ (rev ITR) (Aurnhammer et al., 2012). The sequence of the AAV2 ITRs probe is 5′-6-FAM-CACTCCCTCTCTGCGCGCTCG-TAMRA-3′ (Aurnhammer et al., 2012). After a 95°C activation step for 10 min, a two-step PCR cycle was performed at 95°C for 15 sec and 60°C for 1 min for 40 cycles. The TaqMan Universal PCR Master Mix (Roche Diagnostics) was used in the qPCR. DNA plasmid standards were used to determine absolute titers.
Vector genome titration (dot blot)
After incubation with DNase I to eliminate nonencapsidated DNA, as described for qPCR, for DNA dot blot hybridization, the samples were loaded on a positively charged nylon membrane (Whatman Nytran N Charged nylon membranes; Sigma-Aldrich) using a dot blot manifold (Schleicher & Schuell, Dassel, Germany). After cross-linkage, the membrane was then prehybridized for 30 min at 65°C in 20 ml of hybridization solution (RapidHyb; GE Healthcare Europe GmbH, Milano, Italy) and incubated overnight with a 32P-labeled bGH probe (221-bp Xho/BglII fragment from the plasmid pAAV CMV eGFP) radiolabeled using the Rediprime II Random prime labeling system (GE Healthcare Europe GmbH) and [α-32]-CTP (PerkinElmer, Monza, Italy) according to the manufacturer's instructions, at 65°C for 18–20 hr. Membrane was then washed once in 30 ml of wash solution (2× SSC, 0.1% SDS) at 65°C for 30 min and once in 30 ml of wash solution at 37°C for 30 min. Several dilutions of the pAAV CMV eGFP plasmid were used as standard, and the amount of blotted AAV2/8 DNA was estimated by autoradiography using a PhosphorImager (GE Healthcare Europe GmbH).
The final titer of each prep was calculated as the average between the qPCR and dot blot results.
Vector particle titration (enzyme-linked immunosorbent assay)
To determine the total amount of assembled AAV2/8 capsids, the Progen AAV8 Titration enzyme-linked immunosorbent assay (ELISA; Progen Biotechnik GMBH, PRATV [Heidelberg, Germany]) was used according to manufacturer's instructions. We have used four separate AAV2/8 dilutions from each AAV2/8 lot and a standard curve from a previously titered AAV2/8 preparation.
In vitro infectivity
HEK293 cells were plated in a 96-well plate (7×104 cell/well), and infected with 10-fold serial dilutions of each AAV2/8 CMV eGFP vectors. For AAV2/8 TBG eGFP, HEPA 1–6 cells were similarly infected. After 72 hr, the number of cells expressing eGFP (green forming units) was determined in the well at the dilution with less than 10 positive cells. All transduction assays were done in duplicate, and the number was averaged between the two duplicates.
Genome copies in liver
We extracted total genomic DNA from livers using the DNeasy Blood & Tissue Kit (Qiagen, Monza, Italy). Real-time PCR analysis on genomic DNA extracted from snap-frozen tissues was performed using a set of primers/probe specific for the viral genome. Vector genome copy numbers in livers were expressed as genome copies per diploid genome.
In vivo transduction
All studies on mice for liver and muscle were conducted in strict accordance with the institutional guidelines for animal research and with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
All retina studies on mice were conducted in strict accordance with the institutional guidelines for animal research and with the Association for Research in Vision and Ophthalmology Statement for the Use of Animal in Ophthalmic and Vision Research.
All procedures on mice were submitted to the Department of Public Health, Animal Health, Nutrition and Food Safety, Italian Ministry of Health, on October 17, 2011. The Ministry of Health approved the procedures by silence/consent, as per article 7 of the 116/92 Ministerial Decree.
All surgery was performed under anesthesia, and all efforts were made to minimize suffering.
For analysis of AAV transduction efficiency in vivo, the tibialis anterior muscles of 4-week-old C57BL/6 mice (4 animals per group) were injected with 100 μl of AAV2/8 CMV eGFP (1.0×1011 gc/mouse). Subretinal vector administrations (9 animals per group) were performed as previously described (Liang et al., 2001) using 1 μl of AAV2/8 CMV eGFP containing 1.0×109 gc/mouse. For liver gene transfer, each mouse (10 animals per group) received a retro orbital injection of either 4.0×1010 gc or 2.0×1011 gc of AAV2/8 TBG eGFP vector in 200 μl. Livers were harvested 30 days after injection and lysed for subsequent eGFP ELISA analysis (Bioo Scientific, Austin, TX).
For muscle, retina, and liver histology, tissues were frozen, cryosectioned, and examined by direct fluorescence for eGFP detection.
Statistical analyses
Data are presented as mean±standard error. Two-tailed Student's t-test was used to determine statistically significant differences between AAV2/8 vectors purified from cells and from medium. p≤0.05 was considered significant. In Fig. 2d, p=0.73; in Fig. 3c, p=0.86; and in Fig. 3d, p=0.91.
Results and Discussion
Previous reports have revealed that substantial amounts of complete and infective AAV particles are found in the culture medium of producing cells (Lock et al., 2010; Vandenberghe et al., 2010). This occurs for all serotypes; however, the amount of vector found in the medium relative to that found in cellular lysates varies from serotype to serotype (Lock et al., 2010). For instance, AAV2/1, AAV2/8, and AAV2/9 are those with the highest AAV medium/AAV cell lysate ratios (Lock et al., 2010). This finding is a major discovery in AAV vector biology, as AAV has been considered so far a prevalently intracellular virus, and also opens new avenues for AAV production and purification. Indeed, the medium contains less nonviral impurities than cellular lysates, and can be handled at large amounts for easy-to-scale downstream purification processes.
Here we tested a simple and fast protocol to purify AAV2/8 vectors. Adherent HEK 293 cells in CellSTACK were triple-transfected for AAV2/8 eGFP production. Cells were harvested 3 days later, and AAV vectors were independently purified either from medium by TFF concentration or from the same cell lysates by CsCl2 ultracentrifugation. About 3 hr was required for AAV purification from the medium and about 63 hr for the vectors purified from cellular lysates (Fig. 1).
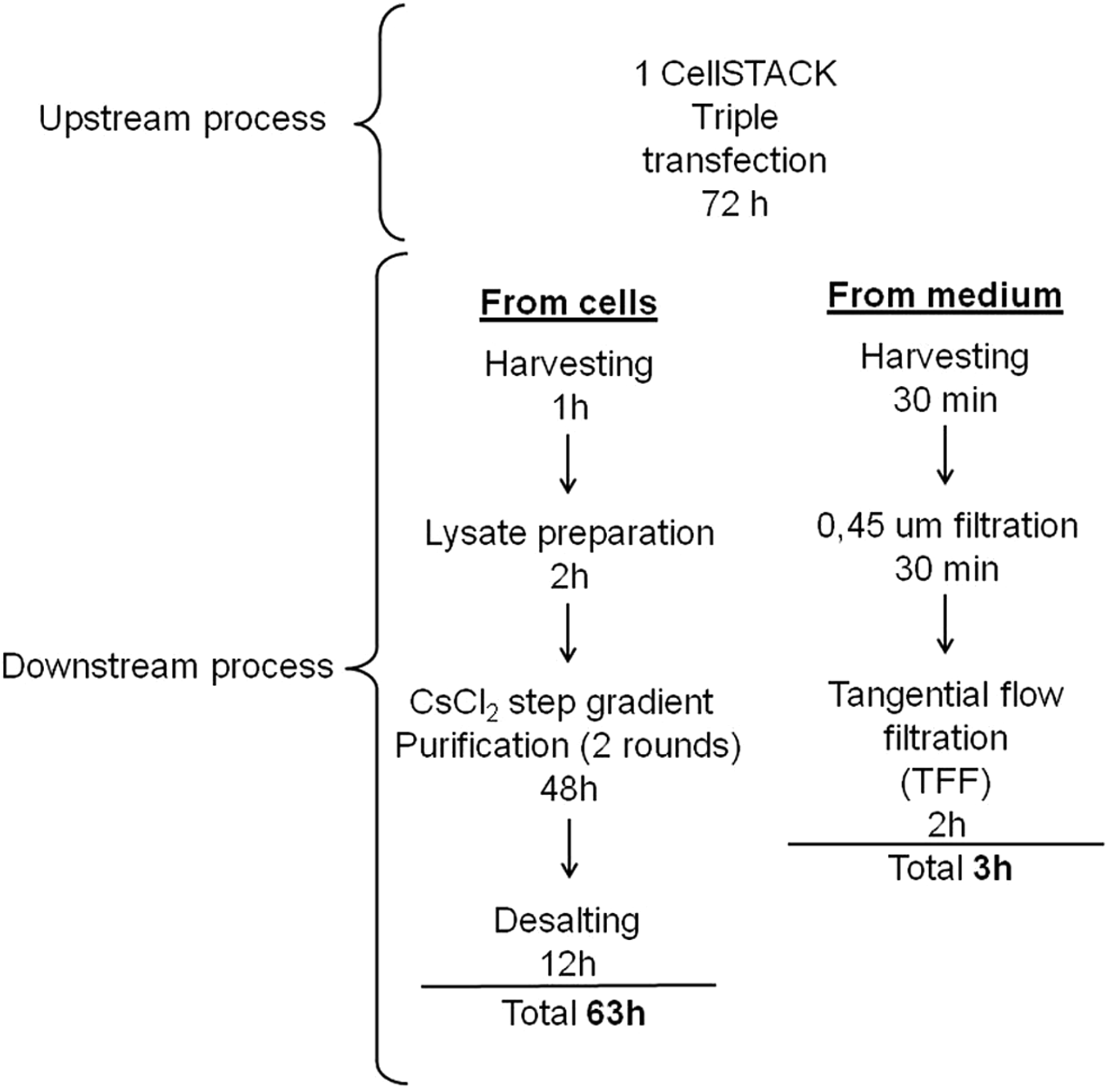
Flow chart of the methods used to purify AAV from either cells or medium. The diagram illustrates the steps of the two purification protocols that were investigated in this study.
The physical yields (Fig. 2a) as well as the infectivity (Fig. 2b; Table 1) of the various AAV2/8 vectors appeared superior when the vector was purified from the medium than from cells. The purity of some preparations was examined by Comassie staining of SDS-PAGE (Fig. 2c): 3.4×1010 gc of either TFF or CsCl2-purified lots were loaded per lane. The VP1, VP2, and VP3 bands of the capsids are clearly visible in both samples; however, there are lower-molecular-weight bands in the samples purified by TFF. Indeed, additional studies, that is, the characterization of the in vivo immune responses to the vectors by a cytokine array, will be required to understand if this simplified TFF-mediated purification method results in vector preps that contain impurities that may affect the safety or efficacy of in vivo gene transfer.
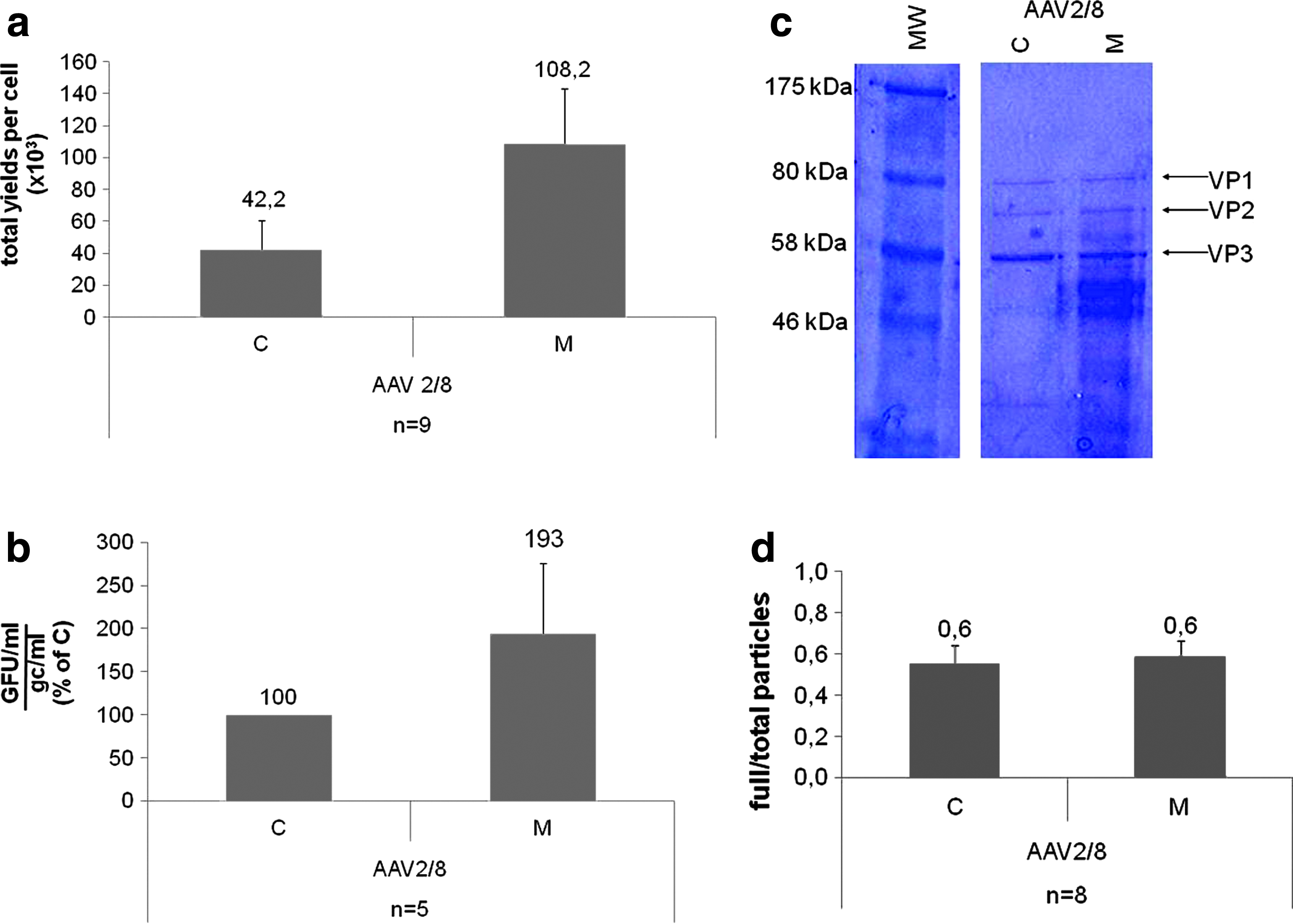
In vitro characterization of AAV2/8 vectors purified from either cells (C) or medium (M).
Infectivity of AAV2/8 Vectors Represented in Figure 2b
AAV, adeno-associated virus; C, AAV vectors purified from cells; Cell line, cells used to test the in vitro infectivity of the various AAV preps; CMV, ubiquitous cytomegalovirus promoter; gc, genome copies; GFU, green forming units; M, AAV vectors purified from medium; TBG, liver-specific thyroxine-binding globulin promoter.
Interestingly, the ratio between AAV2/8 genome-containing particles (full) measured by qPCR+dot blot and total particles (full and empty) assessed by ELISA appears similar (Fig. 2d). This suggests either that our CsCl2-based method is partly inefficient at removing empty capsids (40% of total capsids, Fig. 2d) or that the majority of particles found in the medium are indeed full (60% of total capsids; Fig. 2d).
We then compared the in vivo transducing ability of AAV isolated using the two different methods in three major targets of gene therapy: liver, muscle, and retina.
Two different doses of AAV2/8 TBG eGFP purified either from medium or from cell lysates were administered systemically to 10 C57BL/6 mice. Thirty days after injection, livers were harvested and either cryosectioned or lysed. Tissue sections of livers from treated mice were examined by direct fluorescence (Fig. 3a and b), and the relative eGFP expression was assessed by ELISA in liver lysates (Fig. 3c), whereas the AAV gc numbers in liver were determined by qPCR (Fig. 3d). Both the eGFP levels, AAV gc numbers, and tissue distribution are similar in transduced livers independently of the AAV purification method.
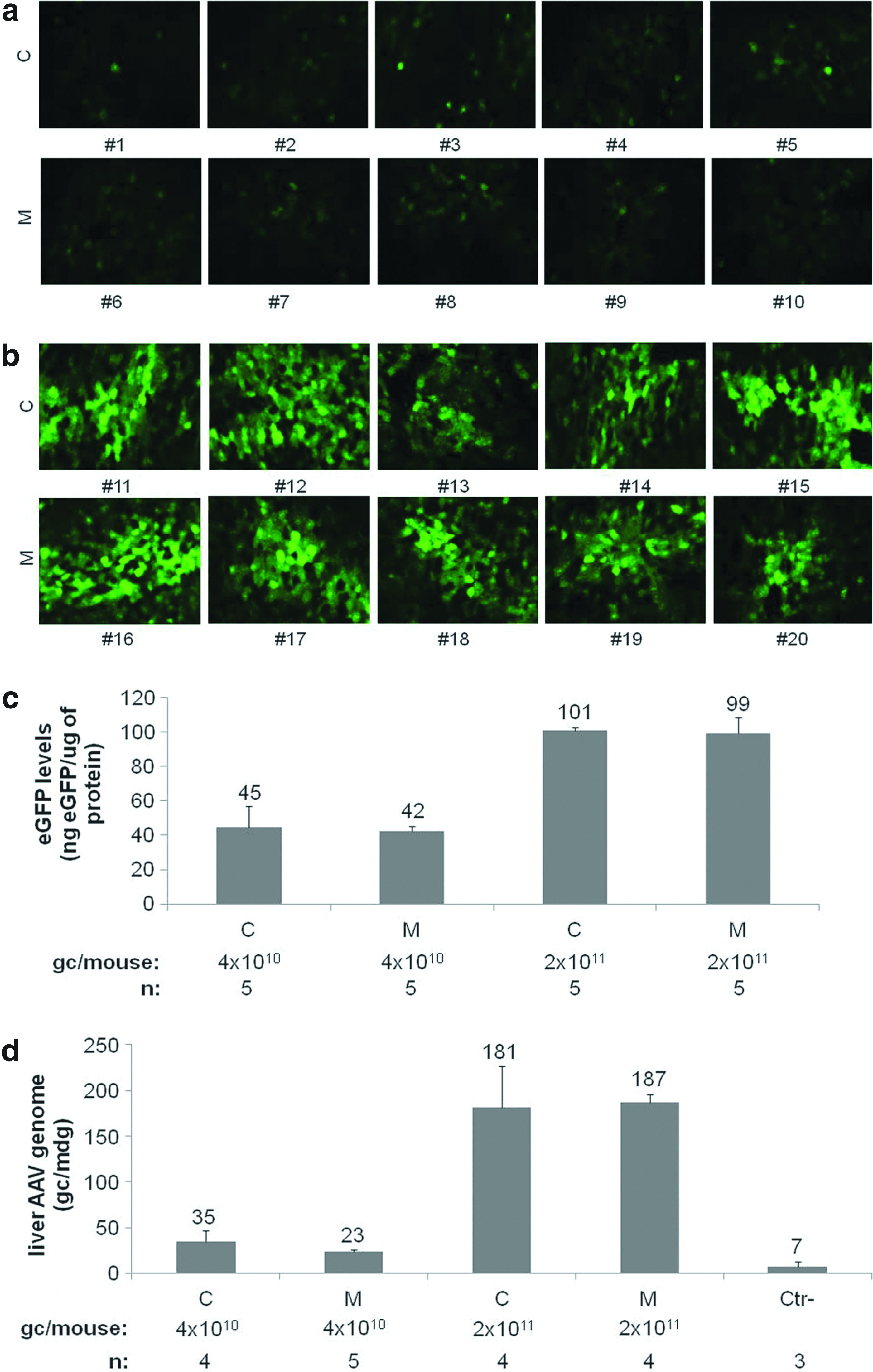
Murine liver transduction by AAV2/8 vectors purified from either cells (C) or medium (M). C57 BL/6 mice were injected intravenously with AAV2/8 TBG eGFP vectors. eGFP expression was visualized 1 month after injection of 4×1010
To assess muscle transduction, the tibialis anterior muscle of four C57BL/6 was injected with AAV2/8 CMV eGFP purified from either cells or medium. Animals were sacrificed 1 month postinjection. The observation of eGFP fluorescence revealed that both vectors transduced muscle fibers similarly (Fig. 4a).
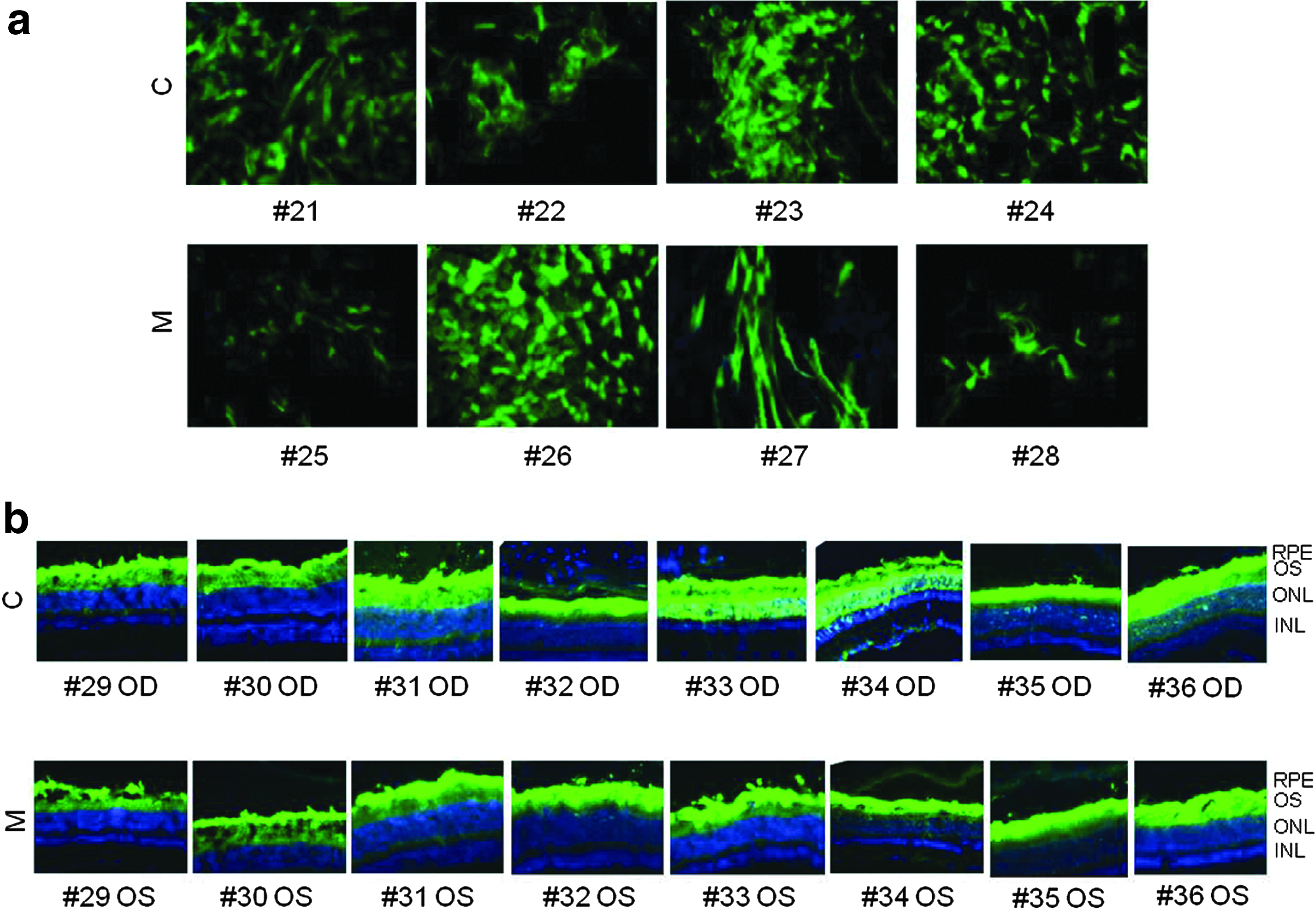
Murine muscle and retina transduction by AAV2/8 vectors purified from either cells (C) or medium (M). C57 BL/6 mice were injected intramuscularly with 1.0×1011 gc/mouse of AAV2/8 CMV eGFP
Finally, to compare the performance of the purified AAV vectors in the retina, AAV2/8 CMV eGFP vectors were injected subretinally in nine C57/BL6, and 4 weeks later, animals were sacrificed and eGFP fluorescence was observed on retinal cryosections. Both retinal pigment epithelial and photoreceptor cells were transduced to similar levels independently of the purification method (Fig. 4b).
In conclusion, AAV vectors purified from the medium by TFF appear reasonably pure and similarly infectious in vivo to those purified from cells by conventional CsCl2 ultracentrifugation. Thus, TFF of AAV from the medium should appeal to basic research labs willing to produce large quantities of AAV in a fast and cost-effective manner, such as when performing in vivo expression screening of multiple vector genome constructs or when comparing the same vector genome construct with several different AAV serotype capsids.
Footnotes
Acknowledgments
We thank Graciana Diez-Roux (Telethon Institute of Genetics and Medicine Scientific Office) for the critical reading of this article. This work was supported by funding from the Telethon Foundation (Grant TGM06Z01).
Author Disclosure Statement
No competing financial interests exist.