
Abstract
Lentiviral vectors (LVs) hold great potential as gene delivery vehicles. However, the manufacturing and purification of these vectors still present major challenges, mainly because of the low stability of the virus, essentially due to the fragility of the membrane envelope. The main goal of this work was the establishment of a fast, scalable, and robust downstream protocol for LVs, combining microfiltration, anion-exchange, and ultrafiltration membrane technologies toward maximization of infectious LVs recovery. CIM® (Convective Interaction Media) monolithic columns with diethylaminoethanol (DEAE) anion exchangers were used for the purification of clarified LV supernatants, allowing infectious vector recoveries of 80%, which is 10% higher than the values currently reported in the literature. These recoveries, combined with the results obtained after optimization of the remaining downstream purification steps, resulted in overall infectious LV yields of 36%. Moreover, the inclusion of a Benzonase step allowed a removal of approximately 99% of DNA impurities. The entire downstream processing strategy herein described was conceived based on disposable and easily scalable technologies. Overall, CIM DEAE columns have shown to be a good alternative for the purification of LVs, since they allow faster processing of the viral bulks and enhanced preservation of virus biological activity, consequently, increasing infectious vector recoveries.
Bandeira and colleagues report on an alternative strategy for the purification of lentiviral vectors using membrane and chromatographic technologies, including depth-filtration, ultrafiltration/diafiltration, and anion-exchange chromatography. This strategy led to infectious vector recoveries of up to 80%.
Introduction
Lentiviral vectors (LVs) provide several advantages over other vectors for gene therapy applications, including long-term transgene expression in target cells, low immunogenic potential, and the possibility to transduce dividing and nondividing cells (Naldini et al., 1996a,b; Schambach and Baum, 2008). Because of these advantages, the number of clinical trials using LVs for gene delivery has been increasing worldwide, comparatively to the use of other viral vectors (Gene Therapy Clinical Trials, 2012).
Despite significant progress in LVs manufacturing, including the generation of safer vectors and inducible producer cell lines (Xu et al., 2001; Ikeda et al., 2003), the particles obtained are still unstable, presenting half-lives of 9 to 12 hr at 37°C (Higashikawa and Chang, 2001). It is known that retrovirus loss of infectivity can be attributed to the fragility of the lipid membrane layer (affected by cholesterol levels, pseudotyping, temperature, and production medium osmolarity) (Beer et al., 2003; Coroadinha et al., 2006a,b, Carmo et al., 2006) and to the rapid loss of reverse transcriptase activity (Carmo et al., 2008; Carmo et al., 2009). Such drawbacks should also represent major challenges in the development of robust and fast downstream processing (DSP) strategies to produce clinical-grade vector preparations since high-titers (from 107 infectious particles (IP)/ml for ex vivo trials up to 109 IP/ml for in vivo trials) (Segura et al., 2006; Rodrigues et al., 2007b) and high-quality samples are mandatory to achieve the required therapeutic effect. There is, thus, a need to establish an efficient and scalable purification process, allowing rapid processing of large volumes of LVs.
Classical purification protocols make use of several ultracentrifugation steps in order to concentrate the vector preparations. However, centrifugation poses scale limitations, is time consuming, and concentrates impurities that may cause inflammatory or immunogenic reactions or inhibit transduction (Baekelandt et al., 2003; Morenweiser, 2005). Current downstream strategies, based on the integration of membrane and chromatography processes, require a combination of several steps to decrease protein and DNA impurities present in the final vector preparations. Microfiltration is the most popular technique used for clarification of retroviral vector (RV) bulks, and recent studies showed that a novel step filtration system, based on a series of decreasing pore size filters, minimizes the decay of retroviral particles associated with the filtration (Reeves and Cornetta, 2000). Membrane-based methods, such as ultrafiltration/diafiltration (UF/DF), are adequate techniques to concentrate viral stocks. Specifically, tangential flow filtration (TFF) has been applied with success in the concentration and partial purification of LVs, offering scalability, efficiency, and a wide range of processable volumes in short periods of time (Geraerts et al., 2005). The purification of viral bulks can be achieved by chromatographic methods, including heparin affinity, gel filtration, and anion-exchange (AEX) (Transfiguracion et al., 2003; Segura et al., 2005; Rodrigues et al., 2006). AEX chromatography (AEXc) has been a method of choice for LVs purification since negatively charged viral particles bind to the positively charged membranes and can then be eluted with the use of desorption agents or high salt concentrations (Rodrigues et al., 2007a; Schweizer and Merten, 2010). Studies using these chromatographic matrices for the purification of retroviral and lentiviral vectors have shown recoveries of 70%, with host-cell DNA removals reaching 99% (Slepushkin et al., 2003; Rodrigues et al., 2006; Merten et al., 2011). Small pores on the chromatographic beads can, however, originate low virus-binding capacities and high shear stresses (Rodrigues et al., 2007a) that, adding to the labile nature of most of the viral envelopes used, drastically reduces yields and infectious particle titers. This problem can be circumvented by the use of macroporous supports like monoliths, a relatively recent chromatographic support for purification of nanoparticles like viruses (Gagnon, 2010). CIM® monoliths combine the advantages of conventional chromatographic columns (separation power, capacity, and sample distribution) with membrane technology (convective mass transport), presenting a matrix composed of methacrylate polymers and highly interconnected flow through pores (Barut et al., 2005; Podgornik and Štrancar, 2005). These structural characteristics have been shown to promote extremely high resolution and binding capacities for viruses, with a low-shear fractionation environment and 10-fold higher operating flow rates compared to particle-based supports (Jungbauer and Hahn, 2008; Gagnon, 2010).
In this study, we present an alternative strategy for the purification of lentiviral vectors, using membrane and chromatographic technologies, including depth-filtration, ultrafiltration/diafiltration, and anion-exchange chromatography (i.e., CIM® DEAE monolithic columns). The several steps involved in the purification process were optimized, and a reproducible purification platform for LVs downstream processing was established.
Materials and Methods
Cell line and culture media
HEK 293T cells (CRL-11268) obtained from the American Type Culture Collection (ATCC) were maintained in Dulbecco's modified Eagle medium (DMEM; Gibco, Life Technologies, Paisley, United Kingdom), supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco), in an incubator with humified atmosphere of 7% CO2 in air at 37°C. For viral production, cells were grown in DMEM supplemented with 10% (v/v) FBS without phenol red (Gibco). Cell concentration was assessed by counting cells with the trypan blue exclusion method using a haemocytometer (Gibco) after trypsin cell detachment.
Plasmids
The HIV-1-derived lentiviral vector particles produced consisted of a transfer vector, pRRLSIN, with an enhanced green fluorescent protein (eGFP), third-generation packaging plasmids pMDLg/pRRE and pRSV-Rev, and the envelope protein plasmid pMD.2G, encoding the vesicular stomatitis virus envelope glycoprotein (VSV-G). All the plasmids were kindly provided by Didier Trono through Addgene (Cambridge, MA).
Lentiviral vector production
HEK 293T cells were seeded at a concentration of 5×104 cells/cm2 in HYPERFlask Cell Culture Vessels (Corning Inc., Corning, NY) using 560 ml of DMEM supplemented with 10% (v/v) FBS. After 24 hr, cells were transfected (at 60–80% of confluence) with the four plasmids referred above, using 5 μg of total DNA per 106 cells. Transfection conditions were first tested at a small scale; plasmid amount and concentration was optimized so that the concentration of the transfer plasmid exceeds that of the others (3 pRRLSIN : 1.5 pMDLg/pRRE: 1.5 pRSV-Rev: 1 pMD.2G). Plasmids were mixed together in 10 ml of culture media without FBS. Linear 25 kDa polyethylenimine (PEI; Polysciences Inc., Eppelheim, Germany), diluted in sterile water to 1 μg μl–1, acidified to pH 2.0 with HCl until dissolution and neutralized to pH 7.0 with NaOH, was used as transfection reagent in a DNA to PEI ratio of 1:3. Transfection complexes (DNA–PEI) were prepared by adding the filtered (0.22 μm) DNA mixture to PEI, also previously diluted in 10 ml culture media, without FBS. This mixture was vortexed, incubated for 15 min at room temperature, and added to 540 ml of culture media. After this, the culture media in the HYPERFlask was exchanged by the new media containing DNA–PEI mixture, and cells were incubated at 37°C. Forty-eight hr after transfection, LV supernatants were harvested and purified.
Clarification of lentiviral vector supernatants
To remove cellular debris from the recovered viral supernatants, two methods were compared: batch centrifugation and depth filtration. Batch centrifugation was performed at 10,000 g for 15 min at 4°C. For depth filtration, supernatants were filtered at room temperature using a disposable depth-filter capsule (Sartopore 2; Sartorius Stedim Biotech, Goettingen, Germany) of 0.8+0.45 μm pore size and with an effective filtration area of 50 cm2. Clarification was carried out at 50 and 100 ml min–1; the clarified supernatants, collected in sterile containers, were then subjected to AEXc. Before clarification, depth-filters were equilibrated either with 10 mM Tris buffer (1 mM MgCl2, at pH 8.0) (Merck, Darmstad, Germany) or with culture media (DMEM, Gibco).
Anion-exchange chromatography
For AEXc, the performance of CIM® DEAE monolithic columns (BIA Separations, Villach, Austria) and Sartobind® D MA75 (Sartorius Stedem Biotech) membrane adsorbers was evaluated. Both matrices were coupled to an ÄKTAexplorer 100 chromatography system (GE Healthcare, Uppsala, Sweden), equipped with UV, conductivity and pH sensors, and a fraction collector, controlled online with the coupled UNICORN software (all from GE Healthcare). Matrices were equilibrated with 10 mM Tris buffer (pH 8.0). After sample loading, the matrices were washed with 10 mM Tris buffer (pH 8.0) to remove the nonbinding material. Stepwise elution started with 10 mM Tris buffer (pH 8.0) with 0.1 M NaCl to release the weakly bonded proteins. Next, elution of the LVs was carried out using 10 mM Tris buffer with 0.65 M NaCl. Finally, in order to regenerate the column and remove stronger bonded impurities from the column, such as DNA, 10 mM Tris buffer with 1 M NaCl was used. This final step is performed with the aim of regenerating the column and evaluating the column's efficiency in the removal of residual DNA from the viral samples.
Three different elution buffers were evaluated: 10 mM Tris (pH 8.0), phosphate-buffered saline (PBS) (Gibco), and 50 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) at pH 7.4 (Sigma-Aldrich, St Louis, MO). The flow rates varied from 5 ml min–1 to 10 ml min–1.
Concentration assays
For the concentration step, UF/DF techniques were used. Three different systems were tested: Vivaspin polyethersulfone membranes of 100 and 300 kDa molecular weight cut-off (MWCO) and Vivaflow cassettes of 100 kDa MWCO (all from Sartorius). Concentration with Vivaspin membranes was performed by centrifugation at 3,000 g at 4°C, in a swing bucket rotor, until the desirable volume was attained. Vivaflow cassettes were previously saturated with 20 ml (4 g/L) of human serum albumin (HSA) (Sigma) for approximately 3 min, at a flow rate of 7 ml min–1, in order to prevent unspecific adsorption of the LVs to the membrane. The applied inlet pressure was approximately 1 bar at 150 rpm. For Vivaflow systems, a continuous recirculation of the concentrate to the feed was performed until 5% of total size exclusion chromatography (SEC) column volume was reached.
Polishing and storage
SEC was performed with a Superdex XK26/60 column (GE Healthcare) with Tris buffer (pH 7.8) in 150 mM NaCl and with a flow rate of 2 ml min–1. The final LV preparation was recovered, subjected to a sterile microfiltration using 0.22 μm Acrodisc Syringe Filters (Pall, Port Washington, NY), aliquoted, and stored at −85°C until further analysis. All the purification steps referred to were performed at 4°C.
A cryoprotecting formulation of 0.5 M of sucrose (Merck) and 0.6 mg/ml of HSA was added to the viral samples before freezing to reduce loss of infectious particles. In order to analyze the effect of the addition of these formulations, several aliquots (with an already known infectious titer) were frozen at −85°C with and without this cryoprotecting formulation. After 24 hr, samples were thawed on ice and their infectious titer was quantified by the method described in the section “Analysis of viral titers.”
Benzonase treatment and DNA impurities
To digest residual nucleic acids from host producer cells and plasmid DNA present in the LVs bulk, samples from different steps of DSP were incubated with Benzonase endonuclease grade II (Merck), at a concentration of 50 U/ml for 30 min at 37°C.
Total DNA content of the samples from each step of the process (including those incubated with Benzonase) was quantified with Quant-iT PicoGreen dsDNA Assay Kit (Molecular Probes Inc., Eugene, OR), using Modulus Microplate Luminometer (Gentaur, Brussels, Belgium) and standard fluorescent wavelengths (excitation ∼480 nm; emission ∼520 nm).
Analysis of viral titers
To determine the infectious titer of the vectors carrying an eGFP reporter gene, 5×104 293T cells/cm2 were seeded and transduced (at 60–80% confluence), with serial dilutions of viral suspensions (from each step of the process), in DMEM (Gibco) containing 10% FBS (v/v) (Gibco) and 8 μg/ml of polybrene (Sigma). Forty-eight hours after transduction, quantification of infectious particles was performed by monitoring the expression of the eGFP reporter gene using CYFlow Space flow cytometer (Partec, Munster, Germany). In order to estimate the stability of the infectious particles in high salt concentrations, samples obtained after AEXc were incubated at 4°C in Tris-HCl with 0.65 M of NaCl for 30 min. After this period, infectious titer of these samples was quantified and compared to the titer obtained when the same samples were diluted with Tris-HCl immediately after elution.
P24 capsid protein quantification was performed to estimate the total viral particles present in the LV samples using the Innotest HIV Antigen mAb kit (Innogenetics N.V., Gent, Belgium), following the manufacturer's instructions.
Results
Downstream processing of lentiviral vectors
The main objective of this work was to establish a downstream processing protocol for the purification of LVs based on AEXc, aiming at high recoveries of infectious particles and faster processing of the viral bulks with scale-up potential. The general experimental design of the LVs downstream process followed is depicted in Figure 1.
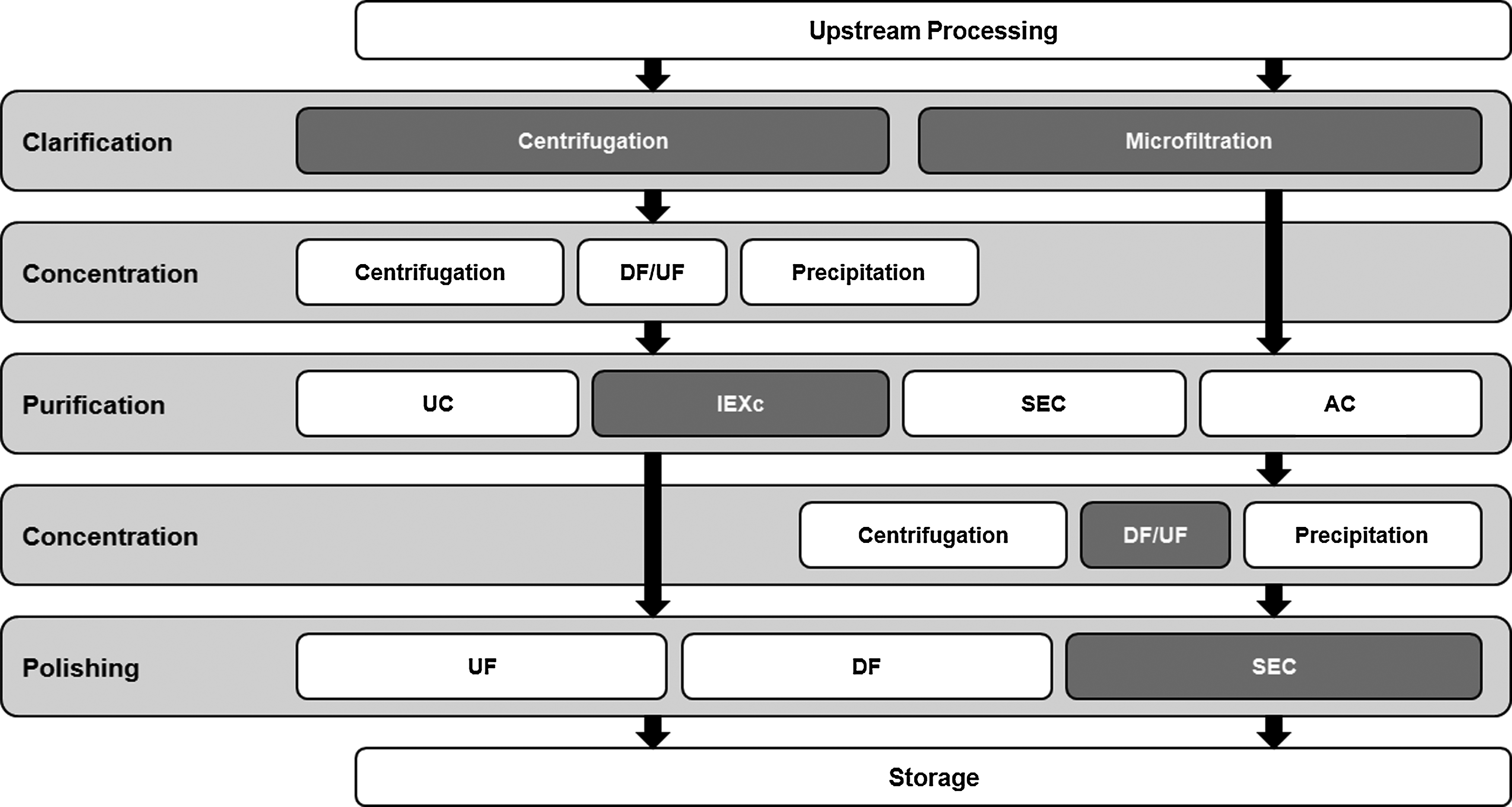
Overview of the downstream strategies described in the literature for the purification of lentiviral vectors. Boxes in dark gray show the different techniques compared and used in this work. UC, ultracentrifugation; IEC, ion-exchange chromatography (including AEXc, anion-exchange chromatography); SEC, size-exclusion chromatography; AC, affinity chromatography; DF/UF, diafiltration/ultrafiltration.
Clarification assays
The first step in a downstream process is the clarification of the viral bulk to remove cells and cellular debris. In this study, the performance of batch centrifugation and disposable depth-filter capsules was compared. For the depth-filters, a flow rate of 50 ml min–1 was used after pre-wetting the membranes with Tris buffer. For both methods, recoveries of 70% were obtained (Table 1). The performance of the depth-filter capsules after pre-wetting the membranes with culture medium (DMEM) or Tris buffer was also analyzed. Comparable recoveries (71±4% vs. 74±2%, respectively) were obtained. Furthermore, an increase of the loading flow rate from 50 to 100 ml min–1 permitted to increase the infectious vector recoveries up to 91±6% when depth-filter capsules were used for clarification (Table 1).
Infectious Titers, Concentration Factors, and Recoveries Obtained at the End of Each Downstream Process Step, Before and After Optimization
Results after optimization are shown for the methods presenting higher yields and chosen to be part of the downstream protocol developed herein due to their advantages.
Recovery efficiency of total infectious particles, obtained after optimization of several conditions in each downstream processing (DSP) step: aincrease of the flow rate from 50 to 100 ml min–1; bimmediate five-fold dilution of viral preparations after elution; cno optimization was performed in this step due to the high recoveries obtained; dincrease of the concentration of the loading material by six-fold; eoverall recovery obtained after using the techniques that gave the best recoveries in each purification step. The errors correspond to standard deviation (n=3).
CF, concentration factor (in volume).
Anion-exchange chromatography
After clarification, a purification step to isolate the LVs from proteins and DNA impurities was performed. Two different AEXc-capture matrices were tested: Sartobind® D MA75 and CIM® DEAE monolithic columns, both eluted with Tris buffer. As shown in Table 1, LVs adsorbed to both matrices. Nevertheless, CIM® DEAE presented higher recoveries in LVs elution, with a yield of 55±2%. Since monolithic columns provided higher performance, best chromatographic buffers for elution were studied in this matrix to obtain optimal purification conditions. Efficient separation of viral particles and contaminant proteins was achieved using 0.65 M of NaCl for elution (Fig. 2). After elution, volumes between 10 and 24 ml were obtained. Given the potential detrimental effect of high salt concentrations on the infectivity of the viral particles, a five-fold dilution step of the LVs samples with Tris-HCl buffer (without NaCl), immediately after elution, was included in our protocol. This dilution allowed the establishment of an optimal pH and low salt concentrations for the LVs samples, therefore achieving infectious titers of 8.0±0.4×107 IP/ml. In contrast, aliquots from the same viral samples, which were not diluted after elution but instead were maintained in a 0.65 M NaCl buffer for 30 min, showed infectious titers of 6.1±0.2×107 IP/ml, a decrease of 24%. The inclusion of this dilution step may explain the higher recoveries obtained, which increased the clarification step yield to 80±5% (Table 1).
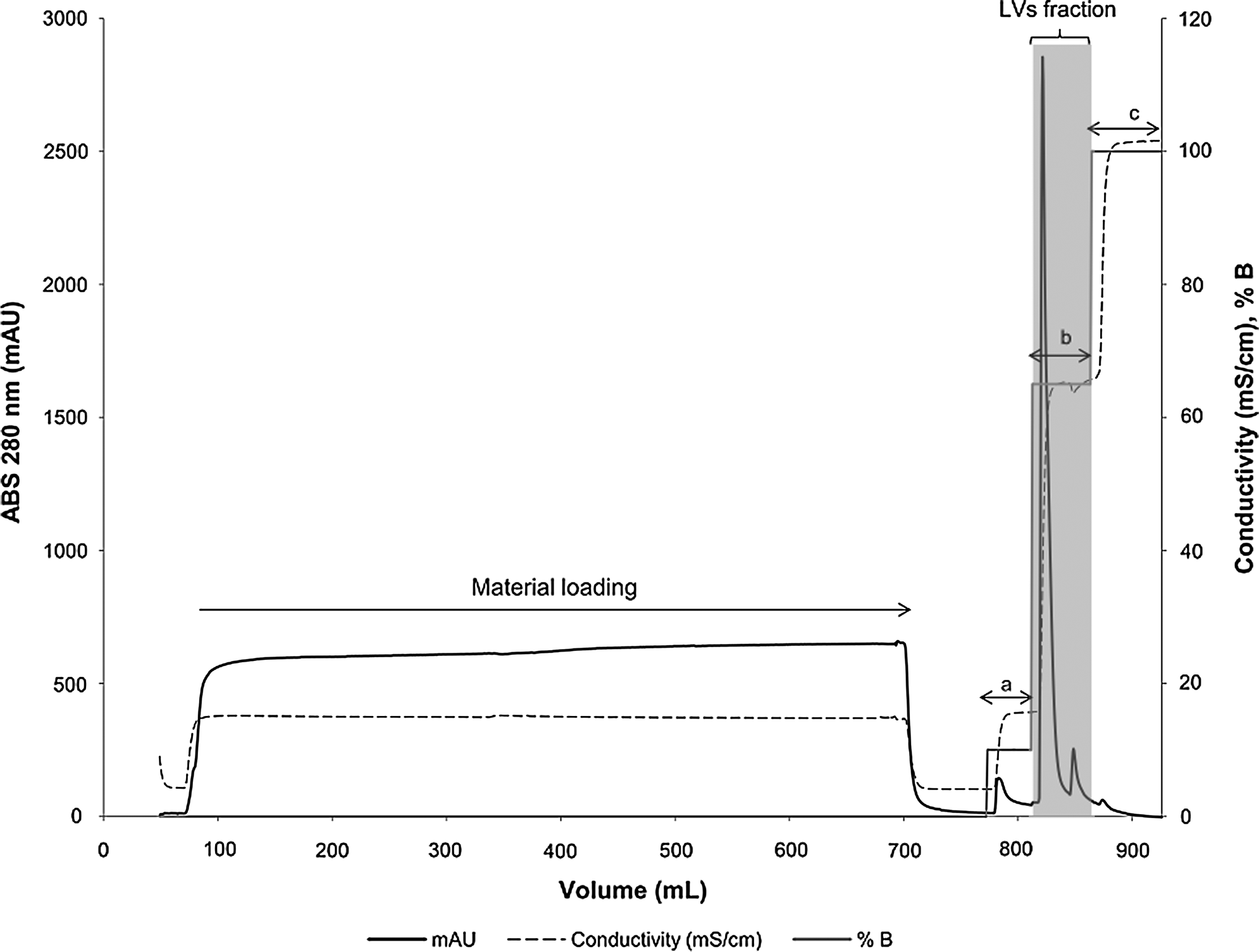
Stepwise elution chromatographic profile obtained after loading the CIM® monolithic column with diethylaminoethanol (DEAE) anion exchangers with 560 ml of clarified supernatant. Matrices were equilibrated with Tris–HCl buffer. Stepwise elution was performed using 10 mM Tris buffer with 0.1 M NaCl (a) to release the weakly bonded proteins, 0.65 M NaCl (b) for lentiviral vectors (LVs) elution (fraction identified in the gray box), and 1 M NaCl (c) to regenerate the column and to remove stronger bonded impurities. Loading and elution was performed at 10 ml min–1. Dashed line represents the conductivity in mS/cm, the bold line the absorbance at 280 nm (mAU), and the gray line the molarity in % of buffer B (Tris–HCl with 1.0 M NaCl, pH 8.0).
An assay comparing the performance of three different chromatographic buffers (HEPES, PBS, and Tris) in the elution of the LVs revealed similar recoveries (between 71±4 % and 80±5%) (Table 2) and high infectivity ranges (>5.2×104 IP/ng p24) for all the buffers studied.
Impact of the Chromatographic Buffers Used For LV Elution (50 mm HEPES, 10 mm Tris, and PBS) in the Infectious Titer, Concentration, and Stepwise Recovery
The errors correspond to standard deviation (n=3).
Concentration and polishing
After elution, the collected and diluted peak fraction was concentrated using UF/DF techniques. The main aim of this step was to concentrate the diluted viral peak to less than 5% (i.e., 16 ml) of total SEC column volume in order to guarantee a high resolution in the SEC step. Vivaspin (of 100 and 300 kDa) and Vivaflow systems (of 100 kDa) were used and compared, showing similar stepwise yields (from 67±6% to 72±1%) (Table 1). Vivaflow cassettes, however, had to be saturated with HSA before concentration, since, without this coating, very low recoveries (8.5±1.3%) were obtained (data not shown). All the systems used were equally efficient, with only 1.5±0.7% of total infectious viral particles being lost in the permeate.
The suitability of the Superdex XK26/60 column for size exclusion chromatography was analyzed for further purification and separation of LVs from remaining low molecular weight contaminants. This specific matrix excludes the viral particles from the matrix pores and separates proteins without excessive dilution of the vectors. Elution profiles were generated after injection of up to 16 ml of viral sample at 2 ml min–1, using Tris buffer with 150 mM NaCl at pH 7.8 (Fig. 3). The void volume of the column is equivalent to approximately 30% of the total column volume (i.e., 106 ml). Therefore, the LVs were eluted as one peak, starting at the column void volume, and indicating that the injected LVs were already highly pure. Stepwise recoveries of purified vectors after this step were in the range of 27±2% of the injected infectious particles (0.40±0.05×107 IP/ml). However, after increasing the concentration (infectious titer) of the loading material by six-fold (to 2.4±0.2×107 IP/ml), yields were enhanced up to 68±7% (Table 1).
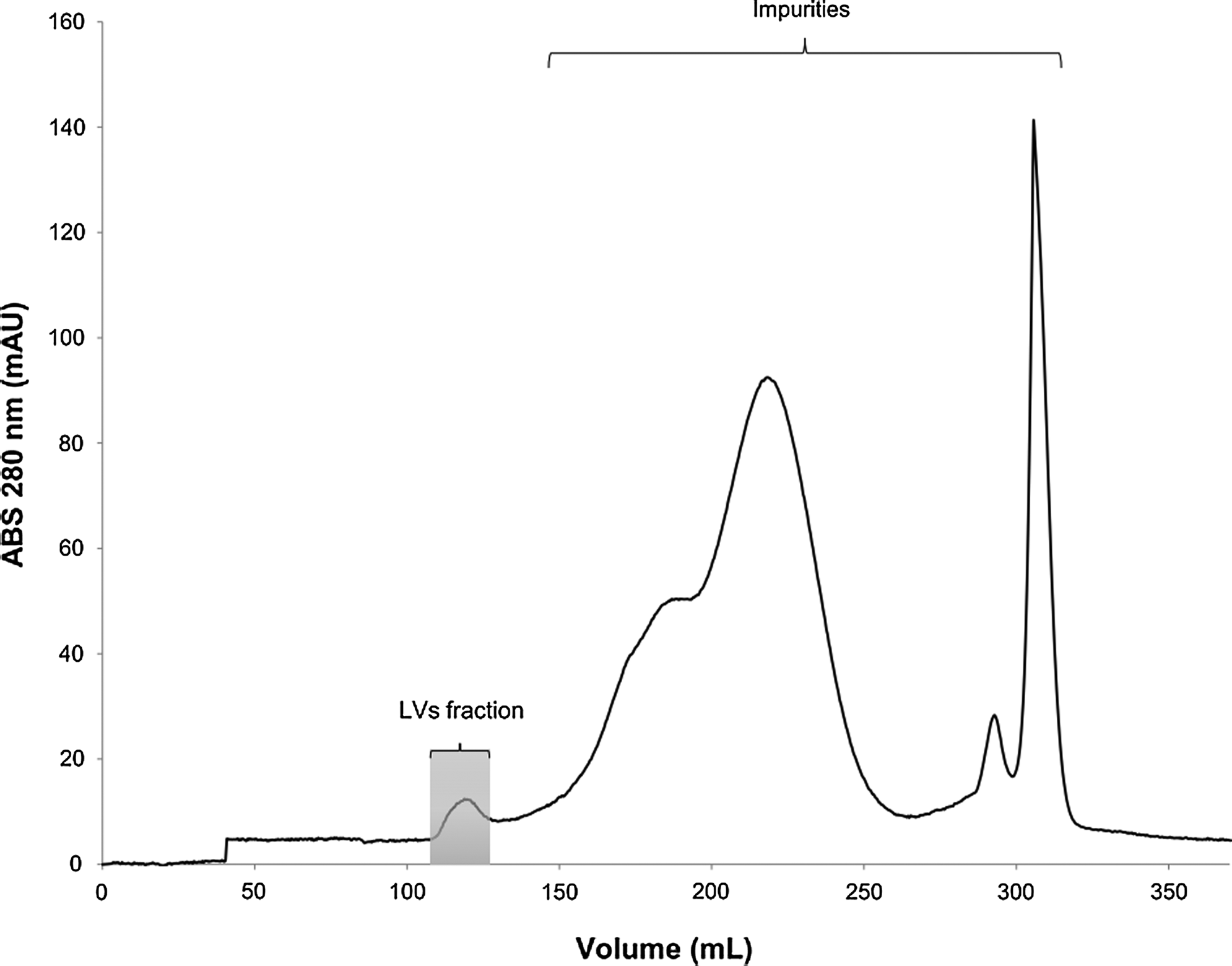
Chromatographic profile of LVs by size-exclusion chromatography using a Superdex XK26/60 column. The viral sample was injected into the column and eluted at a flow rate of 2 ml min–1 using Tris buffer with 150 mM NaCl, at pH 7.8. LVs were eluted as one peak (identified in the gray box), starting at the column void volume (106 ml).
Overall, recoveries of infectious particles of 36% were obtained for the purification protocol described herein after optimization of several parameters used in each step of the process.
Characterization of purified lentiviral vectors
LVs samples from several steps of the process were analyzed for infectious titer, infectivity, stability in the formulation, and for the presence of impurities. The LVs infectious titer from samples of the harvested bulk reached 1.1×107 IP/ml. After performing the purification process herein described, ranges from 1.8×107 IP/ml (including the polishing step) to 4.7×107 IP/ml (without the polishing step) were obtained. The infectivity of the LVs samples showed to be high, reaching 7.3×104 IP/ng of p24.
Analysis of the LVs infectious titer after freeze-thawing revealed a drop to 84±3% (from 1.7±0.1 to 0.28±0.01×107 IP/ml); however, samples stored in a formulation containing sucrose and HSA had a loss of only 26±2% of infectious particles (from 1.7±0.1 to 1.3±0.1×107 IP/ml) after freeze-thawing. The addition of these, or potentially other, stabilizers is, therefore, essential.
Analysis of DNA impurities showed that the amount of DNA present in the clarified supernatant ranged from 2 to 4 μg per 108 IP. Results revealed that the monolithic column, on its own, reduced the DNA content by 88.8% (Table 3). However, in order to further reduce the DNA, 50 U/ml of Benzonase was added either to the clarified supernatant or after the AEXc step. Samples treated with Benzonase before injection in AEXc presented DNA removal efficiencies of 97.7%; samples treated with Benzonase after AEXc showed even higher DNA removal efficiencies (99.9%). Final LV samples had DNA impurities below 0.02 μg per 108 IP (Table 3).
Effect of Benzonase Endonuclease Addition in the Removal of DNA Impurities During the Downstream Process Herein Described
50 U/ml of Benzonase was added after clarification and AEXc steps. Total DNA content of these samples was measured during the clarification, purification, and concentration steps by Quant–iT PicoGreen dsDNA Assay Kit. The errors correspond to standard deviation (n=3).
ND, data not determined.
Discussion
The establishment of a downstream process for virus purification involves the combination and optimization of several steps and parameters toward maximum infectious particle titers and final maximal recoveries. For the specific case of LVs, the challenge is to achieve clinical grade preparations with high titer and high quality in a short period of processing time, since these vectors are very unstable, rapidly losing infectivity.
In the present study, a multistep protocol was developed, combining several chromatographic and membrane-based process techniques (Fig. 1) that had been reported in the literature as promising for the purification of retroviral and lentiviral vectors. CIM DEAE monolithic columns were used for the first time by Lesch et al. (2011) to remove LVs from a baculoviral vectors' bulk, achieving stepwise yields of 65%. Due to the high recoveries obtained, these matrices were used herein and each step of the process was improved to allow higher yields and purer viral preparations.
In the clarification step, recoveries of 91% were obtained using the depth-filter capsules. These results, in accordance with the literature (Segura et al., 2005),were obtained after optimizing the flow rate from 50 to 100 ml min–1. The use of these higher flow rates promoted a lower entrapment of the viruses in the filter pores, which may explain the higher recoveries obtained. Depth-filter capsules are disposable, scalable and easy for good manufacturing practice (GMP) validation and were, therefore, the clarification method of choice for the downstream process herein described. This step could be further optimized depending on the LVs envelope used, which may affect viral stability and vector affinity to the membrane. Moreover, a broad range of sizes (up to 0.45 m2) and formats of filter elements are available, allowing a scale-up of this step.
Fast processing and a reduced number of downstream processing steps are required for minimization of vector losses. It was already known that fixed-bed AEXc (with DEAE adsorbers) offered one of the best binding and elution conditions for RVs and LVs purification (Rodrigues et al., 2006; Lesch et al., 2011). Therefore, AEXc efficiency in LVs purification was herein analyzed using CIM monolithic columns. High stepwise yields of 80% were obtained, an improvement of 10% over the currently reported state-of-the-art values (Slepushkin et al., 2003). The use of different chromatographic buffers for viral elution showed yields in the range of 70%. However, in order to avoid any pH limitations for virus binding to the AEXc matrices and due to PBS lack of buffering capacity at lower temperatures and its incompatibility with the Benzonase step (Eglon et al., 2009) Tris was the buffer chosen for elution. CIM DEAE has advantages over other chromatographic matrices, namely convective mass transport, extremely fast separations, high-resolution power, and high binding capacities for viruses (Jungbauer and Hahn, 2008; Gagnon, 2010) and permitted: i) an improved preservation of viral infectivity during purification; ii) an increase of the step yields, and iii) a reduction in processing time, from five working days when using gradient centrifugation protocols to 3 hr.
Vivaspin and Vivaflow systems were used to concentrate the LVs after AEXc. Both systems showed similar recoveries. However, since Vivaspin's scalability is limited by the size of the centrifuge and is much more time-consuming, Vivaflow was the preferred system because of its higher scalability and ability to perform diafiltration. To assure high resolution in the next step (SEC), only up to 5% (i.e., 16 ml) of the total SEC column volume should be injected. Therefore, the LVs fractions (from 10 to 24 ml in volume), previously diluted five-fold to avoid infectivity losses, were concentrated by UF/DF to 7–13 ml, resulting in concentration factors between 1.1 and 3.4-fold. For a better analysis, larger production and purification scales would be required. Hollow-fibers or flat-sheet cassettes can be used for large-scale processes, without the need for pre-coating, and recoveries of 100%, with concentration factors of 20 to 66-fold, and have already been reported for both RVs (Rodrigues et al., 2007b) and LVs (Geraerts et al., 2005). However, disposable cartridges such as MidGee (of 500 and 750 kDa MWCO) only permit processing of volumes from 25 to 200 ml.
The presence of residual DNA in the final viral preparations, derived from host producer cells and plasmid contamination (Chen et al., 2001), should be reduced in their amount and size to below approximately 200 base pairs in order to avoid the transfer of a residual functional open reading frame (FDA Guidance for Industry, 2010) and to further increase the product's purity. DNAses, such as Benzonase endonuclease, are often used to decrease the DNA load (Sastry et al., 2004) and to reduce the length of oligonucleotide fragments down to 3–8 bp (Janning et al, 1994). In this work, purified and nonpurified samples (after and before AEXc, respectively) were incubated with 50 U/ml of Benzonase for 30 min at 37°C. Although an early Benzonase addition reduces large DNA pieces in size, facilitating their elimination during the following purification steps, higher DNA removals (99%) were obtained for Benzonase addition after the AEXc step (0.02 μg per 108 IP). This result is comparable to those already described by Merten et al. (2011) for GMP runs but reflect a much less expensive approach, since 28 times less Benzonase was used.
Sterile filtration at 0.22 μm was performed as a final purification step, although it is known for being responsible for vector infectivity losses. In this step, losses lower than 30% were observed; however, this could not be avoided because no alternative sterilization method was available. The implementation of a semi-closed system that would allow it to perform all the purification steps in a sterile environment would make this sterile filtration step unnecessary.
In summary, the best DSP approach obtained in this study (Process C) integrates filtration and chromatographic techniques, being closely related to other processes recently described for LVs purification, such as the ones published by Merten et al. (2011) (Process A) and Couture (2008) (Process B) (Fig. 4). In Process C, CIM® DEAE monolithic columns improved the purification step yields to 80%, 42% more than the results obtained by Merten et al. (2011) (Process A) and Zimmermann et al. (2011) (using TSKgel DEAE columns and LentiSelect kits, respectively) and 10% more than obtained by Slepushkin et al. (2003) using Mustang Q Ion Exchange Capsules. Furthermore, a faster process was achieved, with overall infectious vector recoveries of 36%, 12% more than obtained in Process A. Similar yields have only been obtained, until now, in Process B or by Dupont (2008); however, fewer purification steps were used in the former one, which may justify the higher yields obtained.
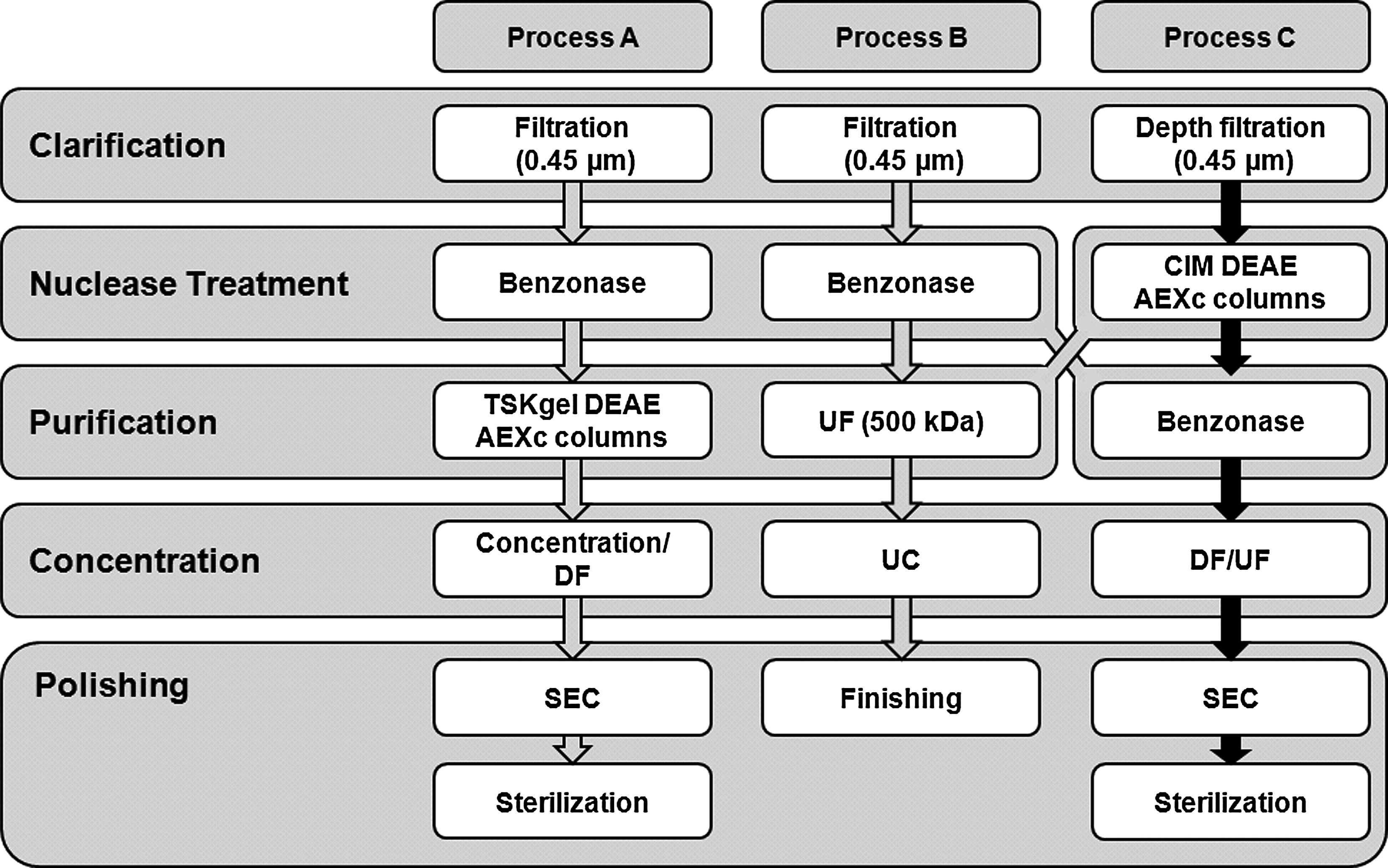
Downstream process developed in this work (Process C), compared with two representative downstream strategies to purify vesicular stomatitis virus envelope glycoprotein (VSV-g) pseudotyped LVs from Genéthon (Merten et al., 2011; Process A) and Beckman (Couture, 2008; Process B). UC, ultracentrifugation. (Adapted from Schweizer and Merten, 2010).
The work hereby presented compared several purification methods and established a novel downstream process strategy for the purification of LVs. The use of CIM® monoliths for LVs elution allowed the achievement of a faster, more efficient, and enhanced purification protocol. Progress in downstream processing will contribute to improve LVs purification toward clinical-scale for gene therapy trials and, hopefully later, for industrial-scale. Additionally, by providing higher quality grade preparations and, thus, increased safety to patients, it will contribute to faster advances in the gene therapy field.
Footnotes
Acknowledgments
The authors would like to acknowledge Sartorius Stedim Biotech and BIA Separations for their technical advice and for providing chromatographic matrices. Financial support received from European Commission Clinigene Network of Excellence (LSHB-CT-2006) and Fundação para a Ciência e Tecnologia—Portugal (PTDC/EBB-BIO/100491/2008, PTDC/EBB-BIO/102649/2008 and SFRH/BD/48393/ 2008).
Author Disclosure Statement
No competing financial interests exist.