
Abstract
SmartDCs (
Pincha and colleagues describe a technique for generating clinical-grade “SMARTDCs” (self-differentiated myeloid-derived antigen-presenting cells reactive against tumors). The authors show that the cells can be cryopreserved and that thawed human SMARTDCs injected subcutaneously into NOD.Rag1−/−IL2rγ−/− mice maintain dendritic cell characteristics and viability for 1 month in vivo and do not cause any signs of pathology.
Introduction
Incidence rates for melanoma have risen faster than for any other malignancy in white populations over the past 30 years (Giblin and Thomas, 2007). The overall median survival from diagnosis of metastatic melanoma has been estimated to be 8 months (Lee et al., 2000), and chemotherapy and radiotherapy are used with palliative intent. The cytotoxic T lymphocyte–associated (CTLA)-4 blocking antibody ipilimumab has received marketing authorization by the U.S. Food and Drug Administration (FDA) after it was shown in phase III trials to prolong the overall survival in approximately 10%–15% of patients (Hodi et al., 2010), although serious immune side effects (liver failure, hormone problems, severe skin reactions, and inflammation of the intestines) were observed in 13% of the patients. In subgroups of melanoma patients with BRAF and KIT mutations, drugs targeting these mutations appear to produce remarkable initial clinical responses that are, however, short term (Chapman et al., 2011; Satzger et al., 2010). Thus, it is currently unclear how many patients will reach long-term remission. Therefore, effective alternative therapies providing long-lasting immunological memory to promote complete tumor eradication are warranted in light of the increasing incidence and highly unmet needs in the medical treatment of metastatic melanoma.
In 2010, a novel cellular vaccine product called Sipuleucel-T received marketing authorization from the FDA for improving the survival of men with advanced prostate cancer in large phase III clinical trials. Men diagnosed with asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer who received Sipuleucel-T lived 4 months longer on average than men in the placebo group of the study (Sims, 2011). Sipuleucel-T consists of peripheral blood mononuclear cells (PBMCs) incubated with a recombinant fusion protein of human prostatic acid phospatase (PAP) and granulocyte-macrophage-colony stimulating factor (GM-CSF). Its use as a cell infusion resulted in immune responses to the immunizing PAP antigen in prostate cancer patients (Sims, 2011). The manufacture of the vaccine product is relatively simple, consisting of a 48-hr procedure (leukapheresis, elutriation, incubation with recombinant protein), but the logistics of production are complicated by the fact that patients are treated for three consecutive cycles with fresh cells. Not surprisingly, Sipuleucel-T is currently one of the most expensive cancer therapies on the market.
On the other hand, for over a decade, numerous clinical trials have been carried out to assess the ability of immunotherapy using ex vivo differentiated dendritic cells (DCs) to induce clinically relevant immune responses in patients with melanoma. An extensive review analyzing a total of 626 patients with malignant melanoma treated with DC therapy demonstrated complete responses in 3%, partial responses in 6%, and stable disease in 21% of the patients with the use of DCs pulsed with peptide antigens (Engell-Noerregaard et al., 2009). Progression to larger DC clinical trials has generally been compromised by the high costs, consistency, and viability of the ex vivo produced and antigen-loaded DC products. DCs are typically cultured for several days ex vivo, and at the end of the culture period a considerable drop in cell viability is observed, potentially affecting in vivo potency and biodistribution of the DCs (Figdor et al., 2004). As a result, until now, ex vivo generated DCs have not received marketing authorization as cellular vaccines either by the FDA or by the European Medicines Agency (EMA).
In addition, several clinical trials have been conducted to explore recombinant vectors for expressing full-length melanoma-associated antigens in DCs in order to direct multifunctional immune responses since several class I and class II epitopes in an antigenic protein are targeted. Phase I and I/II studies have been reported with DCs expressing melanoma antigens delivered by different types of vectors (fowlpox, vaccinia, plasmids, adenovirus) (Ribas, 2005). These studies have demonstrated that the administration of DCs transduced with these vectors are safe and well tolerated. However, these vector systems have also demonstrated low gene delivery efficiency, cytotoxic effects in DCs, or stimulation of bystander immune responses against viral proteins co-expressed in DCs. In light of the difficulties in clinical development of genetically reprogrammed DCs, several groups have recently explored the transfection/electroporation of DCs with messenger RNAs (Michiels et al., 2005), which is relatively unpredictable with regard to the stability of transgene expression in DCs (hours to a few days). The approach was shown in animal models of immunotherapy to be less effective than gene delivery by viral transduction (Dullaers et al., 2004).
Overall, a common perception in the field of DC immunotherapy is that laborious and lengthy DC production protocols are not only time and cost intensive but lead to inconsistent cell quality. In order to circumvent these issues, we have devised a short overnight production approach for ex vivo generation of genetically reprogrammed DCs. We have explored lentiviral vectors (LVs) that transduce DC precursors with higher efficiency than the other tested viral vector systems in the absence of cytotoxic or unwanted immunologic effects and have transgene expression that persists for several weeks (Koya et al., 2007; Jirmo et al., 2010; Pincha et al., 2011). Using LVs to transduce mouse and human DC precursors, we have shown that expression of GM-CSF and interleukin (IL)-4 could actually induce autonomous DC self-differentiation in vivo (Koya et al., 2007; Pincha et al., 2011; Salguero et al., 2011). These lentivirus-reprogrammed cells have been named
Here, we show that, for clinical development of SmartDCs, human monocytes can be obtained from leukapheresis and transduced ex vivo with a tricistronic LV in a closed bag system. After the 28-hr ex vivo procedure, the cells can be cryopreserved. These cells are able to self-differentiate into DCs, the time of cell manipulation is short (1 day), and the consistency, cell recovery, and cell viability are potentially higher compared to the conventional DC preparation procedures. Thus, these aspects facilitate standardized production of genetically reprogrammed DC vaccines for future clinical trials.
Materials and Methods
Construction of tricistronic vectors and lentivirus production
Mouse homologue vector (mLV-G242T)
The self-inactivating (SIN) lentiviral bicistronic vector expressing RRL-cPPT-CMV-mGMCSF-P2A-mIL4 and the monocistronic vector expressing tyrosine related protein 2 (RRL-cPPT-CMV-mTRP2) were previously described (Kimura et al., 2007; Pincha et al., 2011). A tricistronic vector (RRL-cPPT-CMV-mGMCSF-P2A-mIL4-F2A-mTRP2) containing mGM-CSF-P2A-mIL-4 and mTRP2 separated by a Kozak consensus sequence and F2A element was constructed by overlapping polymerase chain reaction (PCR) essentially as previously described (Szymczak and Vignali, 2005). Primers used to generate interspacing F2A were F2A/TRP2 forward 5′- CCGG TGAAACAGACTTTGAATTTTGACCTTCTCA AGTTGGCG GGAGACGTGGAGTCCAAC CCAGGGCCCGCCACCA TG GGCCTTGTGGGATG -3′ and F2A/IL-4 reverse 5′- TGGGT TGGACTCCACGTCTCCCGCCAACTTGAGAAGGTCAA A ATTCAAAGTCTGTTTCACCGGTCCGGACGAGTAATCCA TTTGCATGATGC -3′. The products were digested with SpeI and SalI and introduced into the site of the pRRL-sin-cPPT-hCMV-MCS vector (Kimura et al., 2007).
Human homologue vector (hLV-G242T)
The SIN lentiviral bicistronic vector expressing (RRL-cPPT-CMV-hGMCSF-P2A-hIL4) was previously described (Salguero et al., 2011). The LV backbone contains a mutated, non-encoding oPRE element as previously described (Schambach et al., 2006). The plasmid pcDNA3 encoding human hTRP2 was kindly provided by Dr. Thomas Woelfel. A tricistronic vector (RRL-cPPT-CMV-hGMCSF-P2A-hIL4-F2A-hTRP2) containing hGM-CSF-P2A-hIL-4 and hTRP2 separated by an F2A element was constructed by overlapping PCR. Primers used to generate interspacing F2A were F2A/TRP2 forward 5′- CCGG TGAAAC AGACTTTGAATTTTGACCTTCTCAAGTTGGCGGGAGACG TGGAGTCCAACCCAGGGCCCATGAGCCCCCTTTGGTGG GG-3′ and F2A/IL-4 reverse 5′- TGGGTTGGACTCCACGTCT CCCGCCAACTTGAGAAGGTCAAAATTCAAAG TCTGTTT CACCGGTCCGGAGCTCGAACACTTTGAATATTTCTC-3′. The products were digested with XbaI and SalI and introduced into the site of the pRRL-sin-cPPT-hCMV-MCS vector (Salguero et al. (2011). Amplifications were carried out with initial denaturation at 94°C for 2 min (one cycle), denaturation at 94°C for 30 sec, annealing at 68°C for 30 sec, elongation at 72°C for 10 min (30 cycles), and a final elongation at 72°C for 12 min (one cycle). The structural integrity of all constructs was reconfirmed by restriction digestion and sequencing analysis of all transgenes. Large-scale lentivirus production was performed as previously described (Stripecke, 2009; Pincha et al., 2011). Lentiviral titers assessing p24 antigen concentration were determined by enzyme-linked immunosorbent assay (ELISA) (Cell Biolabs, Inc., San Diego, CA). One microgram of p24 equivalent per 1 ml corresponds to approximately 1×107 infective viral particles per 1 ml.
Cell culture
The human embryonic kidney 293T cells and murine melanoma cell line B16-fLUC (Pincha et al., 2011) were cultured in Dulbecco's modified Eagle medium (Invitrogen, Karlsruhe, Germany) plus 10% fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 mg/ml) (Biochrom AG, Berlin, Germany). K562 cells stably transfected for HLA-A*0201 expression (a kind gift from Prof. Thomas Woelfel) were cultured in RPMI with 10% FBS, penicillin (100 U/ml), streptomycin (100 mg/ml), and geneticin (1 mg/ml) (Biochrom AG).
Western blot
293T cells were transduced with 1 μg p24/ml (multiplicity of infection [MOI] of 200) of each type of indicated LV. Transduced 293T cells were lysed in lysis buffer (with protease inhibitor cocktail) (Bio-Rad, Munich, Germany) for 10 min on ice. Cell lysates and supernatant samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (AnykD gel, Bio-Rad) and then evaluated with rabbit anti-human GM-CSF (Peprotech, Hamburg, Germany), mouse anti-human IL-4 (R&D Systems, Wiesbaden, Germany), or goat anti-TRP2 (T-17) (Santa Cruz, Heidelberg, Germany) antibodies. Protein bands were visualized using secondary antibodies coupled to horseradish peroxidase and the enhanced chemiluminescence kit (Pierce, Thermo Scientific, Bonn, Germany) according to the manufacturer's instructions.
Ex vivo generation and cryopreservation of mouse SmartDC-TRP2
Bone marrow–derived mouse SmartDC-TRP2 were generated as previously described (Koya et al., 2007; Pincha et al., 2011). One day after LV transduction and washing, mouse SmartDC-TRP2 were cryopreserved in freezing medium containing 70% RPMI (Invitrogen, Darmstadt, Germany), 20% FBS, and 10% dimethyl sulfoxide (DMSO; Sigma Aldrich, Munich, Germany).
Ex vivo generation and cryopreservation of human SmartDC and SmartDC-TRP2
PBMCs were obtained from HLA-A*0201–positive, healthy adult volunteers or from melanoma patients who had consented to undergo leukapheresis, and all studies were performed in accordance with protocols approved by the Hannover Medical School Ethics Review Board. CD14+ monocytes were isolated from PBMCs obtained from leukapheresis using CD14 isolation beads (Miltenyi Biotec, Bergisch-Gladbach, Germany). The monocytes were kept in culture with serum-free Cellgro medium (Lonza, Basel, Switzerland) in the presence of recombinant human GM-CSF and IL-4 (50 ng/ml each; Cellgenix, Freiburg, Germany) for 8 hr prior to transduction. For the generation of SmartDC SmartDC-TRP2 or SmartDC-TRP2/fLUC, 5×106 CD14+ monocytes were transduced with 2.5 μg p 24 equivalent of hLV-G24 (Salguero et al., 2011), hLV-G242T, or hLV-G242T/LV-fLUC in the presence of 5 μg/ml protamine sulfate for 16 hr. Conventional DCs were generated from CD14+ cells and supplemented with recombinant hGM-CSF and hIL-4 every 3 days. After transduction, SmartDC-TRP2 were washed twice with phosphate-buffered saline (PBS) and maintained in culture without cytokines or were cryopreserved in freezing medium containing 70% PBS, 20% human serum albumin (Lonza), and 10% DMSO.
Analyses of mouse and human cytokines
Detection of secreted mouse GM-CSF and IL-4 in supernatants of DC cultures was performed with commercially available ELISA kits (R&D Systems, Wiesbaden, Germany). Detection of human Th1/Th2 cytokines in the supernatants of DC cultures and mixed lymphocyte reactions was performed with fluorescent bead–based 14-plex Luminex assay according to the manufacturer's protocol (Millipore, Billerica, MA). The 14-plex assay measured the following cytokines: GM-CSF, IL-4, tumor necrosis factor alpha (TNF-α), IL-6, IL-8, monocyte chemotactic protein-1 (MCP-1), IL-10, IL-1β, IL-5, IL-13, interferon (IFN)-γ, IL-7, IL-2, and IL-12(p70). Detection limit of the assay in the lower range was 3.2 pg.
Flow cytometry analyses of surface and intracellular antigens
The phenotypes of bone marrow–derived mouse DCs and monocyte-derived human DCs were analyzed as previously described (Pincha et al., 2011; Salguero et al., 2011). Immunostaining for mouse cells was performed using the following anti-mouse monoclonal antibodies (mAbs) CD11c, CD11b, CD80, and CD86 (BD Pharmingen, Heidelberg, Germany) and MHCII and CCR5 (eBiosciences, Frankfurt, Germany), with respective isotype controls. Intracellular staining for IFN-γ production was performed as previously described (Pincha et al., 2011). Staining for human cells was performed with anti-human mAb CD14, CD209, CD86, HLA-DR, CCR2, and CCR5 (Becton Dickinson, Heidelberg, Germany), with respective isotype controls. For intracellular staining of human TRP2, cells were permeabilized and fixed with Cytofix/Cytoperm kit (Becton Dickinson) according to manufacturer's instructions, followed by incubation with polyclonal rabbit anti-Dopachrome tautomerase (Sigma Aldrich) and a secondary Dylight649-conjugated donkey anti-rabbit antibody (BioLegend, Fell, Germany). Cells were measured with FACSCalibur cytometer or LSRII (BD Pharmingen) and analysis was done using Cell Quest (Becton Dickinson) or Flow Jo software (Tree Start, Ashland, OR).
Mice, vaccinations, melanoma challenge, and bioluminescence imaging analyses
All procedures involving mice were reviewed and approved by the Lower Saxony State Office for Consumer Protection and Food Safety and followed the guidelines provided by the Animal Facility at the Hannover Medical School. Eight- to 10-week-old C57BL/6 female mice were purchased from Charles River (Sulzfeld, Germany) and NOD.Cg-Rag1tm1MomIl2rgtm1Wjl (Nod.Rag1−/−.IL2rγ−/− [NRG]) mice were bred in house and maintained under pathogen-free conditions in an IVC system (BioZone, London, UK). All cell suspensions diluted in PBS were subcutaneously injected in volumes of 100–150 μl in the hind flanks of mice with a 27-gauge needle. CryoSmartDC-TRP2 were thawed immediately prior to injections. They were frozen at an appropriate concentration to obtain 1×106 cells, 2% DMSO, and 4% FBS in a volume of 100–150 μl after thawing and dilution with PBS. Mice were immunized with 1×105 “fresh” or 1×106 “cryo” SmartDC-TRP2 following different schemes indicated for each experiment type. Melanoma challenge was performed with 5×104 B16-fLUC cells, injected subcutaneously on the same side. Monitoring of tumor growth and in vivo bioluminescence imaging analyses were performed as previously described (Pincha et al., 2011; Salguero et al., 2011).
Carboxyfluorescein succinimidyl ester labeling and immunofluorescence analyses
Human SmartDC-TRP2 cells were incubated with 5 μM carboxyfluorescein succinimidyl ester (CFSE, Invitrogen) at 37°C for 10 min after transduction. After incubation, cells were washed twice and resuspended in PBS. Cell suspensions were subcutaneously injected in volumes of 50 μl in the ear with a 27-gauge needle. Ears were harvested 7 days after injection and embedded in optimal cutting temperature compound (O.C.T. Sakura Finetek, Torrance, CA) for cryopreservation. Frozen sections (5 μm) were fixed in acetone and stained with rabbit monoclonal anti-human CD11c (eBioscience, San Diego, CA) and Cy5-conjugated goat anti-rabbit IgG (Sigma Aldrich). Immunofluorescence analyses were performed in an Axio Imager microscope (Carl Zeiss GmbH, Jena, Germany) and images were analyzed using AxioVision 40 software (Carl Zeiss).
Mixed lymphocyte reaction
Human SmartDC-TRP2 or conventional DCs were harvested from culture on day 7 and irradiated with 30 Gy and used as stimulators. CD3+ T cells were positively selected from PBMCs with CD3 isolation beads (Miltenyi Biotec). Different numbers of DCs were co-cultured with 1×105 autologous or allogeneic CD3+ T cells at various ratios (1:2, 1:5, and 1:20) in round-bottom 96-well plates with a total volume of 200 μl of Cellgro medium. Triplicate wells were set up for each ratio and reaction condition. The reactions were incubated for 6 days at 37°C and 1 μCi/well of [3H]thymidine was added for the last 18 hr. [3H]Thymidine incorporation was measured on a β-scintillation counter. The stimulatory capacity was determined with stimulation index (SI)=cpm of (T cells+stimulator cells)/cpm of unstimulated T cells. The allogeneic mixed lymphocyte reactions were performed with CD3+ T cells isolated from PBMCs of the same donor.
Human TRP2-specific T-cell responses in vitro
PBMCs obtained from a melanoma patient were used for the production of SmartDCs and SmartDC-TRP2, harvested on day 7, and used directly for the first T-cell stimulations or cryopreserved for subsequent restimulations. A control group consisted of SmartDCs loaded with the TRP2 overlapping peptide mix (JPT Peptide Technologies, Berlin, Germany; 10 μg/ml). After 2 hr, excess unloaded peptides were washed off. CD8+ T cells from the patient were isolated following the manufacturer's protocol (Miltenyi Biotec). Purity and viability of the enriched cells were checked by FACS. The stimulations were carried out in a microculture setting in 96-well round bottom plates. CD8+ cells were seeded into a 96-well U bottom plate at 1×105 cells/well and autologous DCs were added at a 10:1 ratio (T cells to DCs). In addition, 2×105 autologous feeder cells (CD14 and CD8 negative) gamma-irradiated with 40 Gy were added to the CD8+ T cells. X-Vivo 15 (Lonza) supplemented with 5% human AB serum (Lonza) and cytokines were added (IL-2: 25 IU/ml [Proleukin], IL-7: 5 ng/ml and IL-15: 5 ng/ml [Cellgenix, Gladbach, Germany]), to a final volume of 200 μl per well and kept at 37°C for 7 days. The medium with cytokines was replenished every 2 days. Restimulations were performed every 7 days by adding the corresponding numbers of cryopreserved/thawed DCs to the T cells at a ratio of T cells to DCs of 10:1. For enzyme-linked immunospot technique (ELISPOT) assay, stimulated T cells were seeded at a density of 30,000 cells per well in 96-well ELISPOT plate coated with anti-human IFN-γ (Mabtech AB, Büro, Germany). The cells were incubated for 20 hr at 37°C and 5% CO2 with 5×104 target cells: K562/HLA-A*0201 cells alone, K562/HLA-A*0201 transduced with LV-hTRP2, or K562/HLA-A*0201 loaded with CEF peptide pool (this pool contained 32 HLA class I–restricted T-cell epitopes from human cytomegalovirus, Epstein-Barr virus, and influenza virus) (JPT Peptide Technologies) as positive recall control. After incubation, cells were washed and plates were further incubated with biotin-conjugated anti-human IFN-γ antibody followed by alkaline phosphatase–conjugated streptavidin. Plates were developed using NBT/BCIP liquid substrate (Sigma) and analyzed with an ELISPOT reader (CTL Immunospot® analyzer), and the quantification was done with ImmunoSpot® Software for ELISPOT Analysis (CTL-Europe GmbH, Bonn, Germany).
Tetramer analysis
T cells expanded in vitro were stained with the following anti-human antibodies: FITC-conjugated CD8 (BD Pharmingen), PECy7-conjugated CD3 (BD Pharmingen), and APC-conjugated HLA-A*0201 anti TRP2 180–188 or 360–368 tetramer (a kind gift from Dr. Philippe Guillaume and Prof. Immanuel F. Luescher, Ludwig Institute for Cancer Research, Lausanne). An irrelevant tetramer (PE-conjugated negative tetramer) was purchased from Beckman Coulter. Stimulated T cells (2×105) were incubated for 30 min at room temperature with tetramer, washed once, stained with surface antibodies for 20 min at 4°C, washed once, fixed, and analyzed in a flow cytometer (LSRII, BD Pharmingen). At least 10,000 CD3/CD8 double positive viable cells were analyzed. Tetramer positive cells are expressed as the percentage of CD3+/CD8+ T cells.
Scaling up production of human SmartDC-TRP2 under good manufacturing practices compliant conditions
CD14+ selection was performed with PBMCs obtained from leukapheresis of healthy donors by magnetic-activated cell separation (CliniMACS cell separation system; Miltenyi Biotec) in B cleanroom of the good manufacturing practices (GMP) center under laminar air flow conditions (CliniMACS in laminar air flow field). CD14+ cells (200×106) were cultured in a cell differentiation bag 250 (Milteny Biotec) and preconditioned for 6 hr with 5 μg each of GMP grade cytokines rhGM-CSF and rhIL-4 (Cellgenix, Freiburg, Germany) in Cellgro medium at 37°C (5% CO2). These procedures were performed in an authorized GMP facility. Transduction was performed with hLV-G242T (100 μg/ml) and protamine chloride (500 μg; Valeant Pharmaceuticals, Duesseldorf, Germany) for 16 hr at 37°C (5% CO2) in a total volume of 100 ml of culture, after which SmartDC-TRP2 were washed twice with PBS and cryopreserved at 2×106/ml/vial aliquots. Cryopreservation was done by resuspending SmartDC-TRP2 in 20% human serum albumin and mixing with equal amounts of freezing medium containing 20% human serum albumin, 20% DMSO, and 10% glucose (Braun, Nürnberg, Germany).
Quantification of the LV copy number in DCs by quantitative real-time PCR
Total genomic DNA was extracted from 1×106 cells using the QiaAmp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. DNA concentration was measured by using a Qubit Fluorometer (Invitrogen) and normalized to 4 ng/μl with deionized and distilled water (ddH2O; Roche, Mannheim, Germany). Ten-fold serial dilutions of a PCR product consisting of one fragment derived from a sample transduced with the LV vector of interest were used as standard references. Absolute quantification of vector copy numbers and analysis were performed with the Light Cycler LC 480 System (Roche) using primers located in the long terminal repeat (LTR) (SKLTR3 AGCTTGCCTTGAGTGCTTCA) and in the backbone (qPCRLV2 revB GAGTCCTGCGTCGAGAGAGC) of the LV vector. Amplification of the housekeeping human myoglobin gene was used as a two-copy gene standard to quantify the exact amount of genomic DNA present in each sample using the following primers Myo forward (GGACTGCCACCCACTTTGCC) and Myo reverse (GACAAGCTGCTTAACCTCTG). Each reaction contained 20 μl: 4 μl of ddH2O, 0.5 μl of each primer (10 pmol/μl), 10 μl of SYBRGREEN I Mix (Roche), and 5 μl (4 ng/μl) of DNA. The thermal cycling protocol was the following: 95°C for 15 min, 95°C for 10 sec, 58°C for 5 sec, and 72°C for 25 sec for 45 cycles; melting curve program: 95°C for 5 sec, 65°C for 1 min, 97°C continuous; and cooling for one cycle: 40°C for 30 sec. Standard references and samples were analyzed in triplets to octets. Furthermore, a sample containing a defined vector copy number was used as an internal positive control. Copy numbers were determined by calculating the ratio between the quantity of vector copies and host genome copies. The following formula was used for the calculation: vector copy number=(quantity mean of LV sequence/quantity mean of myoglobin sequence)×2. Multiplying by a factor of 2 reflects the myoglobin sequence being present in two copies per genome.
Statistical analyses
For statistical analyses of tumor challenge studies, the probability of disease-free survival was estimated using the Kaplan–Meier method; disease-free survival was plotted and compared using Mantel Cox test. The p-values were calculated by Student's t-test. All tests were two-sided and p<0.05 was considered significant. Graph Pad Prism 5 Software (La Jolla, CA) was used for the statistical analysis.
Results
Tricistronic vectors driving efficient expression of GM-CSF, IL-4, and TRP2
We previously demonstrated that mouse bone marrow precursors and human monocytes self-differentiate into cells with typical DC characteristics after LV-mediated gene delivery of murine/human GM-CSF and IL-4 genes (Kimura et al., 2007; Koya et al., 2007; Pincha et al., 2011; Salguero et al., 2011). SmartDCs co-transduced for expression of a melanoma antigen (TRP2) were highly viable in the injected skin, migrated to lymph nodes, and generated potent and long-lasting immune responses against melanoma (Koya et al., 2007; Pincha et al., 2011). Compared with conventional DCs produced with recombinant cytokines, SmartDCs co-expressing TRP2 were more effective in protection and therapy of the highly aggressive B16 melanoma implanted subcutaneously in C57BL/6 mice (Koya et al., 2007; Pincha et al., 2011). In the current study, we have developed tricistronic LVs encoding for human or mouse GM-CSF, IL-4, and the full-length TRP2 antigen (hLV-G242T and mLV-G242T) (Fig. 1a). Human and murine GM-CSF and IL-4 are not cross-reactive. On the other hand, human and murine TRP2 are highly homologous (83% homology at the amino acid level) with several shared immunodominant epitopes, such as the TRP2180-188 CTL epitope presented both by the mouse H-2Kb and human HLA-A*0201 (Supplementary Fig. S1; Supplementary Data are available online at www.liebertonline.com/hgtb); thus, TRP2 is a unique conserved antigen, relevant for predicting efficacy and safety in humans (Cassady and Sturm, 1994; Parkhurst et al., 1998). 2A elements interspacing the transgenes were used to enable translation of individual protein products at relatively similar stoichiometric levels in the cell (Fig. 1a). Heterologous 2A elements from porcine teschovirus (P2A) and foot and mouth disease virus (F2A) were used to avoid homologous recombination in the LV. Viral titers were routinely determined by measuring the concentration of p24 (the core protein of HIV), which is correlated with the infective viral titer, measured by analyses of the number of integrated viral copies in the 293T standard cell line (Fig. 1b). One microgram of p24 equivalents of the hLV-G242T vector corresponds to approximately 1×107 infective particles. We consistently obtained 20–30 μg of p24 equivalents (2×108 to 3×108 infective viral particles) for laboratory-scale production runs of human and mouse LV-G242T, which is comparable with the standard LV-GFP vector (Fig. 1c). Expression of the human TRP2 antigen could be directly assessed in >90% transduced 293T cells by intracellular staining and flow cytometry (comparable with the LV-GFP expression analyses) (Fig. 1d). GM-CSF and IL-4 produced by transduced 293T cells were detectable by ELISA at high concentrations (μg/ml) in the cell supernatants (data not shown). In order to confirm the expression of the processed protein products at the molecular level, Western blot analyses were performed. Protein extracts prepared with 293T cell lysates and cell culture supernatants demonstrated that human GM-CSF and IL-4 were detectable both intracellularly and in the medium. 293T-produced GM-CSF and IL-4 proteins ran at higher molecular weights than recombinant proteins produced in bacteria, due to glycosylation and the additional P2A and T2A elements (Fig. 1e,f). Secreted GM-CSF was detected as a wide smeary band due to heavy glycosylation. Expression of fusion products possibly caused by translational read-through of the 2A elements was detectable in cell lysates, but these products were not secreted. Mouse and human TRP2 proteins were detectable in 293T cells transduced with mLV-G242T or hLV-242T both as single and larger fusion proteins (Fig. 1g).
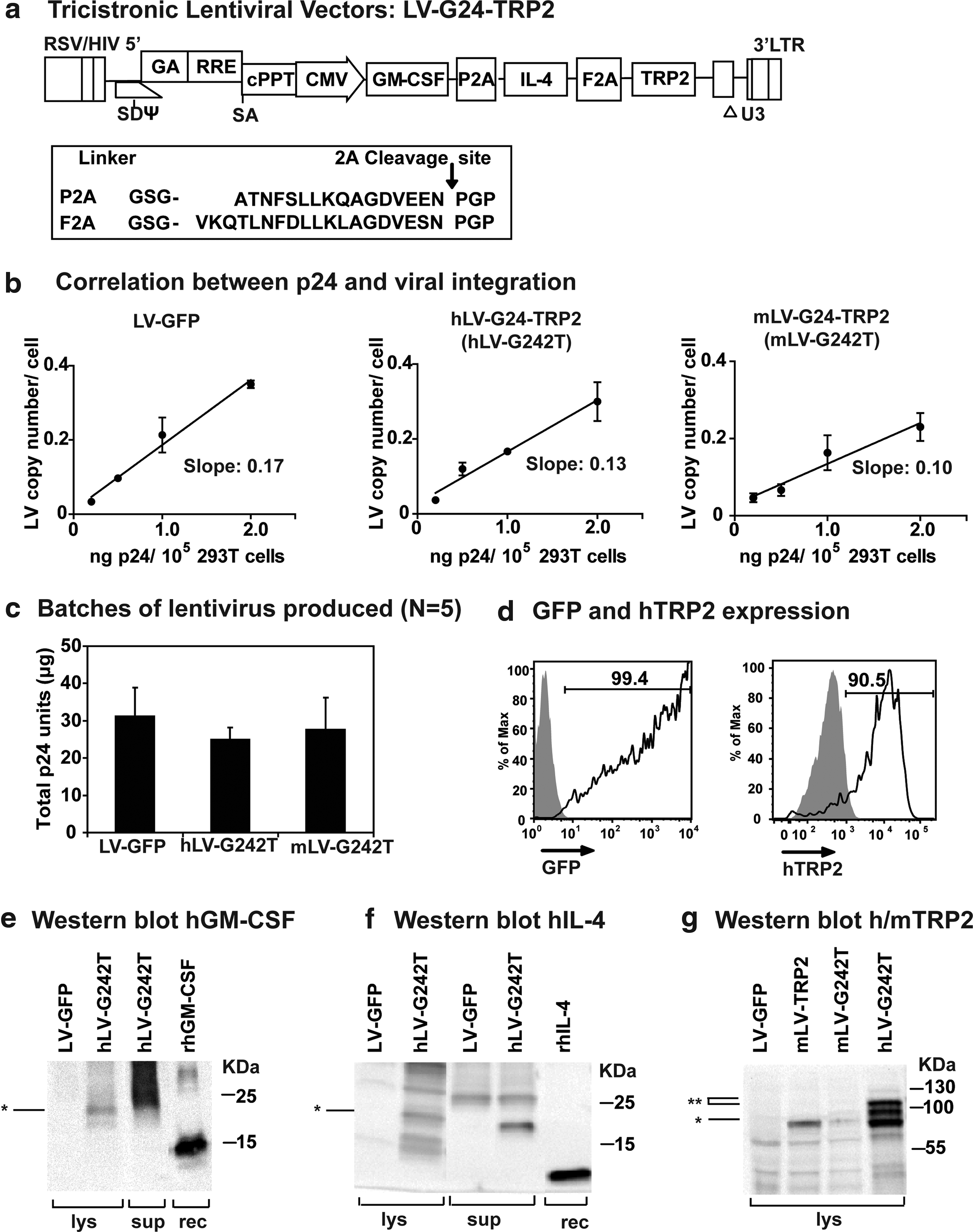
Testing of human and mouse homologous lentiviral vectors: titers and transgene expression.
Cryopreserved mouse SmartDC-TRP2 is an effective vaccine against melanoma
We confirmed the generation of functional mouse SmartDC-TRP2 using the mLV-G242T mouse tricistronic vector. Following established protocols for transduction of bone marrow cells with LV vectors (MOI of 5) and for analyses of phenotype and transgene expression of mouse SmartDCs, we confirmed their previously described characteristics (Koya et al., 2007; Pincha et al., 2011): high viability in vitro (up to 3 weeks), stable expression of GM-CSF/IL-4, typical DC immunophenotype, and stimulation of TRP2-specific CD8+ T-cell responses in C57BL/6 mice (Supplementary Fig. S2a–e).
Subsequently, characterization of SmartDC-TRP2 cryopreserved 1 day after transduction (cryoSmartDC-TRP2) was performed (Fig. 2a). Recovery of viable cryoSmartDC-TRP2 cells was 60%–80% immediately after thawing, which decreased to about 10%–13% after 7 days of culture (the recovery of mouse fresh SmartDC-TRP2 after 7 days of culture was 20%–25% of cell input). One day after in vitro culture, cryoSmartDC-TRP2 secreted detectable levels of GM-CSF and IL-4 (80 and 50 pg/106 cells/ml/24 hr, respectively) (Fig. 2b), and on day 7 cells expressed typical myeloid DC markers and costimulatory markers, comparable to SmartDC-TRP2 that were not frozen (Fig. 2c). For in vivo vaccination experiments, 1×106 cryoSmartDC-TRP2 were injected subcutaneously in a biweekly prime/boost vaccination schedule (Fig. 2d). Vaccinated mice elicited significant levels of TRP2180-188–specific CD3+ CD8+ IFN-γ+ T-cell responses (vaccinated: 2%, baseline control: 1%) (Fig. 2e), which were correlated with protection against a lethal melanoma challenge (5×104 B16-fLUC melanoma cells injected subcutaneously on the same side as vaccine). Optical imaging studies on day 14 showed that four out of five control mice developed tumors, whereas only one out of five vaccinated mice developed tumors (Fig. 2f). The protective effect was maintained long-term as shown by observation for 4 months after tumor challenge and Kaplan–Meier survival analyses (vaccinated: 80% survival, control: 20%) (Fig. 2g,h). Long-term survivors that were vaccinated with cryoSmartDC-TRP2 and challenged with tumor displayed patches of vitiligo (all four mice) and alopecia (three out of four mice) on the body (Fig. 2i). Since melanocytes present in the dermis and hair follicles express TRP2, this confirms autoimmune responses generated against TRP2, corroborating autoimmunity findings during melanoma immunotherapy trials in mouse studies and in humans (Shibagaki and Udey, 2003; Bridle et al., 2010). Other than these autoimmune effects, long-term survivors vaccinated with cryoSmartDC-TRP2 did not demonstrate signs of progressive autoimmunity or behavioral changes. Histopathological analyses of different organs (spleen, liver, kidney, lung, skin, inguinal lymph nodes, bone marrow, and brain) did not reveal any pathology. Histological examination of skin (vaccination and distal sites, also including skin specimens with alopecia and vitiligo) demonstrated occasional occurrence of dermatitis typically characterized by mild to moderate lympho-histiocytic infiltrates (data not shown).
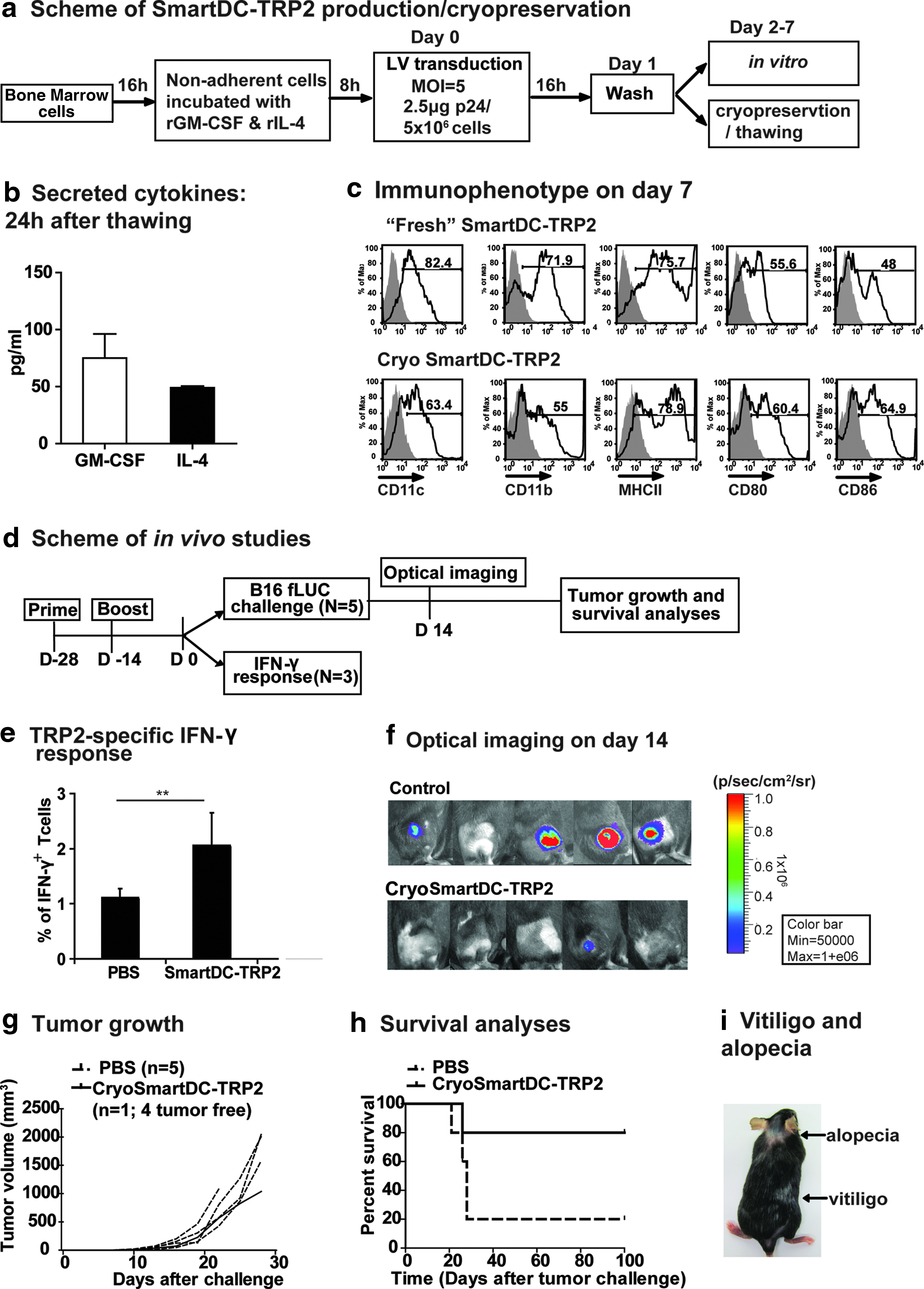
Feasibility of cryopreservation of mouse SmartDC-TRP2 after transduction.
Generation and testing of human SmartDC-TRP2 from melanoma patients
Our next goal was to confirm the feasibility of human SmartDC-TRP2 generation using the human tricistronic vector design. Since the cell targets for gene transfer and functional testing could potentially differ between healthy donors and cancer patients, we used cells obtained from stage IV melanoma patients in clinical remission, who kindly consented to undergo leukapheresis. CD14+ monocytes isolated from the banked cryopreserved leukapheresis samples were transduced with hLV-G242T at MOI of 6 using our routine procedures to generate SmartDC-TRP2 (Fig. 3a). Microscopic observation of SmartDC-TRP2 7 days after transduction showed cells with typical DC morphology (Fig. 3b). Bead-array analyses of SmartDC-TRP2 culture supernatant demonstrated the accumulation of high levels of transgenic GM-CSF (average day 7: 0.4 ng/ml; day 14: 2.4 ng/ml) and IL-4 (average day 7: 0.9 ng/ml, day 14: 10 ng/ml) (Fig. 3c). Among different Th1/Th2 cytokines measured, high levels of endogenous TNF-α, MCP-1, and IL-8 were also detectable on day 7, which further accumulated until day 14 (Fig. 3c). On day 14, IL-6 was also detectable. Th2 cytokines, which could theoretically prime the DCs to immune tolerance, were either very low (IL-10: 10 pg/ml) or undetectable (transforming growth factor β) (data not shown). Conventional DCs (i.e., produced in parallel in the presence of recombinant cytokines) and SmartDC-TRP2 harvested on days 7 and 14 after initiation of culture were characterized for immunophenotype; both cell types showed down-regulation of CD14 and expressed high levels of DC markers (HLA-DR, CD86, CD209) and relevant immunologic markers (CD80, CD83, CCR5, CCR2) (Fig. 3d). Quantification of the level of DC surface markers by median fluorescent intensity quantification showed similar levels of expression in SmartDC-TRP2 and in conventional DC, except for significantly higher levels of CD86 and consistently higher HLA-DR expression in SmartDC-TRP2 (Fig. 3e). CD86 is a DC activation marker and highly expressed in mature DCs, indicating SmartDC-TRP2 had a more activated status, which might result from endogenously produced TNF-α and IL-6 (Fig. 3c,e).
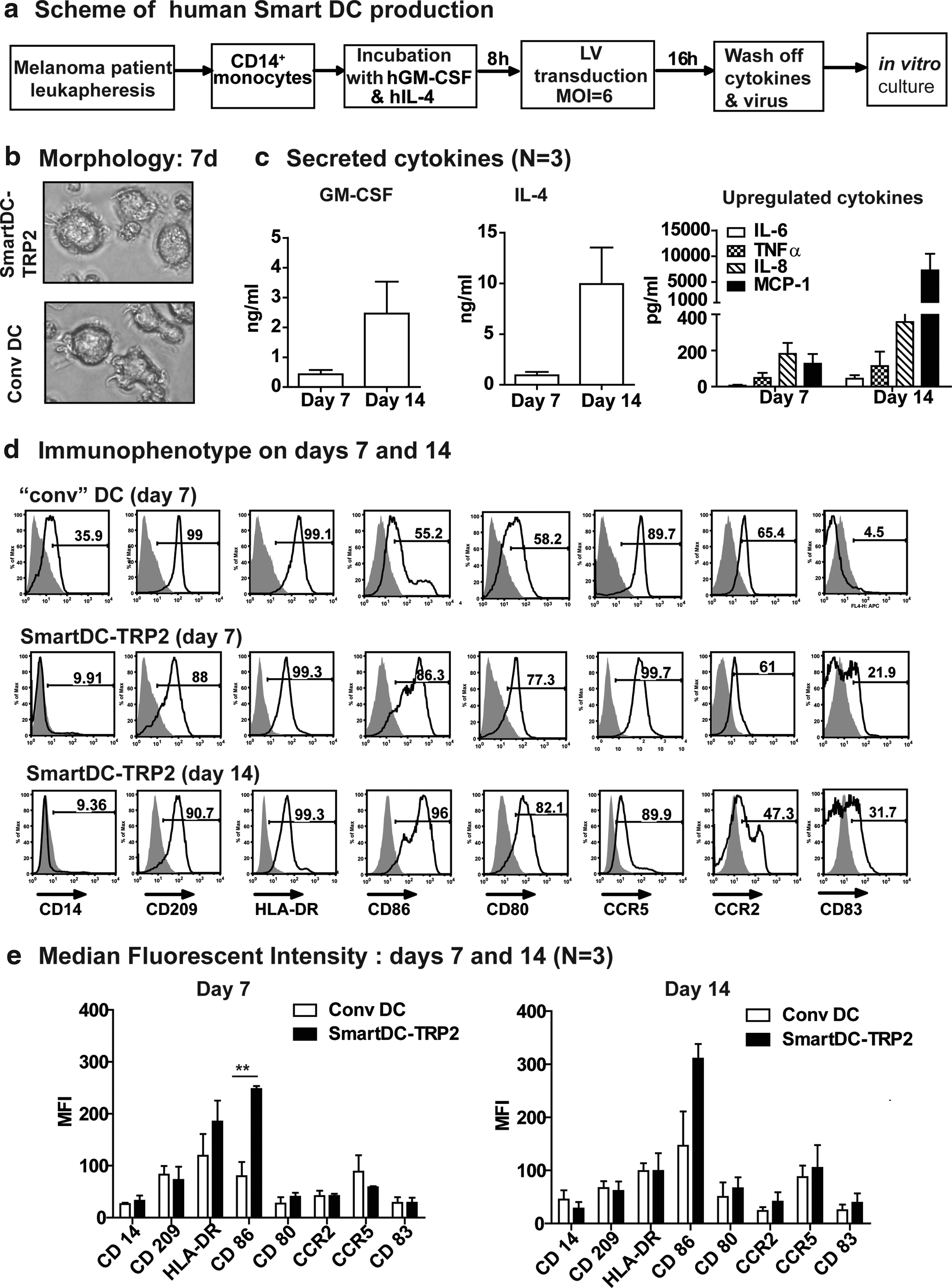
Human SmartDC-TRP2 generated from CD14+ monocytes from melanoma patients display a DC phenotype.
Immunologic potency of human SmartDC-TRP2 from melanoma patients
In order to validate their immunologic potency in a clinically translatable assay, SmartDC-TRP2 and conventional DCs generated from three melanoma patients were compared with regard to their capacity to prime autologous or allogeneic CD3+ T-cell expansion in mixed lymphocyte reaction assays. At the lowest nonsaturating DC:T cell ratio (1:20), SmartDC-TRP2 demonstrated significantly higher T-cell stimulatory effect than conventional DCs for the allogeneic system (Fig. 4a). Analyses of the cytokines secreted in the medium when SmartDC-TRP2 were incubated with T cells for 6 days at a ratio of 1:20 demonstrated detectable levels of the Th1 cytokine IFN-γ; Th2 cytokines IL-5, IL-13, and IL-10; and inflammatory cytokines TNF-α, MCP-1, and IL-8 (Fig. 4b).
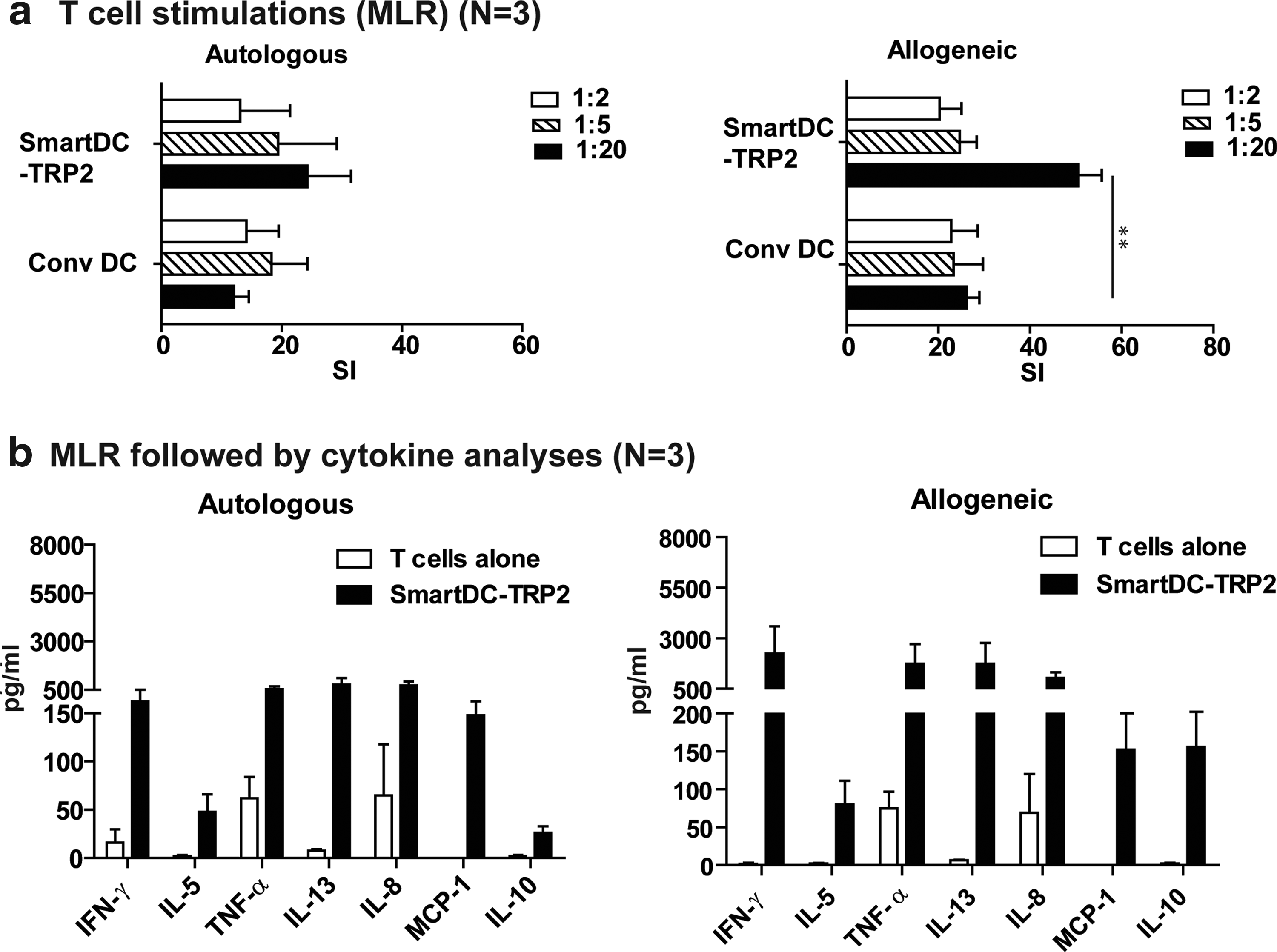
Human SmartDC-TRP2 from melanoma patients were potent stimulators of T cells.
In vitro stimulation of CD8+ T cells with SmartDC-TRP2 from melanoma patients
We evaluated whether CD8+ T cells obtained from a HLA-A*0201–positive melanoma patient during remission stimulated in vitro with SmartDC-TRP2 resulted in the expansion of T cells and correlated with TRP2-specific responses (see experimental scheme in Supplementary Fig. S3a). CD8+ T cells from the patient showed baseline immune reactivity against TRP2360-368 epitope (Supplementary Fig. S3b). SmartDC-TRP2 derived from this patient showed normal immunophenotype (CD14−, CD209+, HLA-DR+, CD86+, and CD80+) (Supplementary Fig. S3c). SmartDCs and SmartDC-TRP2 were produced with the monocytes obtained from the melanoma patient and maintained in culture for 7 days. The day 7 SmartDCs, SmartDCs loaded with a TRP2 overlapping peptide mix (positive control), and SmartDC-TRP2 were either cryopreserved (for use in later rounds of restimulations) or used fresh to stimulate autologous CD8+ T cells. Irradiated CD8− T cells were used as feeder cells. After co-culture for 7 days, CD8+ T cells were harvested for analyses. After three rounds of stimulations, SmartDC-TRP2 stimulated the highest expansion of the T cells (35-fold relative to the number of T cells used for the culture), followed by SmartDC pulsed with TRP2 peptides (22-fold) (Fig. 5a). The expanded T cells (restimulated two or three times) were then co-cultured with target cells K562/HLA-A*0201 not expressing antigen or transduced with LV-hTRP2 to express full length TRP2 (67% TRP2 expression, Fig. 5b) prior to ELISPOT. Untreated T cells or T cells treated with the CEF peptides were used as negative and positive controls, respectively. After two stimulations, a high frequency of TRP2-reactive cells were found in the T cells stimulated with SmartDC-TRP2, resulting in about a 10-fold difference when compared with the SmartDC group, not expressing the antigen (Fig. 5c). This specific reactivity persisted for the T cells stimulated three times (Fig. 5d). Subsequently, the microcultures of CD8+ T cells stimulated with SmartDC-TRP2 that showed high reactivity by ELISPOT were analyzed for the presence of the T-cell receptor reactive against the HLA-A*0201 restricted TRP2360-368 TLDSQVMSL epitope by tetramer analyses (Fig. 5e). The baseline frequency of CD8+ T cells reactive against this epitope prior to stimulation was approximately 0.17% (Supplementary Fig. S3b). The average frequency of TRP2-reactive T cells increased to 2.5% for T cells stimulated three times with SmartDC-TRP2, whereas the control groups (SmartDC alone or SmartDC loaded with peptides) showed lower reactivity (0.5%). Tetramer analyses of individual T cells microcultures showed two highly reactive lines stimulated with SmartDC-TRP2 and identified by ELISPOT showed a high frequency (6%–7%) of T cells reactive to TRP2360-368 (Fig. 5e).
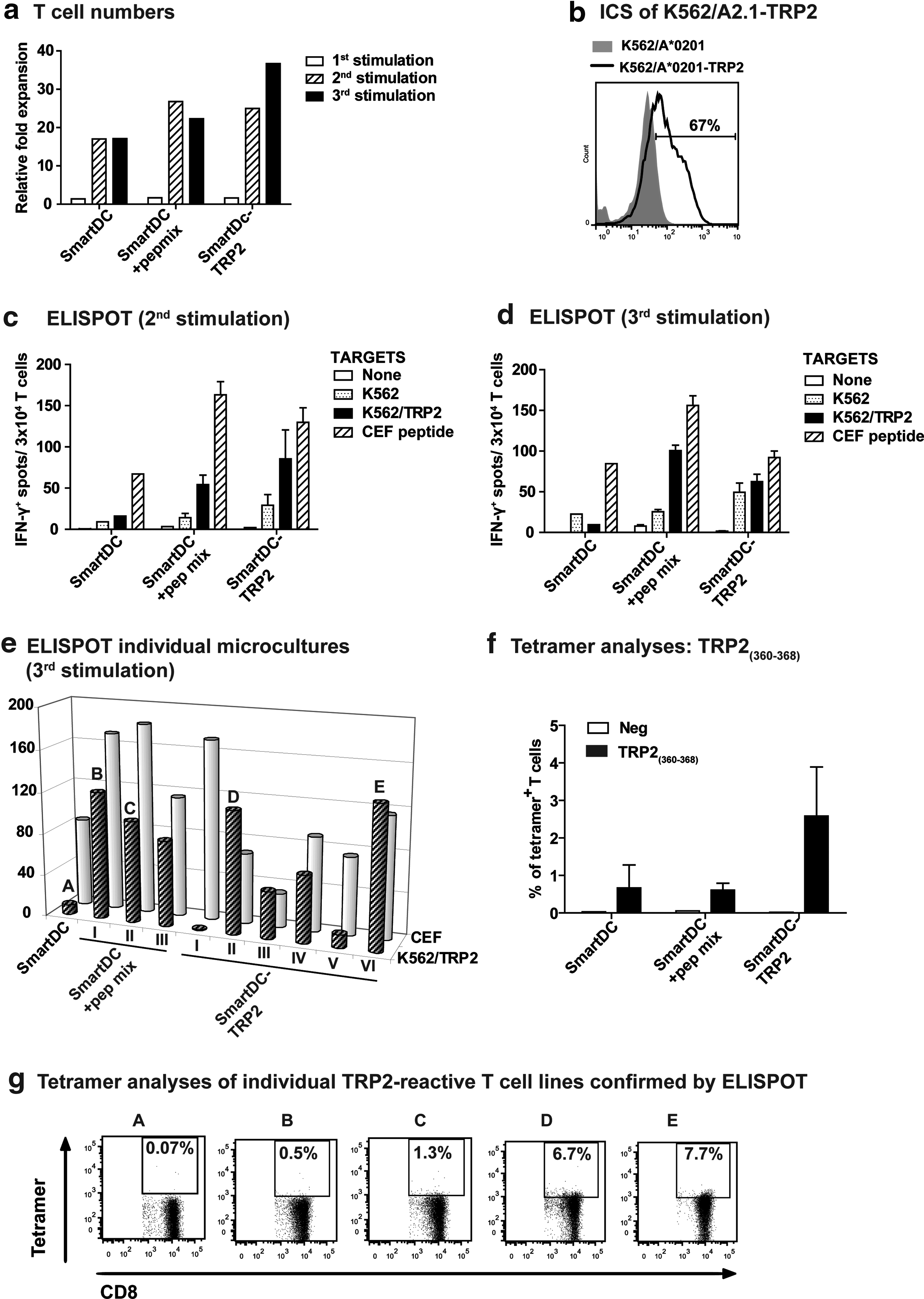
Stimulation of TRP2-specific CD8+ T cells in vitro.
Thawed human SmartDC-TRP2 demonstrate high viability for several weeks, stable DC characteristics in situ, and no malignancies in vivo in NRG mice
Past DC clinical trials identified poor DC viability and biodistribution after injection into patients as a major limitation for successful vaccination, and thus, meaningful approaches for allowing human DC tracking in vivo preclinically and in the clinics are currently in development. Using techniques we had previously described (Salguero et al., 2011), preclinical analyses were performed to confirm engraftment and viability of human cryopreserved SmartDC-TRP2 in vivo using the xenograft NRG mouse model and optical imaging analyses. CD14+ monocytes were co-transduced with hLVG242T/LV-fLUC (SmartDC-TRP2/fLUC) and were maintained fresh in culture for 7 days or immediately cryopreserved for subsequent analyses in vitro and in vivo (Fig. 6a). One day after thawing, cryoSmartDC-TRP2 showed down-regulation of CD14 and expression of the DC markers CD209, HLA-DR, and CD86 (Fig. 6b). On day 7 after thawing, their immunophenotypic identity was similar to fresh SmartDC-TRP2/fLUC (Fig. 6b). CryoSmartDC-TRP2/fLUC were thawed and 1×106 cells were injected subcutaneously in the flanks of NRG mice. Optical imaging analyses showed efficient engraftment and high viability for up to 1 month (Fig. 6c). Quantification of luminescent signal at several time points showed significant loss of viability on day 60, which further decreased by day 90, indicating gradual loss of viability over time (Fig. 6d). Mice to which cryoSmarDC-TRP2/fLUC had been administered were maintained for up to 6 months and showed no bioluminescence signal or pathologies.
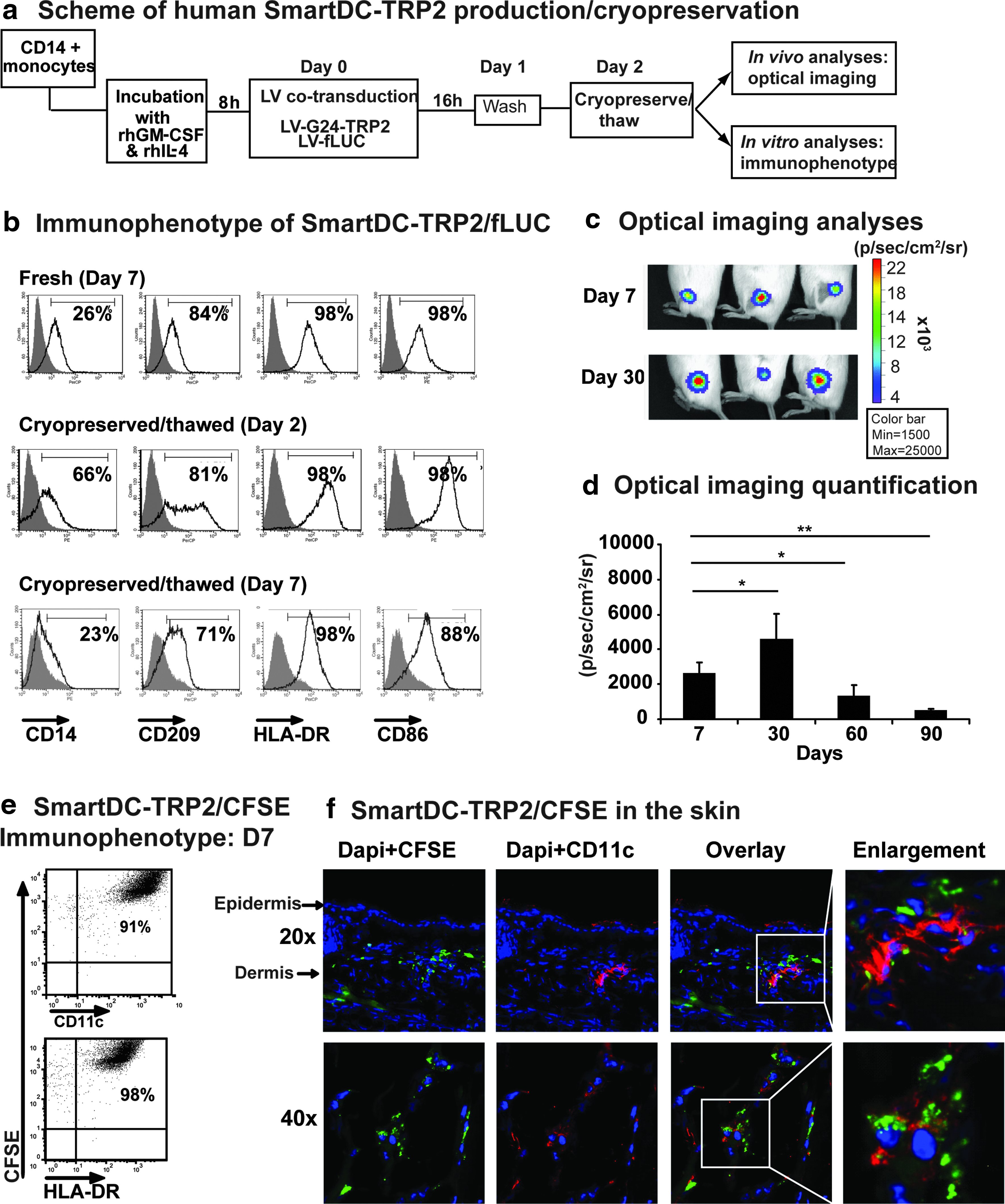
High viability of cryopreserved human SmartDC-TRP2 in vitro and in vivo in Nod.Rag1−/−.IL2rγ−/− (NRG) mice.
In another set of experiments, and as an additional procedure to preclinically characterize the stability of the DCs in situ in the area of the injected skin, we developed a new simple method, based on CFSE labeling of human DCs in vitro. Our method is based on the previous observations that CFSE labels long-lived intracellular molecules with a highly fluorescent dye, carboxyfluorescein, allowing the tracking of lymphocytes for months in vivo (Quah et al., 2007). The persistent intracellular fluorescence signal is correlated with cell integrity and viability. Upon T-cell replication, the fluorescent molecules are divided among the cell progeny, and thus, the fluorescent signal gradually fades away. Since differentiated DCs do not actively replicate, we hypothesized that this would be a convenient method to be combined with immunofluorescence analyses of tissues for direct assessment of the administered cells. Labeling of SmartDC-TRP2 with CFSE in vitro, did not affect their immunophenotypic characteristics (Fig. 6e). SmartDC-TRP2/CFSE that were labeled with CFSE after the washing step were administered subcutaneously in the ears of NRG mice, in order to facilitate skin tissue retrieval (on day 7 after injections). Cryosections of the ears were processed by immune staining against human CD11c (DC marker) and analyzed by immunofluorescent microscopy. Clusters of large CFSE-positive cells were detectable in the dermis, which were also costained for CD11c, confirming the human DC identity (Fig. 6e). Thus, this practical method directly demonstrated the viability and stability of SmartDC-TRP2 in the skin of the animal.
For pilot biosafety testing of human SmartDC-TRP2 cells, nonclinical safety experiments were performed under good laboratory practices conditions using a small cohort of immunodeficient NRG mice (n=3, for each time point). After subcutaneous administration (day 0) with 1×106 cryoSmartDC-TRP2, mice were sacrificed on days 7, 28, 60, and 210 for complete macroscopic pathology examination. No pathologies were detected.
Scaling up production of human SmartDC-TRP2 in a closed bag system
Another major challenge in conventional DC-based vaccination approaches has been scaling up and standardizing production, which has been hampered by variable and low cell product recovery after the lengthy ex vivo manipulation procedures. In order to demonstrate that SmartDC-TRP2 could be produced in a short time at a clinically relevant scale and to allow highly standardized techniques, we explored cell transduction in a closed bag system. Selection of CD14+ cells from leukapheresis samples obtained from two healthy donors was performed by CliniMACS under GMP-compatible conditions. CD14+ monocytes (200×106) were transferred to a cell culture bag and preconditioned with rhGM-CSF/rhIL-4 for 6 hr in serum-free culture medium. This was followed by transduction with hLV-G242T at MOI of 5 for 16 hr (Fig. 7a). After extensive washing steps, 45%–54% of SmartDC-TRP2 was recovered from the initial input of monocytes used for transduction (Table 1). SmartDC-TRP2 were cryopreserved at a density of 2×106 cells/ml per vial, and after thawing, 33%–49% of the cells remained viable. SmartDC-TRP2 cultured for 7 days after thawing in the absence of exogenously added cytokines showed 21%–24% viability (Table 1). The recovery of viable SmartDC-TRP2 was thus consistent for both runs, resulting in theoretically sufficient number of cells per vials for quality control analyses and for clinical administration. We aimed at setting up stringent yet simple analytical criteria to monitor sterility, efficiency of gene transfer, identity, and potency. Sterility tests conducted after thawing SmartDC-TRP2 showed that they were negative for bacterial and fungal contamination (data not shown). LV copy number determination by quantitative real-time PCR performed with cells cultured for 7 days after thawing (in order to avoid pseudo-transduction artifacts) showed an average of two integrations per cell, confirming success of LV-mediated gene transfer in a range that is considered safe regarding insertional mutagenesis effects (Fig. 7b). LV gene transfer efficiency was correlated with detectable levels of secreted GM-CSF (average range 30–50 pg/ml) and IL-4 (average range 50–100 pg/ml), which in general accumulated further in the culture as the SmartDC-TRP2 progressed towards day 7 (Fig. 7c,d). As expected, thawed SmartDC-TRP2 displayed a typical DC morphology on day 7 of culture, which was associated with expression of a basic panel of human DC identity markers (CD209, HLA-DR, and CD86) (Fig. 7e,f). Finally, potency markers were attributed to endogenous inflammatory cytokines highly expressed by SmartDC-TRP2 and detectable on days 1 and 7 in culture supernatants: IL-6, TNF-α, IL-8, and MCP-1 (Fig. 7g).
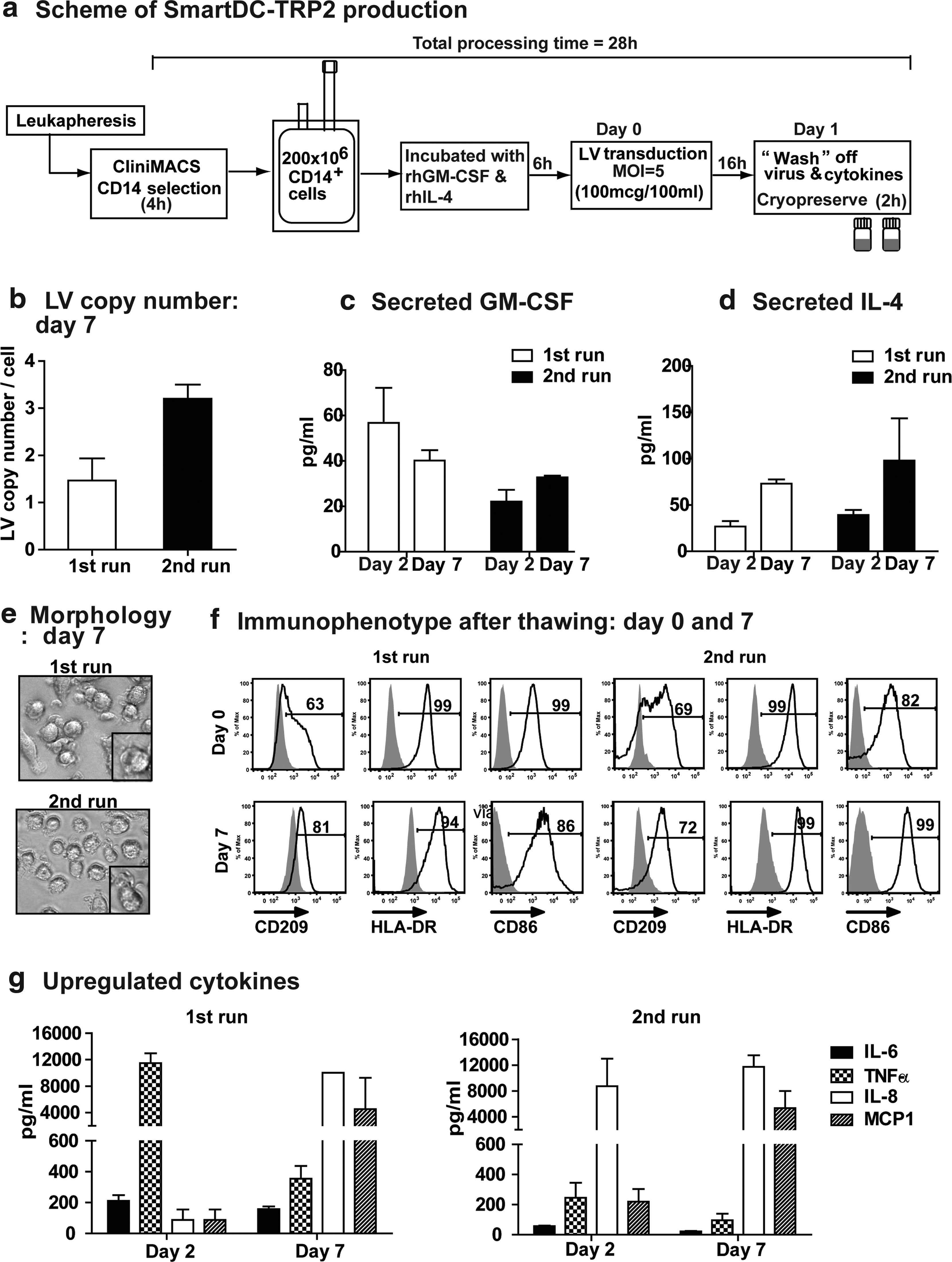
Scaling up SmartDC-TRP2 production in a closed bag system, cryopreservation, and quality control analyses.
Scaling Up Production of SmartDC-TRP2: Results from Two Independent Runs
Discussion
Several preclinical studies have demonstrated the use of LVs for genetic manipulation of mouse DCs leading to expression of tumor antigens (Breckpot et al., 2004; Dullaers et al., 2004; Lizee et al., 2004; Lopes et al., 2006). With this present work, we report a novel generation of multicistronic LVs co-expressing cytokines and antigens for an overnight genetic reprogramming of human DC vaccines for clinical translation. The lentiviral vector LV-G242T expressing GM-CSF, IL-4, and TRP2 was derived from a multiply attenuated third generation four-plasmid split genome packaging system (previously designed by Prof. Luigi Naldini and colleagues [Zufferey et al., 1998]), which is currently the most employed HIV-derived lentiviral packaging system in research and clinical development (Cornetta et al., 2011). The main features attributed to this designation are the following:
(1) Tat and the four accessory genes of HIV were deleted from the viral packaging system, consisting of four plasmids used to transfect the 293T packaging cell line (Zufferey et al., 1998). (2) A 400-nucleotide deletion was included in the 3′ LTR of the backbone vector, which is copied to the 5′ LTR upon reverse transcription, thereby abolishing the 5′ LTR promoter activity and reducing risk of vector mobilization with the wild-type virus (Dull et al., 1998); (3) Because the original lentivirus envelope protein (gp120) restricts the host range, is unstable, and makes production of LVs more complex, vectors are usually pseudotyped (i.e., encoded with a heterologous envelope protein) with vesicular stomatitis virus glycoprotein.
In our previous studies, we had demonstrated that mouse and human DC precursors transduced overnight with LVs mediating gene delivery of GM-CSF and IL-4 self-differentiated into highly viable SmartDCs in vivo. SmartDCs co-transduced with an additional LV expressing the murine melanoma antigen TRP2 led to protection and therapeutic benefits against B16 melanoma tumors in mice (Koya et al., 2007; Pincha et al., 2011). In this current study, we devised a simplified concept with a single LV mediating codelivery of GM-SCF, IL-4, and full-length TRP2 to generate SmartDC-TRP2, which proved to be an effective vaccine in mouse and human experimental systems. Both hLV-G242T and mLV-G242T vector designs enabled consistently high titer virus (followed by p24 concentration as routinely done in the field [Cornetta et al., 2011]), and transductions of DC precursors at LV concentrations of 2.5–3 μg p24/ml (MOI 5–6) led to efficient gene delivery and stable co-expression of transgenes and ultimately induced autonomous differentiation of highly viable mouse and human SmartDC-TRP2. We were able to demonstrate in both mice and humans that SmartDC-TRP2 stimulated cytotoxic T lymphocytes that react specifically against the TRP2 melanoma-associated antigen.
The explored TRP2 antigen (also known as dopachrome tautomerase) is a key enzyme in melanogenesis, and its function is correlated with lowering the oxidative stress in melanocytes. Melanoma cells show higher levels of TRP2 expression than normal melanocytes, which seems to promote their higher resistance to chemotherapy and radiation (Pak et al., 2004). Immunity against TRP2 has been reported in melanoma patients as CD8+ T-cell responses (Khong and Rosenberg, 2002), CD4+ T-cell responses (Paschen et al., 2005), and antibody responses (Boasberg et al., 2006). A recent study using alphavirus to express several melanoma antigens showed that TRP2 was the most effective in a therapeutic viral vaccine against B16 mouse melanoma and successfully induced both effector and humoral immune responses (Avogadri et al., 2010). In our current studies with samples obtained from melanoma patients, we could demonstrate TRP2-specific CD8+ T-cell effector responses (by IFN-γ ELISPOT) and also show that SmartDC-TRP2 stimulated the secretion of cytokines typically produced by T helper cells. This indicates that central tolerance to the auto-antigen TRP2 is not complete. Indeed, autoimmunity in form of vitiligo and alopecia may be a correlated side effect, predictive of antimelanoma responses (Boasberg et al., 2006). The findings obtained herein demonstrated that persistent co-expression of the full-length TRP2 protein in mouse or human SmartDCs did not affect their differentiation, viability, or immune potency.
The ability to freeze and thaw SmartDCs without altering their functional properties is a basic requirement for future immunotherapy clinical trials because, as recommended by EMA, quality control evaluation of gene therapy medicinal products (GTMPs) has to be performed prior to administration into humans. Also for practical purposes, several clinical studies routinely employ cryopreserved DCs to facilitate the logistics of production and allow more flexible scheduling of multiple vaccination cycles. After thawing, cryopreserved mouse SmartDC-TRP2 acquired typical DC immunophenotype in vitro, and although cryopreservation resulted in lower levels of secreted GM-CSF and IL-4, subcutaneous administration of SmartDCs after thawing resulted in TRP2-specific immune response and successful protection against B16 melanoma growth in vivo.
Human SmartDC-TRP2 generated from monocytes obtained from melanoma patients were also highly viable, expressed typical relevant DC markers, and were functionally superior to conventional DCs in stimulating T-cell proliferation in vitro. SmartDC-TRP2 expressed high levels of endogenous pro-inflammatory cytokines such as TNF-α, IL-8, and MCP-1. Thus, although expression of the GM-CSF/IL-4 transgenes does not lead to terminal DC maturation, endogenous TNF-α can potentially promote further DC activation and is routinely included in DC maturation cocktails (Berger et al., 2002). Of interest, a study with ex vivo cultured mouse DCs incubated with TNF-α demonstrated that, upon subcutaneous administration, they matured in vivo, converting from tolerogenic to immunogenic DCs (Voigtlander et al., 2006). In addition, murine studies have shown that TNF-α–induced maturation sustained expression of Bcl-2 in DCs, protecting them from melanoma-induced apoptosis (Esche et al., 2001). Interestingly, expression of MCP-1 (also known as chemokine [C-C motif] ligand 2, CCL2), which is known to be a potent chemoattractor of memory CD8+ T cells (Wang et al., 2008), was coordinated with the high expression of its receptor CCR2 on SmartDC-TRP2, indicating that a feedback loop or bystander effects might be operational. Altogether, these data suggest SmartDC-TRP2 have several autocrine and paracrine mechanisms to create a cytokine milieu promoting multivalent immune responses.
Implantation of human SmartDC-TRP2 subcutaneously in immunodeficient NRG mice confirmed the high viability of the cryopreserved/thawed cells in vivo, with the highest viability up to 1 month after administration. This robust viability of SmartDCs can potentially allow prolonged antigen presentation, which has been reported by others to enhance TRP2-specific antitumor immunity in mice (Wang and Wang, 2002). Underscoring these results, previous studies in our laboratory performed with human SmartDCs expressing a cytomegalovirus antigen (pp65) and administered subcutaneously into NRG mice prior to administration of antigen-reactive human T cells resulted into a long-lasting (30 days) massive recruitment of the T cells to the vaccination site and subsequently systemic biodistribution and generation of antigen-specific human CD8+ T-cell responses (Salguero et al., 2011). In addition to the optical imaging methods, the simplified CFSE method for labeling human DCs ex vivo and subsequently tracking them in situ after subcutaneous administration by immunofluorescence analyses provides valuable preclinical quality control information for prediction of the viability of cells that will be used clinically. To our knowledge, this type of quality control is usually not performed in DC immunotherapy trials.
Finally, our concluding proof-of-concept studies were designed with consideration of the EMA guidelines for development, clinical testing, and marketing authorization of GTMPs (www.ema.europa.eu/pdfs/human/genetherapy/67163908en.pdf). These guidelines are focused primarily on quality, safety, and efficacy requirements of genetically modified cells developed as medicinal products. The following steps are usually taken in account regarding development of GTMPs: (1) methods for selection or isolation of cells from suitable donors; (2) procedures for preparing cells for gene transfer; (3) properties and characteristics of a suitable vector to transfer the target gene into the cells; (4) processing, formulation, storage, and characterization of the genetically modified cells for batch release criteria.
Manufacture of clinical grade DCs generated after immunomagnetic selection of CD14+ monocytes by CliniMACS and their differentiation in cell culture bags has previously been shown to be feasible for clinical trials (Babatz et al., 2003; Motta et al., 2003; Dietz et al., 2004). Nevertheless, cells genetically modified ex vivo by viral vectors face additional regulatory demands, not only due to the risk of the patients, but also due to environmental risks (Wilson and Cichutek, 2009), and thus, monocyte transduction in a bag system is also a plus in order to minimize risks of viral spread. Our protocol required only 28 hr of ex vivo culture for the generation of SmartDC-TRP2 and generated number of cells in quantity sufficient for clinical development (considering 1×106 to 20×106 cells as the usual range used for DC vaccinations). Except for the vector batches used in our studies, which were research grade up to this point, the remaining procedures (leukapheresis, CliniMACS separation, transduction, washing, and dispensing) were performed under GMP, following standard operation procedures. The production of DC-based vaccines under standardized, reproducible, and clinical good manufacturing procedures (cGMP)-compliant conditions is an essential prerequisite for the production of meaningful clinical data. Therefore, the cell culture bag system combining standard technology of transfusion medicine with biotechnology provides the elements for a more standardized and safer development of the SmartDC production process.
Quality control assays performed under good laboratory practices obtained from two independent runs of cryopreserved SmartDC-TRP2 production, were shown to be consistent for the following parameters obtained on days 1 and/or 7 after thawing: viable cell count (by counting viable cells by trypan blue exclusion); sterility testing (using standard microbiological plates for the detection of bacteria and fungus); transduction efficiency (using HeLa cells harboring a specified number of LV integrations as a positive control); identity assay by analyses of secreted GM-CSF and IL-4 (using bead array analyses); identity assay by surface marker characterization (flow cytometry analyses of CD209, HLA-DR, CD86); and potency assay by analyses of endogenously produced cytokines associated with DC activation/maturation (bead array analyses of IL-6, TNF-α, IL-8, and MCP-1).
In conclusion, the central element in our SmartDC technology is the simplified 1-day genetic reprogramming of monocytes by lentiviral codelivery of a combination of factors, resulting in highly viable antigen-loaded DCs that can be cryopreserved and then, upon subcutaneous administration, reach terminal differentiation in vivo. The future impact of SmartDCs is broad since the cytokines and antigens expressed in the LV can be exchanged in a disease-specific manner (such as co-expression of GM-CSF/IFN-α for immunotherapy of chronic virus infections [Daenthansanmak et al., submitted]). Compared with existing DC therapies, this approach is unique because the DCs that self-differentiate in vivo are longer lived and produce paracrine adjuvant effects for immunization (through secretion of transgenic GM-CSF, IL-4, and endogenously up-regulated TNF-α, IL-8, and MCP-1). In addition, this approach provides sustained intracellular expression of the tumor antigen TRP2 allowing the processing of the protein into multiple peptide epitopes, necessary for multivalent cellular and humoral immune responses. Altogether, with this work we are advancing the concept of DCs programmed ex vivo with LVs from the bench to the bedside, using standardized methods that could be easily transferred to several laboratories and medical centers worldwide.
Footnotes
Acknowledgments
We would like to thank Prof. Thomas Woelfel for providing us with the plasmid pcDNA3 encoding human TRP2, K562/A2.1 cell line, and valuable technical assistance; Dr. Axel Schambach for providing us with a lentiviral vector backbone containing the mutated oPRE element; and Dr. Annette Paschen for technical protocols and discussions regarding TRP2. We thank Dr. Sandra Kuhs for completing the construction and sequencing of the hLVG242T vector, Prof. Schwinzer for technical assistance in setting up the mixed lymphocyte reaction assays, Prof. Dolores Schendel's lab for the optimized cryopreservation protocol, and Prof. Baumgaertner for discussing the pathological analyses. We thank the melanoma patients and healthy volunteers who kindly provided us with leukapheresis samples and Dr. Florian Schenk and Dr. Lillia Goudeva for all the coordinations. We are very grateful to the participants of the “SmartDC consortium” (Prof. Farzin Farzaneh, Prof. Dolores Schendel, Dr. Klaus Kuhlke, Dr. Hermann Bohnenkamp, Dr. Marc Barthold, and Dr. Ruediger Hacker) for their useful suggestions for development of this study. We thank the representatives of the Paul Ehrlich Institute for our first Scientific Advice Meeting. This work was supported by funds from the Deutsche Krebshilfe, the Excellence Cluster Rebirth (DFG), and the SFB738 (to R.S.).
Author Disclosure Statement
The authors have no conflict of interest to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.