
Abstract
A frequent topic of biomedical research is the potential clinical use of non-coding (nc) RNAs as quantitative biomarkers for a broad spectrum of health and disease. However, ncRNA analyses have not been pressed into widespread diagnostic use. Strong preclinical evidence suggests obstacles in the translation and reproducibility of this type of biomarker which may result from preanalytical and analytical variation in the non-standardized processes used to collect, process, and store samples, as well as the substantive differences between small and long ncRNA. We performed a narrative review of selected literature, through the lens of key laboratory-developed test (LDT) regulations under the Clinical Laboratory Improvement Amendments (CLIA) in the United States, to study critical gaps in ncRNA validation studies. This review describes the leading candidate ncRNA subclasses, their biogenesis and cellular function, and identifies specific pre-analytical variables with disproportionate impact on testing performance. We summarize these findings with strategic recommendations to clinicians and biomedical scientists involved in the design, conduct, and translation of ncRNA biomarker development.
Introduction
Identification of noncoding RNAS
The success of genomics research in identifying root causes of single-gene disorders is a defining moment in modern medicine and may be attributed to focused study of causal gene variants in protein coding areas. To delve deeper into a better understanding of the human genome, research is pivoting to include noncoding regions, including sequence variation, and the regulatory elements that control their impact. Genes representing regulatory RNAs outnumber genes coding for proteins, and identification of diverse RNA-RNA interacting networks have recognized subclasses of noncoding (nc) RNA. These include small nuclear (snRNA), small nucleolar (snoRNA), micro (miRNA), short interfering (siRNA), long noncoding (lncRNA), and circular RNA (circRNA) (Guil and Esteller, 2015).
Other, more nuanced forms of small ncRNA also include RNAs exclusively located in cajal bodies (scaRNA), with piwi-associated Argonaute proteins (piwi) RNA, transcription initiation (tiRNA), or splice site RNAs (spliRNA) (Morris and Mattick, 2014). This diverse array of noncoding RNAs display substantial differences in biogenesis, stability, and structural characteristics. Detectable differences in their levels are highly touted as new classes of biomarkers.
Appreciation for this added complexity of the human transcriptome, and the recognition of its potential impact in understanding health and disease is apparent in the immense biomarker research productivity in this field in the recent past. As of the time of this writing, ∼99.2% of 36,200 publications listed in PubMed identified with the search terms “ncRNA,” “biomarker,” and “human” occurred with or after the launch of The Encyclopedia of DNA Elements (ENCODE) project in 2003. Funded by the National Human Genome Research Institute (NHGRI), the ENCODE project is an international effort incorporating diverse assays and methods to identify functional elements in the human genome beyond genes coding for proteins.
Research activity and the discovery of specific ncRNA biomarkers demonstrates promising potential in a number of different diseases, such as prostate cancer (Hessels et al., 2003) and pulmonary arterial hypertension (PAH; Schlosser et al., 2016) to name a few, as well as aging-related disorders (Fehlmann et al., 2020). However, diagnostic tests incorporating a quantitative ncRNA measurement rarely receive authorization for use as a medical device by the U.S. Food and Drug Administration (FDA) indicating either a concerning difficulty with regulatory approval or a lack of successful validation studies (Goossens et al., 2015).
Recognition of new types of ncRNAs can be traced to the evolution of molecular analytical techniques. In particular, the shift from low-resolution physical separation and radioactive isotope labeling by biochemists and physicists to the high-resolution sequencing methods used today (Cobb, 2015), as well as the bioinformatic approaches that accompany them has sped the molecular revolution. Whole-genome sequencing (WGS) testing has identified a large number of ncRNAs implicated in human diseases. However, a lack of corresponding functional annotations explaining biological relevance, either in general (Uszczynska-Ratajczak et al., 2018) or in cancer-specific cases (Nakagawa and Fujita, 2018), limits its usefulness in the current clinical setting as tools for assessing quantitative differences. Furthermore, the lack of focus on the key issues that confine the development of ncRNA-based laboratory tests will continue to detract from the possibilities of more personalized care (Glinsky, 2013).
Purpose
In this narrative review, we examined the current state of molecular diagnostics in relationship to establishing analytical validity for quantification of the leading candidate subclasses of promising ncRNA biomarkers. We focused on current regulatory considerations for laboratory-developed tests (LDTs), as this is the most likely path ncRNA biomarkers will follow in the near future. New biomarkers require the establishment of both clinical validity and analytical validity for clinical use (Hayes, 2015). Steps required to establish clinical validity are outside the scope of this review (Pletcher and Pignone, 2011). Establishment of analytical validity requires assessment of specific performance characteristics defined by a regulatory norm, such as those described in the United States 42 Code of Federal Regulations (CFR) 493.1253(b)(2).
Under this framework, we focused herein on knowledge gaps in preanalytical and analytical system variation specific to quantitative biomarker testing for ncRNAs. Consideration of these key areas is critical to wider and faster test adoption and should be of interest in preclinical discovery work to improve biomarker clinical validation studies. It is important to note most clinical laboratory errors occur as a result of preanalytical variation, and this is a recognized issue in biomarker discovery in clinical studies (Kellogg et al., 2015).
United States Regulatory Authority for Laboratory Biomarker Testing
Manufacturers seeking regulatory approval for marketing and distributing a new medical test in the United States must register the test as an in vitro medical device with the FDA, who evaluates the test's safety and effectiveness for its intended use before approval. Depending on the risk classification, this process can take years at great expense. Most biomarker tests are instead developed by independent laboratories for their own use as LDTs as laboratory services, which currently fall under regulation by the Centers for Medicare and Medicaid Services (CMS) in accordance with Clinical Laboratory Improvement Amendments of 1988 (CLIA) (Allen, 2013).
CLIA sets the basis for laboratory proficiency under 42 CFR 493 and includes personnel qualifications, laboratory documentation, categorical levels of testing complexity, as well as quality, preanalytical, analytical, and postanalytical systems minimally required for certification. For the purpose of this review, we narrow these considerations to specific components found in 42 CFR 493.1242 (preanalytical), 42 CFR 493.1252-1256, and .1281 (analytical). Issues related to broader CLIA laboratory aspects pertaining to general laboratory proficiency not specific to ncRNAs are not included.
Methods
We assessed literature using published interpretative guidelines for laboratories provided by the Centers for Medicare and Medicaid Services in the State Operations Manual Appendix C (som107ap_c_lab, Rev. 166, 02-03-17) accessed May 17, 2021. We selected standards deemed specific to ncRNA biomarker testing and development under presumed high-complexity requirements, using our interpretation defined in Table 1.
Selected Standards from the U.S. Code of Federal Regulations Used to Evaluate Selected Literature
ncRNAs: Brief Overview
Knowledge of noncoding (nc) nucleic acids as subtypes of ribonucleic acid (RNA) is not new. Putative roles for transfer RNA (tRNA) (Hoagland et al., 1958) and ribosomal RNA (rRNA) (Roberts, 1958) predate the theoretical discovery and naming of protein-coding messenger RNA (mRNA) in 1961 (Cobb, 2015). Although individual functions were not conclusively defined at the time of these discoveries, it established the idea that gene code for RNA products whose roles are not solely limited to producing proteins, a fundamental part of the central dogma of molecular biology. This central dogma theory is built on the foundation that genomic DNA is transcribed to RNA, which then guides protein synthesis and serves as a template for amino acid arrangements. Substantial evidence from decades of research has thus advanced this principal tenet toward understanding the role of genes in health and disease.
A steady progression away from this simplified, historical version of molecular biology is driven to a large extent by the identification of additional subtypes of ncRNAs that regulate both transcription itself and its products through complex upstream and downstream mechanisms. It is helpful when looking at the considerable diversity of ncRNAs to group them based on length. Small molecule ncRNAs, <200 nucleotides in length, include rRNA (smaller subunit forming), tRNA, snRNA, snoRNA, miRNA, siRNA, and piwiRNA. The lncRNAs may be grouped as any ncRNA larger than 200 nucleotides. Consideration of small molecule ncRNAs with particular relevance to clinical research in humans is focused mostly on miRNAs, whereas siRNAs occur naturally in plants and lower animals and may be encountered as a tool useful for in vitro modeling.
Endogenous production in higher mammals and humans is unknown, but may be detected in humans from external sources (i.e., viral or bacterial infections). siRNA has great potential for therapeutically controlling gene expression, but as it must be exogenously introduced to the patient, it also has some significant challenges with delivery and efficacy (Fujita et al., 2015). We therefore focus on three leading candidate ncRNA subclasses for biomarker assays: miRNA, lncRNA, and circRNA.
Micro RNA
MicroRNAs (miRNAs) have an important function in the regulation of gene expression by sequestering mRNA for degradation or repressing its translation. Mature miRNAs are single-stranded, noncoding RNAs that range in size from 19 to 25 nucleotides (nts) in length, but commonly exist at ∼22 nts in their mature form. A total of 1913 unique miRNAs have been identified according to the latest version of miRBase (v22.1 October 2018) (Fig. 1). miRNAs have a different biogenesis from all other ncRNAs. Genomic coding areas for miRNAs are often located within intronic regions, called mirtrons, in close proximity with the exonic regions encoding the same mRNA transcripts that they repress. Mirtons are transcribed into large primary molecules called pri-miRNAs, which are processed in the nucleus into pre-miRNAs (Vishnoi and Rani, 2017). These transcripts are exported into the cytoplasm, where they are cleaved into a miRNA duplex (∼22 nts in length) by endoribonuclease Dicer.
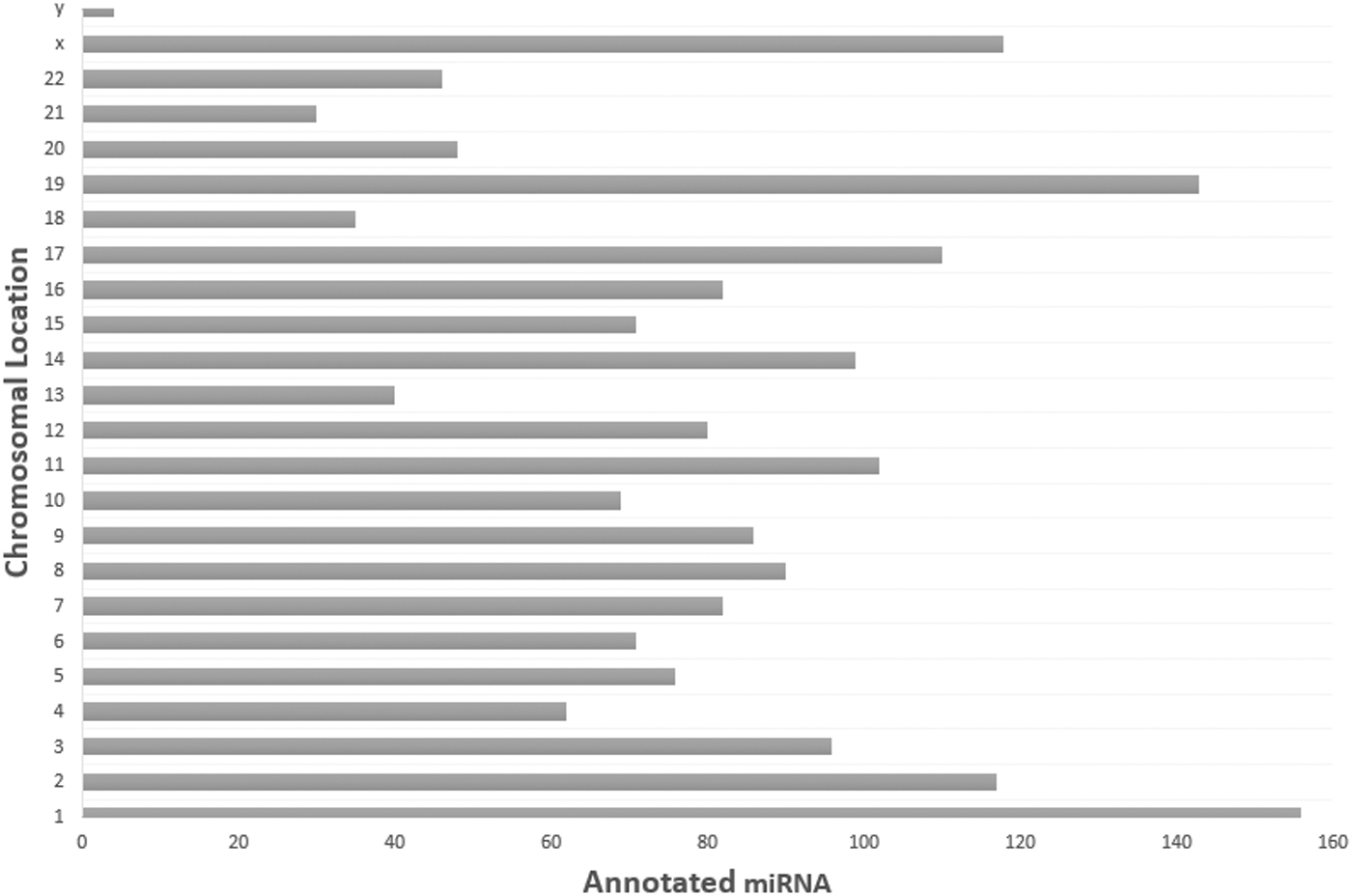
Total annotated miRNA reference sequences by chromosomal location according to miRBase—version 22.1 (October 2018). Accessible at www.mirbase.org/
This duplex then interacts with the ribonucleoprotein complex RNA-Induced Silencing Complex (RISC), which further cleaves it into two complementary single-stranded mature miRNAs through the action of a secondary endonuclease called Argonaute (Bottini et al., 2017). One of the complementary miRNA strands persists in RISC as a guide, using a short seed region of 2-8 nts on the 5′ end of the mature miRNA to recognize a complementary site on the 3′ untranslated region of a mRNA. The other miRNA is believed to be rapidly degraded before it may act in a biological role (Bartel, 2009). General stability of miRNA is widely believed to be greater than messenger RNA; however, individual miRNAs may exhibit distinct differences in degradation due to specific regulatory mechanisms, such as target-directed miRNA degradation (TDMD) (Han et al., 2020), and it is yet unclear how these mechanisms may persist in ex vivo conditions.
Preanalytic concerns for miRNAs
The stability and handling of miRNAs is a pressing preanalytical concern, as it will determine the suitability of potential biospecimens for biomarker testing, either as a singular target or as part of more comprehensive profiling (Witwer, 2015; Nik Mohamed Kamal and Shahidan, 2019; Abramovic et al., 2020; Precazzini et al., 2021). In our own review of 28 studies involving or comparing miRNA quantitation (Table 2), 79% identified preanalytical variation as a potential confounder to their findings. Some simply lack appropriate measures to control for starting volumes or tissue sizes. Most others raise questions that require a deeper delineation and investigation using controlled studies to address wide gaps in knowledge.
Literature Reviewed to Characterize Gaps in MicroRNA Research Under the Framework of 42 CFR Part 493—Selected Laboratory Standards Required for Clinical Utilization of MicroRNA Biomarker Testing
Cellular contamination and hemolysis are two major known sources of variation in plasma and serum samples (Blondal et al.
Moreover, the relationship between miRNA and sample container retention has not been examined, either directly between unbound miRNA and polyethylene containers or indirectly between miRNA-binding molecules and the same containers. These issues can partly be controlled by using specialized vacutainers that simultaneously lyse all cells and vesicles uniformly, while preserving RNA integrity (Stellino et al., 2019).
Nonstandardized interim handling conditions after collection also create variation. Several studies show miRNA is detectable and stable to varying degrees at prolonged storage intervals at common laboratory storage temperatures for fluid-based samples (4°C, −20°C, and −80°C) (Grasedieck et al., 2012) or at ambient for formalin-fixed, paraffin-embedded (FFPE) tissues (Szafranska et al., 2008). These types of studies have focused on extraction of miRNA from primary samples after prolonged storage, whereas in comparison, specific examination of isolated miRNA stability over time demonstrates exaggerated loss, such as in a case using unbound exogenous spike-in miRNAs (Brunet-Vega et al., 2015). Moreover, increased GC content has been shown to positively increase miRNA and mRNA stability in archival FFPE tissues, indicating direct comparisons between FFPE and flash frozen samples may not be feasible (Kakimoto et al., 2016), but the relationship between GC content and stability in non-FFPE samples is not yet known.
This study also demonstrates the need to standardize FFPE fixation methods specifically to include miRNA, as has been demonstrated broadly for proteomic and genomic studies (Bass et al., 2014). Therefore, robust comparative studies examining the stability of isolated and unbound miRNAs are necessary to determine special precautions required for the proper handling of control miRNAs and best practices for long-term storage of isolated miRNA versus its stability in association with primary sample materials. These studies may show that individual miRNAs have distinctive preanalytical considerations and may not be generalizable. The finding that global RNA quality scores designed to identify degraded messenger RNA, such as the RNA Integrity Number, have no measurable association on small RNA sequencing (Lopez et al., 2015), reinforces the need for new quality assessments tailored to detect factors of degradation specific to smaller ncRNAs.
Sources of preanalytical variation also include miRNA isolation and library preparation, areas described as accounting for more than 50% of miRNA intra-assay variation in prostate cancer research (Abramovic et al., 2020). A study comparing five commercially available miRNA isolation kits found high inter-sample variability of exogenous spike-ins, attributable to the isolation itself and not to the qPCR analysis (Brunet-Vega et al., 2015). Interestingly, endogenous miRNAs demonstrated a different trend and were relatively consistent across all five kits. The authors suggest that this is evidence against the use of exogenous miRNA spike-ins to normalize circulating miRNAs; however, this should be carefully considered after ruling out other confounding issues, such as the potential for nonuniform loading of tiny microvolumes of spike-in material, tip retention, and differences between manual and automated processes.
With the exception of a few hybridization assays, direct measurement of miRNAs typically does not occur because of sensitivity challenges and the rarity of miRNAs. Most assays prepare a library of miRNA converted to double-stranded cDNA using two-step reverse transcription and PCR-amplification with various modifications depending on the method (Precazzini et al., 2021). Foremost issues in this step involve the efficiency differences when ligating adaptors onto small RNA and the formation of adaptor dimers, which leads to significant differences even in widely used commercial kits (Baran-Gale et al., 2015). Alternative methods to reduce this bias have been proposed, including circularization of target miRNA before or after reverse transcription, leading to increased sensitivity for rarer miRNA (Barberán-Soler et al., 2018).
Inefficiencies with converting miRNA to cDNA as a surrogate marker are very important to consider, as even single nucleotide error or nontemplate additions can lead to misidentification due to the short length of miRNA. Nucleotide and structural differences, especially GC content, will also affect reverse-transcription and subsequent amplification; therefore, researchers must be cognizant of the potential bias involved when choosing a reverse transcriptase (Minshall and Git, 2020). Second, direct RNA sequencing to study nonsequence-based miRNA modifications is currently not possible if the only available library preparation methods require two-step reverse transcription and amplification.
Analytical steps for miRNA testing will also require careful consideration, starting with the suitability of manufacturer's requirements for the proper handling of reagents designed with research use only intentions. Proper storage and expiration of exogenous miRNA controls and spike-ins are not well defined, likely leaving individual laboratories responsible for documenting proper performance of manufacturer provided materials. In addition, laboratories should be prepared to demonstrate their methodology meets expected performance characteristics of controls despite any modifications that may be planned in preanalytical steps, such as different tissue preparation (Paulsen et al., 2021), or inherently present due to different environmental conditions.
Those aspects relating to establishment and verification of performance specifications in different patient populations may also be prohibitively challenging, particularly those associated with establishment of reference internals for specific testing groups. Specific gender differences in circulating miRNA profiles are observed agnostic of hormonal or fasting status (Max et al., 2018), and given the specificity of miRNA expression to circulating cells and development, it is highly unlikely that pediatric reference ranges can be inferred from adult information. A meaningful consideration for patient status at the time of sample collection, especially in oncology, is the finding that radiation exposure leads to widespread upregulation in the human miRNAome (Templin et al., 2011). This finding indicates that miRNA biomarker testing requires pretreatment sample collection strategies to avoid treatment bias.
Finally, those laboratories developing their own miRNA biomarker tests should be aware that performance differences exist between analytical instruments (Godoy et al., 2019). These considerations are important when comparing test results between two or more sites, or if more than one instrument is used for testing. This may raise issues when comparing control results from proficiency testing between different laboratories as part of accreditation requirements from the College of American Pathologists (CAP) and other compliance-related activity required for clinical testing.
In summary, miRNA biomarker testing must utilize a methodological approach that accounts for sample amount, collection method (venipuncture, heel/finger/stick, and arterial vs. venous central line), sample quality (hemolysis, etc.), fractionation details (force, time, and temperature), isolation methods, storage conditions, library preparation, testing platform considerations, and carefully describe patient conditions that may bias testing. There is a strong indication that miRNA preanalytical conditions must be standardized to a greater extent than conventional sample collections, such as those involved in clinical chemistry panels, as once isolated, miRNA appears to be just as labile as messenger RNA, and differences likely exist between individual miRNAs based on their sequence and association with RNA-binding proteins.
Long Noncoding RNA
LncRNA is the easiest ncRNA to classify from all other RNAs due to its length. It is perhaps the hardest to fully characterize biologically and define by its origins, due to its diversity and alternative isoforms (Ponting et al., 2009). LncRNA is commonly defined as any RNA transcript >200 nts that does not code for a protein. These distinctions serve as more of a categorical cutoff from all the smaller ncRNAs and typically larger mRNAs. Yet, controversy surrounds classification of some lncRNAs that contain sequence motifs (i.e., open but untranslated reading frames, etc.) consistent with protein coding mRNA (Quinn and Chang, 2016).
Shared structural features with mRNA complicates bioinformatic analysis (Wen et al., 2019), particularly because lncRNA may sometimes share characteristic mRNA features, such as a 5′ cap and a 3′ polyadenylated tail. Nearly 16,000 lncRNA genes are identified by the ENCODE project (GENCODE v26) (Marchese et al., 2017). Like mRNA, lncRNA are also transcriptionally regulated at many levels; however, lncRNA have functions defined by a modular structure that confers an ability to interact with DNA, RNA, and proteins (Yoon et al., 2014).
Analytic challenges with lncRNA
There are challenges in lncRNA analysis, however, due to the low expression levels of lncRNA transcripts (Zampetaki et al., 2018). This can create issues for quantification using next-generational sequencing technologies. These complications are exemplified by a recent effort to validate 84 of the most promising circulating lncRNAs as potential biomarkers for PAH. This study failed to distinguish differential changes and was unable to detect many of these lncRNAs despite robust quality measures (Schlosser et al., 2016). Until these technical issues are addressed, lncRNA testing is currently limited to research-only association studies or otherwise in the examination of changes in expression levels of candidate genes (Wu and Du, 2017).
A lone exception stands out; in 2012, the FDA approved the PROGENSA PCA3 Assay as an ancillary measure in “at-risk” men with previous negative prostate biopsies to gauge likelihood of a positive biopsy, if a repeat biopsy may be required. The test quantifies prostate-specific antigen (PSA) mRNA and the lncRNA prostate cancer gene 3 (PCA3), a biomarker long-studied for its role in prostate cancer cell survival (Lemos et al., 2019). The FDA approval includes testing requirements stipulating that urine must be added to Urine Transport Medium (UTM) to protect RNA after collection and the inclusion of PSA to normalize the PCA3 signal and verify RNA is prostate-specific.
In our narrative review of 11 articles published in 2012 or later with considerations for lncRNA biomarker stability (Table 3), we only identified one that incorporated an RNA stabilizing step immediately after collection, indicating that many researchers are not aware of this key component of the process.
Literature Reviewed to Characterize Gaps in Long Noncoding RNA Research Under the Framework of 42 CFR Part 493—Selected Laboratory Standards Required for Clinical Utilization of Long Noncoding RNA Biomarker Testing
Factors affecting stability of lncRNAs are diverse. Some shared lncRNA characteristics, such as subcellular location, splicing arrangements, and RNA-binding protein or miRNA associations; as well as unique isoform traits, such as genomic location, GC percentage, and secondary structure; correlate with stability, whereas others, such as the numbers of exons, do not (Clark et al., 2012; Shi et al., 2021). The observation in humans that lncRNAs with less exons are more stable than those with more is opposite to findings in mice (Shi et al., 2021), suggesting different regulatory pathways for lncRNA degradation that exist between species and some discoveries are not directly translatable.
In addition, ncRNA with housekeeping functions have longer half-lives compared to those with regulatory functions, and short-lived ncRNA are particularly affected by external stimuli, such as retinoic acid treatments (Tani et al., 2012). A subset of lncRNA localized to the nucleus was also found with induced expression after exposure to genotoxic agents, such as doxorubicin (Mizutani et al., 2012). These findings have particular significance in establishing patient characteristics and demographics, wherein different treatment strategies occurring before testing, especially in oncology, may influence stratification of patients and reported differences in lncRNA expression between groups. Finally, recent identification of sex-specific differences in gene expression profiles in healthy tissues and their regulatory networks (Lopes-Ramos et al., 2020) warrants careful consideration when comparisons are made between groups with disproportional sex representation.
Analysis of lncRNA includes additional considerations not generally applicable to mRNA, specifically less efficient splicing and a larger degree of interindividual expression heterogeneity (Kornienko et al., 2016). If this holds true, there will be an explicit need for more diverse public data sets created using analytical platforms capable of sequencing all possible lncRNA isoforms from larger cohort studies than would typically be required for mRNA analysis. In addition, isoforms from select genomic regions, particularly intronic spans in the same orientation as protein-coding RNA, may be difficult to classify and may display less stability than their antisense isoforms, in part, because of less 5′ capping (Ayupe et al., 2015).
Other lncRNA with mRNA-like structural characteristics, such as a 3′ poly(A) tail, are reported to be stable up to a year of long-term storage at −80°C when stored in RNA-stabilizing solutions like those found in PAXgene vacutainers as soon as possible after collection (Wylezinski et al., 2020).
Circular RNA
The idea that only highly nuanced (too small or transiently expressed) or overly rare ncRNA subclasses remain to be identified and classified was recently challenged by the recognition of circular RNA as ncRNA with prevalent functions in mammals (Rybak-Wolf et al., 2015). A circRNA forms when the 3′ and 5′ ends of a linear pre-mRNA transcript are covalently bonded in a process known as backsplicing. This occurs with considerable frequency in humans (Salzman et al., 2012). circRNA is considered a lncRNA subtype, but have significant differences from linear lncRNA counterparts in terms of function and stability (Quinn and Chang, 2016). Although reports of circular RNAs appeared decades ago, they were expressed at very low levels, and like other lncRNAs thought to be transcriptional noise with little functional consequences (Chen and Huang, 2018).
Improved bioinformatics approaches and molecular applications, such as RNA-seq, increased the recognition of circRNA through identifying expressed transcripts from thousands of genes in humans. These proved to be quite a bit more stable than their linear lncRNA isoforms, allowing for greater extracellular potential as a biomarker of cellular states (Barrett and Salzman, 2016). Normal biological relevance of circRNA came from a study of pediatric acute lymphoblastic leukemia (ALL), in which samples from both healthy and ALL study participants contained substantial levels of circRNA, demonstrating that circRNAs do not arise as a result of genomic rearrangements seen in cancer (Salzman et al., 2012).
A critical step in the identification of circRNAs includes the sequencing of the splicing junction, which must be accurate; therefore, circRNAs share many traits with both small and long RNAs wherein the preanalytical handling of samples must be carefully considered to avoid degradation. Although circRNAs are widely viewed as highly stable due, in part, to their structure and a lack of 5′ and 3′ terminal modifications, which provides protection from endogenous RNA degrading enzymes, they may be susceptible to certain clearance mechanisms such as loading into extracellular vesicles, which may concentrate circRNAs in extracellular spaces or fluids (Lasda and Parker, 2016). Therefore, collection of tissues and fractionation of cells from fluids such as blood, urine, or cerebrospinal fluid may disproportionally affect analysis if they exclude these types of vesicles.
Analytical Issues in circRNA
A unique challenge in circRNA analysis comes from the required sample preparation before creating a library. Eluted RNA must be treated with RNase R and/or Ribo-Zero after isolation to enrich the samples by opening up circRNAs (while degrading linear ncRNAs and mRNA) and removing ribosomal RNA, thus limiting the characterization of relationships with other elements of the same transcriptome (Vo et al., 2019). It is unclear how these harsh treatments will impact the study of any bound miRNA or other molecules in association with circRNA. In addition, treatment with RNase R does not fully eliminate all linear RNAs (Panda et al., 2017), nor is it known if certain circRNAs are sensitive themselves to RNase R degradation.
Any analysis requiring reverse transcription also may introduce bias, as template switching or random hexamer-based priming based methods may produce artifacts falsely attributing possible backsplicing regions (Jeck and Sharpless, 2014), particularly in degraded samples with poor quality or in samples enriched with degraded linear RNA after RNase R treatments. For this reason, identified circRNAs must be further validated for circularity using an appropriate technique like Northern blotting, as a bioinformatic-based approach is not sufficient, however, very few biomarker studies are performing these types of confirmatory studies (Pfafenrot and Preußer, 2019).
Conclusion
Comprehensive examination of ncRNA subtype characteristics reveals substantial differences in their biogenesis, stability, and spatiotemporal presence. These inherent properties indicate that a one-size-fits-all preanalytical approach is not appropriate, and common practices developed for mRNA sample handling will likely be insufficient for ncRNA. Preanalytical variables, such as the RNA extraction method, sample type (e.g., blood, saliva), and processing deviations (e.g., incorrect preservation, fixation/preservation delays), are always significant issues when working with RNA but become even more critical when working with ncRNAs. If not accounted for, they will severely impair the interpretability of ncRNA use as biomarkers. Size and structural differences between each type of ncRNA further complicate analytical phases such that different diagnostic platforms incur bias and require a careful understanding of the pros and cons to limit incomplete or erroneous conclusions.
Furthermore, tailored molecular applications to study mature ncRNAs have lagged behind next-generation sequencing technologies, which heavily focus on identifying protein-coding genes possibly due to historical stigmas considering noncoding regions of the genome to be “junk.” It is a particularly pervasive finding that many ncRNA share a biogenesis rooted in transcription from intronic regions or other regions of the human genome not typically part of targeted gene and whole-exome panels used in routine diagnostic testing for diseases with suspected genetic etiologies.
Hence, genetic sequence or structural variation in these areas will be missed in most genetic testing algorithms, and their significance in disease is likely underestimated. This perhaps provides a theoretical gap in knowledge partly responsible for the difficulties in functionally characterizing structural and copy number variants with an associated phenotype despite in silico predictions that fail to account for complex and redundant biological pathways simply because genetic testing is not comprehensive enough.
It is important to consider neither clinical testing nor research testing should exclude preanalytical variation, yet, research testing is often done under a lesser degree of scrutiny and regulation and is therefore not sufficient to ensure reproducibility. Peer review does not possess the same level of rigor as the clinical validation process established by CLIA because gold standards for all phases of ncRNA do not yet exist. A lack of reproducibility in RNA research has been demonstrated in the past by others, such as the Extracellular RNA Communication Consortium finding that disparate methods contributed to varying degrees of reproducibility across biofluids and biotypes (Das et al., 2019). The sentinel inaccuracies linked to preanalytic variation, diagnostic thresholds, and interpretation in estrogen and progesterone receptor testing in breast cancer resulted in recommendations, which now serve as a template for improving diagnostic algorithms (Hammond et al., 2010).
The preclinical knowledge for ncRNA is not sufficiently informed, however, to follow such a template yet. Instead, the Biospecimen Reporting for Improved Study Quality (BRISQ) guidelines may be the most useful guide (Moore et al., 2011), with a few updated recommendations specific to ncRNA.
These concluding recommendations are provided to encourage improved reporting of ncRNA testing conditions currently under emphasized. In particular, patient exposures and RNA modifications (i.e., reverse transcriptase type used for conversion to cDNA, amplification efficiencies, adapter ligation bias, and so on) are highlighted as poorly understood sources of confounding variation in preanalytic and analytic areas, respectively. It is also apparent that there is a need for new workflows that incorporate splitting of samples at collection into identical aliquots of high-quality RNA from each test subject, so each type of ncRNA to be determined may be routed to a specialized workflow permitting simultaneous characterization of specific ncRNAs and coding RNA. The inherent heterogenic variability of ncRNA in individuals suggests that a comprehensive transcriptome analysis requires parallel consideration of all ncRNA subtypes to appreciate their potential influence on the functional transcriptome because of the contextual relationships that exist through interrelated biological pathways.
Footnotes
Authors' Contributions
W.S.S. and J.M.D. conceived the project. W.S.S. wrote the article, with writing contributions and editorial review provided by S.M.S. and J.M.D. All authors approved the final submission.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.