
Abstract
Synthetic biology has a natural synergy with deep learning. It can be used to generate large data sets to train models, for example by using DNA synthesis, and deep learning models can be used to inform design, such as by generating novel parts or suggesting optimal experiments to conduct. Recently, research at the interface of engineering biology and deep learning has highlighted this potential through successes including the design of novel biological parts, protein structure prediction, automated analysis of microscopy data, optimal experimental design, and biomolecular implementations of artificial neural networks. In this review, we present an overview of synthetic biology-relevant classes of data and deep learning architectures. We also highlight emerging studies in synthetic biology that capitalize on deep learning to enable novel understanding and design, and discuss challenges and future opportunities in this space.
Introduction
Synthetic biologists are beginning to take advantage of deep learning methods, powered by advances in synthesis and sequencing and the promise of automation. As a field, synthetic biology applies engineering principles to the design and construction of biological components and systems for use in a wide variety of industrial, agricultural, pharmaceutical, and environmental applications. 1 For synthetic biology domains where it is currently possible to generate large high-quality data sets, emerging results demonstrate the potential of coupling deep learning with engineering biology. Deep learning is a class of machine learning methods that commonly uses models with multiple layers of artificial neurons to “learn” the relationship between input and output data. Examples include models that predict the activity of parts such as promoters based on sequence data or highly accurate protein structure prediction algorithms.2–5
The goal is to develop a model that, when trained on many representative examples, can generalize to make predictions about output given an input it has never seen before. A key feature of deep learning models is that by passing information sequentially between layers in an artificial neural network (ANNs), these models can progressively extract information from the input data. 6 For instance, when analyzing a microscopy image, early layers in the network may extract low-level features such as horizontal or vertical edges, whereas later layers synthesize this information to recognize patterns or shapes of cells within the image.7,8 In addition, deep learning networks can encode complex nonlinear relationships between input values. For example, a deep learning model that predicts a protein's function from its sequence, can learn that certain combinations of amino acids act synergistically, increasing activity over the levels expected given the contributions of individual amino acids. 9
To drive future progress at the interface of synthetic biology and deep learning, there are several challenges that need to be overcome. From a training standpoint, synthetic biologists are not traditionally taught deep learning methods and it can be difficult to keep pace with two rapidly advancing fields in parallel. In addition, synthetic biology data sets have field-specific constraints. In some domains there is an abundance of data, as in the case of natural sequence information, but these data sets are limited in their diversity, as nonfunctional sequences or those that result in very high expression are typically underrepresented. In contrast, other applications are severely limited in the amount of data available due to practical constraints in implementation and testing of synthetic biology constructs.
Despite these challenges, recent results hint at the excellent potential of applying deep learning to synthetic biology, and the goal of this review is to cover topics in deep learning through the lens of engineering biology applications. Although we focus in this review explicitly on deep learning, we note that only a subset of synthetic biology data currently satisfies the volume requirements necessary for training these models. Furthermore, not all synthetic biology problems demand hierarchical or nonlinear models. For an excellent review that covers machine learning more generally and its application to various aspects of biology, we direct readers to the recent article by Greener et al. 10 For reviews of machine learning applied to metabolic engineering, we refer the interested reader to Kim et al, Presnell and Alper, and Lawson et al.11–13 Finally, for the use of machine learning in microbiome studies, we recommend the recent review by Marcos-Zambrano et al. 14
This review aims to support synthetic biologists in understanding and utilizing deep learning approaches in their research, both by providing an overview of methods and also summarizing the state of the art at the intersection of engineering biology and deep learning. We begin by describing classes of data that are relevant to synthetic biology and explain how they can be represented mathematically so they can serve as inputs for a deep learning model. Next, we review common deep learning network architectures that are relevant for engineering biology applications. We then present recent advances that use deep learning to enable synthetic biology, highlighting examples from parts design, structure-based learning, imaging, and other domains. We also review recent works that engineer biomolecular implementations of deep learning networks. Finally, we discuss synthetic biology-specific challenges and potential approaches for mitigating these issues.
Classes of Data Relevant to Synthetic Biology and Their Representation
A properly trained deep learning network can take an input and use it to accurately predict an output. Input data are typically represented as matrices or vectors of numbers. These mathematical representations are essential for converting biological problems into ones that are amenable to model training. Identifying the optimal data representation for a particular problem is critical to the development of high-performing and generalizable models, as the representation codifies which information is fed into the model, and constrains the set of learning algorithms which can be applied.
Practitioners must make careful choices about data representations to ensure that the independent variables pertinent to the problem are represented, whereas limiting the number of irrelevant or confounding variables, which a model would have to learn to ignore. Furthermore, smart selection of data representations can allow the practitioner to leverage the structure of these representations to reduce the problem space and increase data efficiency. Here, we describe common types of synthetic biology-relevant data and how they can be represented numerically.
Sequence data
Thanks to the rapid expansion of sequencing capabilities, 15 one area where we have vast quantities of data is in sequence space. This can include DNA, RNA, or amino acid sequences. These data are typically represented as matrices using embeddings, or functions that map sequence elements to vectors. The most basic embedding is one-hot encoding, so-called because in each embedding vector only a single element is “hot,” taking on a value of one, whereas all the rest are zero. For example, a sequence given by a string of nucleic acids (e.g., ATTGGTCA) is converted into a matrix where the rows represent the possible values, such as A, T, G, or C, and the columns represent the position within the sequence (Fig. 1A).
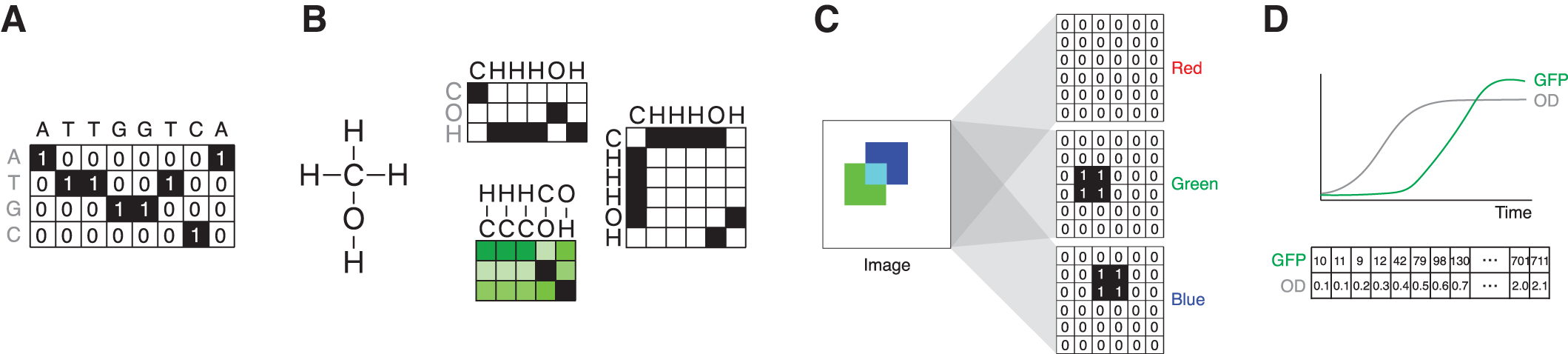
Thus, a 50-mer can be represented by a 4 × 50 matrix. Equivalently, protein sequence data can be represented using one-hot encoding for each amino acid, such that a 300 amino acid sequence is represented as a 20 × 300 matrix. One-hot encoding is straightforward, but in certain cases it can limit the representational power of the model by disregarding the idea that certain amino acids might behave similarly at a given point in the sequence, for example, in terms of their hydrophobicity. Embeddings of amino acids learned from large unlabeled protein data sets have been shown to outperform one-hot encodings in certain protein engineering tasks.16,17
Sequence representations can be used on their own, or coupled with additional biophysical features. For example, in a promoter calculator model, La Fleur et al 18 used a combination of one-hot encodings of −35 and −10 sites in addition to features corresponding to the energetic contributions from distinct parts of the sequence as input to the model.
Molecular structure data
The structure of molecules, at both the small and macromolecular scales, can be described geometrically in several ways, either in a string-based representation such as SMILES 19 or derivatives, 20 or a learned embedding thereof. 21 Alternatively, a molecule can be represented through its structural formula, and this formula can be encoded as a graph (Fig. 1B) upon which graph-based learning methods can be directly applied. Nodes in the graph are the atoms in the molecule, with a set of node features defining the atomic identity and properties, such as atomic mass and charge. 22 Edges can be defined as the bonds between atoms in the molecule, optionally with edge features and edge weights.
For example, edge features might include bond type, 22 and edges might be weighted by bond length. 23 This allows the structural formula of a molecule to be fully defined as a set of node features, a set of edge features, and an adjacency matrix, which encodes which nodes are connected to each other. This approach has the advantage of explicitly including important concepts such as atomic locality into the learning framework and imposing the molecular geometry onto the algorithm throughout, and has led to significant recent progress in the drug discovery field, summarized nicely by Wieder et al. 24
Alternatively, molecules can be treated as objects in three-dimensional space by giving their constituent atoms explicit coordinates alongside their existing node features. These coordinates and node features can be used in machine learning workflows. Note that these concepts can be abstracted to a higher-level view of molecular structure, for example, by defining nodes as nucleotides in DNA and RNA structures, and amino acids in proteins.
Image data
Synthetic biology experiments can also generate image data, such as microscopy files. In this case, the pixels are represented in the rows and columns in the matrix, where the numerical entries in this matrix correspond to grayscale values within the image (Fig. 1C). If the image contains multiple color channels, the dimensions expand to include these values. For example, a color image that is 400 × 600 pixels is represented by a 400 × 600 × 3 object that has data associated with the red, green, and blue color channels.
Time-series, -omics, and other data
Time-series data, such as readouts from a plate reader, can also be fed into deep learning networks. In this case, data points are represented as a vector of numbers, with each entry in the vector corresponding to the value at a specific time point (Fig. 1D). It is also possible to expand this to include multiple types of data. For example, plate reader data measuring green fluorescent protein expression and optical density for 100 time points can be represented as a 2 × 100 matrix.
More generally, synthetic biology applications can generate a variety of “-omics” data sets, such as genomic, transcriptomic, proteomic, or epigenetic data, which can be represented using categorical encodings, or with numerical values such as those associated with biochemical properties, of which the one-hot encoding described earlier is one example.
Network Architectures and Common Building Blocks for Deep Learning Models
ANNs are a type of machine learning algorithm inspired by the way biological neural networks work. In ANNs, an artificial neuron is a mathematical function that is used to simulate the behavior of a biological neuron. ANN models are used to recognize patterns, classify data, and perform other specific tasks. Deep learning is a subset of machine learning that uses networks with multiple layers of artificial neurons to learn complex patterns in data. There are a myriad of deep learning architectures, and in this review we introduce some of the more common representations that have been used in synthetic biology applications to-date. These include traditional deep learning networks such as multilayer perceptrons, convolutional neural networks (CNNs) that are widely used in computer vision and have also proven useful for sequence analysis, and networks that take into account the order or importance of information such as recurrent neural networks (RNNs) and transformers.
For additional information about these topics, Goodfellow et al 6 offer a comprehensive treatment and LeCun et al 25 provide a concise review. We also discuss networks that are well suited for exploiting the geometry of biochemical structures, such as graph neural network approaches. We note that this is not a comprehensive list of modeling approaches–for example, techniques such as reinforcement learning 26 and generative models 27 also have great potential for synthetic biology. In addition to the network architecture, there are key considerations about model training, validation, and testing that are critical to performance. We briefly introduce these ideas in the Supplementary Information S1 and provide citations to more in-depth content.
Multilayer perceptrons
A traditional ANN architecture uses a set of “neurons,” where each neuron takes in a series of numerical inputs. These inputs are multiplied by parameters called weights, and a constant known as the bias is added. This number is then passed through a nonlinear function to become the output of the neuron (Fig. 2A). Historically, researchers used a sigmoid for the nonlinear function, but most modern implementations of deep learning networks use a rectified linear unit (ReLU) for the neurons within the hidden layers of the network for reasons of computational efficiency. Typically, there are many neurons, where the same inputs are multiplied by different weights for each neuron. For example, if the inputs correspond to DNA sequence information, the weights adjust how each nucleotide factors into determining the ultimate output, such as transcriptional activity. In cases where the input is a multidimensional array, it can be unwrapped into a vector (e.g., a 4 × 50 matrix unwrapped into a 200-dimensional vector).
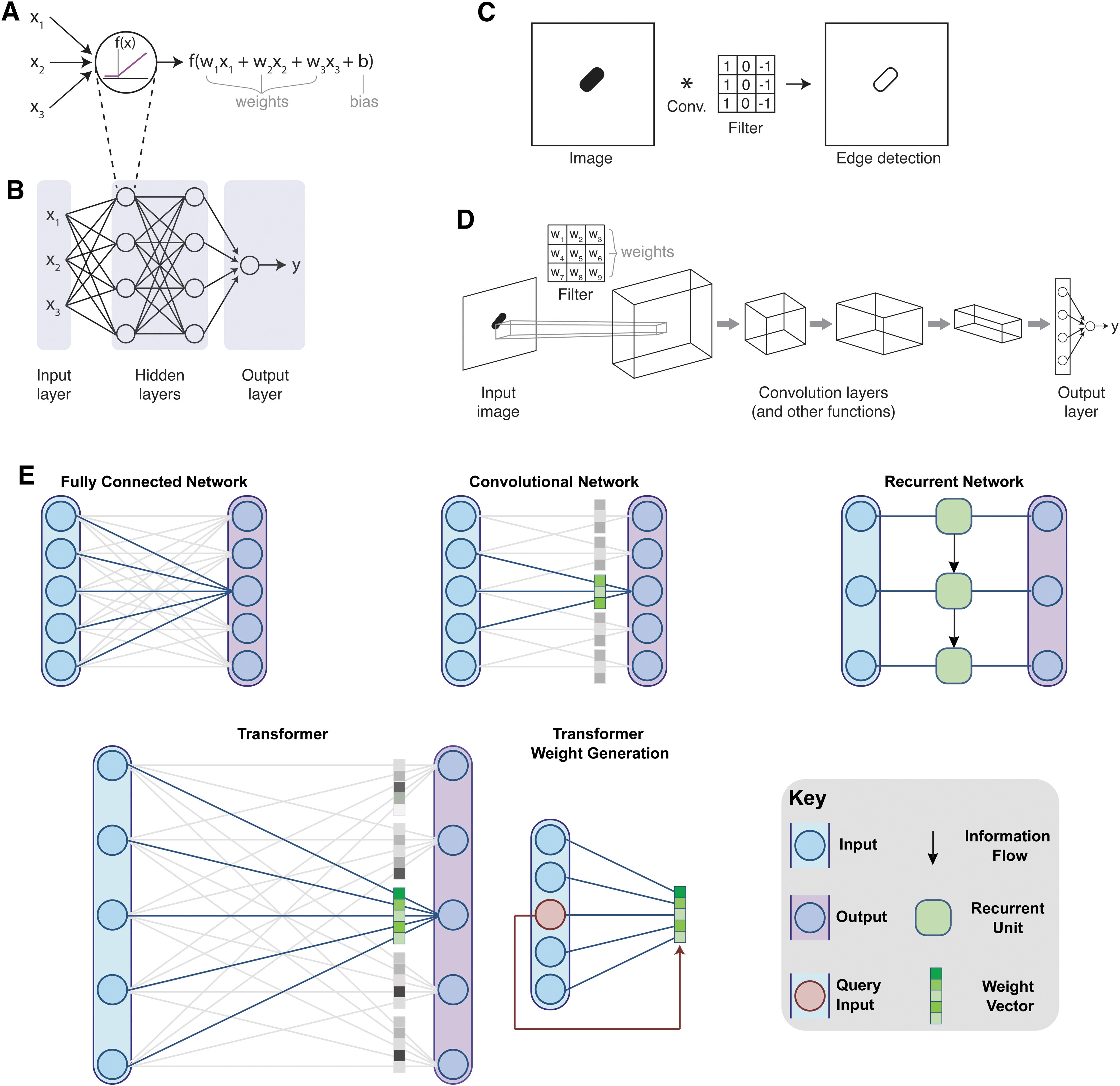
Multilayer perceptrons stack sets of neurons together in fully connected networks such that the output of one layer feeds into the next layer (Fig. 2B). This hierarchical structure allows for identification of low-level features in early layers and more complex features in later layers. The depth contributed by multiple sequential layers is where the term “deep” comes from in the name “deep learning.” In the multilayer perceptron architecture, each output of a neuron is fully connected to all the nodes in the next layer downstream. Internal layers in the network are known as hidden layers, and the final layer is referred to as the output layer. The output layer is special in that it often collapses to a single value or a small number of values, in contrast to the earlier layers which have many outputs. For example, in a network that maps promoter sequence data to a transcriptional activity, the output could be a single number quantifying transcriptional activity.
Convolutional neural networks
CNNs can preserve local positional information about how nearby data are organized in relation to each other. In addition, they use a parameter sharing structure, where the same model weights are applied across the entire input. Because of this, CNNs are especially appropriate for tasks such as image processing, where adjacent pixels contain related information, and where operations such as edge detection should be performed uniformly across the image. For each convolutional layer in the network, the input is convolved with a filter (or filters), and then passed through a nonlinear activation function. Filters can be used to detect specific patterns.
In traditional filter-based analysis tasks the numerical values in the filter are hand-selected to specify properties that a user deems likely to be important, for example, edge detection (Fig. 2C). In contrast, CNNs use filter parameters as the weights of the model, which are learned by the network (Fig. 2D). CNNs typically use a series of convolution steps to perform sequential analysis operations that can abstract features, such as patterns or color gradients. Convolution layers are often interspersed between layers that perform other mathematical operations, such as pooling, which is used to concentrate information by reducing the dimensionality of the data. CNNs can also include elements of other network architectures, such as following convolutional layers by fully connected layers.
Recurrent neural networks
RNNs are a class of models designed for use with sequential data. They operate by iterating over the data sequence, and recursively updating the model's internal state (or memory) based on the content of the internal state and the next value in the input sequence (Fig. 2E). Classically, these networks have been used for language processing, where the order of words is important for context and meaning. Similarly, these networks are appropriate for processing biological time-series data or sequence information. For example, when processing DNA sequences, the relative positioning of start and stop codons is highly significant for determining protein expression. However, the recursive nature of these networks presents several limitations. Most importantly, simplistic RNNs do not learn long-term dependencies between elements that are far apart in sequence space due to the vanishing gradients problem, 28 and their recursive nature prohibits parallelism in implementation, limiting their scalability.
A critical improvement to the performance of RNNs came with the development of long short-term memory (LSTM) networks. 29 LSTM models were designed with the aim of improving the limited temporal memory of RNNs by adding a long-term memory state to the model, where the model must make explicit decisions to remove or add information to the long-term memory. For example, if a model is trying to predict whether a protein will be translated from a given mRNA, the presence of a stop codon would likely be placed into long-term memory and kept there until a downstream start codon is identified. Further details about LSTM models can be found in the review by Van Houdt et al 30 and an example of their application in synthetic biology is included in Angenent-Mari et al. 31 Numerous variants of the LSTM exist 32 ; of notable mention for the interested reader is the gated recurrent unit. 33
Transformers
The transformer is a more recent model developed for sequential data, which eliminates the problems of limited memory encountered with RNN variants, whereas also being more computationally efficient and parallelizable through the elimination of recurrence. The transformer has demonstrated paradigm-shifting performance on sequence-based tasks, outperforming RNNs and LSTMs across the board. 34 Transformers have even demonstrated the ability to outperform CNNs on computer vision problems, 35 although they were not originally designed for such tasks. This transformative performance is achieved by avoiding the concept of model memory, and instead allowing the model to view and generate outputs for every position in the entire sequence of data at once.
For each output, the model selects which parts of the sequence to draw information from. This is achieved through what is known as an “attention” mechanism, where the model can learn what information is important at each point in the sequence, and focus on passing that information forward (Fig. 2E). For example, a model predicting the behavior of a small RNA that might form secondary structures is likely to place a lot of attention on sequences that are complementary to the sequence of interest (e.g., outputs for “CGA” will include a lot of information from another part of the sequence containing “UCG”). The mathematical details of the attention mechanism are beyond the scope of this review, but the reader is encouraged to read Vaswani et al 34 and Chaudhari et al 36 for further details.
Graph neural networks and geometric approaches
Methods for learning on sequence and image data take advantage of the regular Euclidean structure of the data and the natural concept of spatial locality that it imparts. Other structured data, such as structural formula graphs of molecules, secondary structure graphs of DNA and RNA, or atomic coordinate data for proteins, do not have these same structural properties. However, they do have their own symmetries and definitions of locality, which can be used to construct learning frameworks. Graph neural networks were designed specifically to generalize the information flow in Euclidean neural networks to graph structure, defining a scalable and generalizable approach for passing information between nodes through the irregular edge connections between them, which act to encode the structure's locality.
This allows for the learning of high-quality representations of structured data, which can be used to perform node labeling or edge prediction tasks, or pooled together across the structure and fed into a multilayer perceptron to perform classification or regression at the molecule scale. Bronstein et al 37 present a comprehensive and detailed primer for viewing machine learning from a geometric perspective, and Zhou et al 38 provide a breakdown of the specifics of graph neural network development.
Synthetic Biology Applications
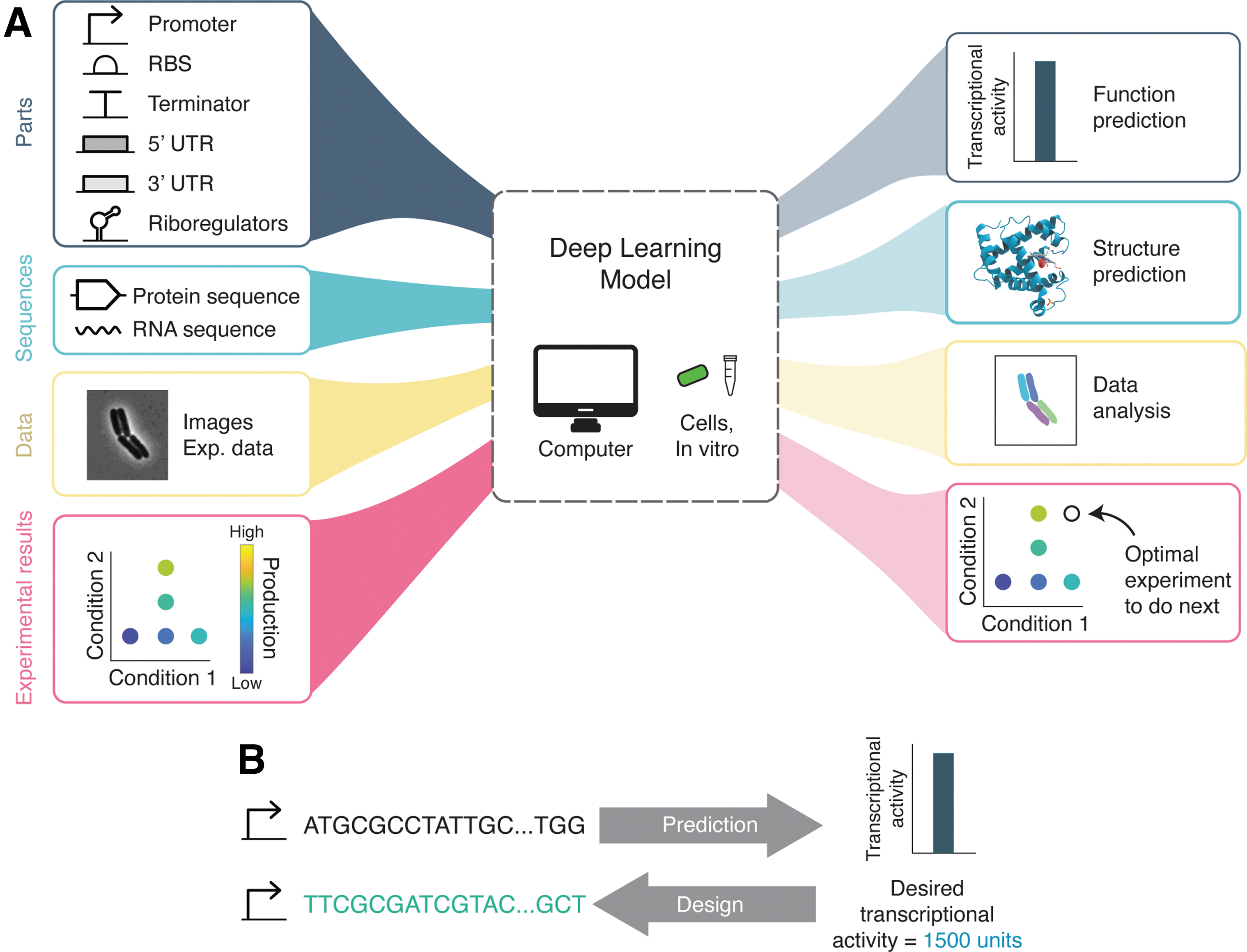
In this section, we turn to examples of deep learning as applied to synthetic biology research (Fig. 3A). We review recent progress on the design of biological parts, structure-based learning, imaging applications, optimal experimental design, and biomolecular implementations of neural networks.
Design and modeling of biological parts
Researchers have made significant recent progress in using deep learning to predict the function of biological “parts,” such as promoters, ribosome binding sites (RBSs), and 5′ and 3′ untranslated regions (UTRs).2,31,39–46 Because these parts are often constrained in length—for example, ∼50 nucleotides for a 5′ UTR sequence or ∼300 for a promoter—it is possible to use DNA synthesis to generate large randomized or semirandomized libraries, where function can be measured with massively parallelized reporter assays coupled with next-generation sequencing. The ability to synthesize large libraries represents an ideal example of how synthetic biology approaches can generate the training sets required for data-hungry models.
For example, Sample et al 41 developed a deep learning model, Optimus 5-Prime, which accurately predicts how the 5′ UTR sequence controls ribosomal loading. Although data sets from endogenous human 5′ UTRs do exist that relate sequence to translation efficiency,47,48 these natural data sets are not ideal for model training because sequences with deleterious effects are likely to be underrepresented in natural examples, and endogenous transcript data are not sufficiently diverse to capture a broad range of expression levels.
To circumvent these issues, Sample et al synthesized and analyzed data from a 280,000-member library consisting of random 50-nucleotide 5′ UTR sequences upstream of the coding sequence for green fluorescent protein. Data from transfected HEK293T cells were used to train the Optimus 5-Prime model, where the inputs to the model were one-hot encoding representations of the 5′ UTR sequence and the output was the mean ribosome load values. The researchers used a CNN and the model had excellent performance, explaining up to 93% of the mean ribosome loading values in the test set.
Similar approaches, which combine DNA synthesis, massively parallel reporter assays, and deep learning, have been used for promoter designs. Synthetic biologists have traditionally used a relatively small number of native promoters in their construct designs. Although artificial promoter libraries exist,49–51 they are often derivatives of existing sequences, such as those achieved through mutagenesis, which limits diversity. Furthermore, there is a dearth of very strong promoters, as they are underrepresented in natural contexts. Using massively parallel reporter assays, Kotopka and Smolke 2 characterized a library of promoter variants. The design preserved conserved sequences within the promoter and randomized the remainder (∼80% of the sequences).
This highlights a potential approach for accessing larger sequence spaces, by using a combination of rational and randomized designs. The researchers used a combination of fluorescence-activated cell sorting and high-throughput DNA sequencing (FACS-seq) to bin cells by expression levels and then sequenced the promoter regions within each bin. These data were used to train a CNN, where the model accepts DNA sequence as input and outputs an activity prediction. Overall, the model predictions generalized well to test data, achieving R2 values >0.79 for all libraries, representing a significant achievement given these complex sequences. This approach of using massively parallel reporter assays is widely generalizable. For example, Jores et al 52 built synthetic promoters for plant species, including Arabidopsis, maize, and sorghum, and trained a CNN to predict promoter strength.
Massively parallel reporter assays are not the only way to generate large data sets, and other approaches may be less prone to biases that can be introduced in processing. For example, Hollerer et al used genetic reporters to create a large data set that directly links sequence to function, applying it to develop a deep learning model that predicts translation activity of an RBS with high accuracy. 42 The researchers built a library of 300,000 bacterial RBSs and placed them upstream of a site-specific recombinase that flips a particular DNA sequence located in a region adjacent to the recombinase.
By sequencing the region containing both the RBS and the recombinase sites, the researchers could assess function by measuring what proportion of constructs had undergone recombination for each RBS sequence. They used this data set to train a ResNet 53 (a CNN variant), ultimately yielding a model that predicted RBS function with high accuracies (R 2 = 0.927). It is worth noting that the general approach used to create a physical DNA-recorded link between gene regulatory element function and DNA sequence is not restricted to RBS optimization, and could be employed for a variety of other tasks including transcriptional or translational biosensor design or the optimization of promoter sequences. Despite the excellent potential of using synthetic sequences to generate diverse libraries, there are some limitations to this approach. Studies using deep learning to generate novel synthetic parts have frequently encountered the issue that using purely randomized sequences results in many parts that do not work. The flip side of natural elements being biased in their representation is that purely random parts are also likely to have low rates of success.
Researchers have circumvented this problem by employing semi-rational methods, such as interspersing regulatory elements known to yield functional promoters with randomized sequences 2 and using model predictions to select libraries that are enriched for elements with intermediate or strong function. 42 In addition, the length of the sequence will ultimately place limits on the diversity of the library. The ability to synthesize and sequence longer regions may lead to decreased coverage and data quality that is biased in the case of longer sequences. In addition, researchers must make trade-offs between the length of sequencing reads, library size, and sequencing depth.
The benefits of focusing on specific sequence regions as “parts” needs to be balanced with the fact that gene regulation is complex. For example, Zrimec et al 54 showed that interactions between coding and noncoding regions are important for determining gene expression levels. Although they demonstrated that DNA sequences can be used to predict mRNA abundance directly with some accuracy (R 2 = 0.6 on average across a broad range of model organisms, including Saccharomyces cerevisiae, Arabidopsis thaliana, Homo sapiens, and others), what was notable from their deep learning results was that it was the interaction between regulatory motifs and not necessarily the motifs themselves that determined mRNA abundance. These results serve as a critical reminder that biological parts do not exist in isolation.
Generative approaches for new synthetic parts
For synthetic biology applications it is often desirable for a model to be not just predictive (e.g., from sequence to predicted performance), but also generative (e.g., from desired performance to sequence, Fig. 3B). Non-deep learning examples have proved very valuable to the engineering biology community. For example, the RBS calculator 55 can generate novel designs based on a thermodynamic model, and synthetic 5′ UTR sequences have been successfully made based on genetic algorithms. 41
Mechanistic modeling approaches are very powerful; however, they require expert knowledge of which features contribute to performance. Generative approaches based on deep learning models are an exciting area for development, as these tools are moving toward the ability to work backward, such as from specifications about translation efficiency to candidate sequence designs. For example, in their study on yeast promoters, Kotopka and Smolke 2 used a CNN model to implement sequence-design strategies, ultimately showing that the best algorithms yielded strong synthetic constitutive and inducible promoters.
However, traditional approaches to design optimization can be prone to practical pitfalls, including computational inefficiency and a propensity to get stuck at local optimization minima. Furthermore, these algorithms have no constraints on sequence diversity, which can be problematic in cases where many distinct library variants need to be generated. Deep generative models have the potential to address these gaps, and include models such as variational autoencoders, autoregressive models, and generative adversarial networks.56,57 In an example of this approach, Linder et al 58 developed a deep exploration network framework. Their approach optimized fitness for the desired function, whereas also explicitly maximizing sequence diversity by using a similarity metric that penalizes sequence similarities that exceed a threshold.
Generative models have also begun to see success in the field of peptide engineering for simple problems dealing with short-chain peptides, including the design of antimicrobial peptides.59,60 We point the interested reader toward the recent review by Wan et al 61 for further details.
Structure-based applications
Rapid progress in the field of geometric deep learning has facilitated an explosion of research into structure-to-function learning in biotechnology. Perhaps the most high-profile case is that of DeepMind's AlphaFold 2 protein structure prediction model,3,62 which boasts protein structure prediction accuracy high enough to be usable as a replacement for costly and time-consuming protein crystallography. The model takes the protein sequence and multiple sequence alignments to similar proteins as inputs, and performs learning on three different data structures: a sequence-level representation, a pairwise nucleotide interaction representation, and the atom-level 3D structure of the protein generated by the model.
The 3D structure is represented by a cloud of unconnected nodes corresponding to the backbone elements of each of the nucleotides, each with their corresponding amino acid side chains. A geometrically equivariant attention mechanism is used to take advantage of the rotational and translational symmetries inherent in the geometry of 3D space. Other applications of interest in the protein space are protein engineering and sequence-function mapping.9,63–67 For example, Gelman et al 9 demonstrated that deep networks, such as convolutional networks, can accurately make predictions about function for new uncharacterized sequence variants when trained on data from deep mutational scanning assays.
The problem of predicting 3D RNA structure is made more difficult by the lack of existing structural data as compared with the protein folding problem. Although >100,000 protein structures have been characterized, high-fidelity structures only exist for a handful of RNA structures. One interesting technique for overcoming this limitation has been deployed by Townshend et al, 68 where they reframe the problem not as one of predicting the RNA structure end-to-end with a deep learning model, but instead using deep learning to score the structural predictions generated by the FARFAR2 algorithm. This facilitated a massive amplification of the available data set, which consists of only 18 RNA structures. It is trivial to generate thousands of candidate structures for each of the RNA molecules in the training data set, and instead learn to predict the similarity between candidate structures and the ground truth. The structural scoring function learned, dubbed the Atomic Rotationally Equivariant Scorer (ARES), achieves significantly improved accuracy compared with existing non-machine learning techniques.
Structural learning on small molecule graphs has expanded rapidly in recent years in the fields of drug discovery69,70 and drug repurposing. 71 For example, Stokes et al 72 employed graph neural networks to predict antibiotic behavior in small molecules in combination with screening assays, identifying a novel drug called halicin as an effective antibiotic in mouse models. Other fields, such as those concerning the simulation of molecular dynamics, have seen similar growth and interested readers are pointed to Noé et al 73 for further details.
Imaging and computer vision applications
Computer vision is an area where deep learning has enabled exceptional progress. 74 In the context of synthetic biology, imaging applications can include automated detection of properties within an image, such as colony formation on a plate or analysis of microscopy data. Two examples of image analysis tasks include classification (e.g., identify if a colony exists or not) and segmentation (e.g., identify the sets of pixels corresponding to each cell in an image). Classification is the simpler of these tasks and classic CNN algorithms from computer vision were developed for this type of task, such as LeNet-5, 75 AlexNet, 7 and ResNets. 53 These classic algorithms traditionally involved deep neural networks with many parameters (e.g., AlexNet uses ∼60 million parameters). More compact versions, such as MobileNetv2 76 (∼3 million parameters) have emerged to reduce this complexity, offering a practical alternative.
Segmentation, or identifying the exact location of an object within an image, is a more complex task but is particularly helpful for quantification. For example, segmentation is useful for detecting the location of cells within a microscopy image so that fluorescence values can be extracted. The field experienced a major breakthrough with the introduction of the U-Net algorithm, 77 a CNN that performs exceptionally well on biomedical data. Examples of notable deep learning algorithms that are relevant for single-cell resolution data include DeepCell, 78 DeLTA,79,80 YeaZ, 81 MiSiC, 82 and CellPose. 83 Image analysis algorithms are also capable of handling more advanced analysis tasks, such as tracking cells from frame-to-frame within time-lapse images and working with 3D image data. For more comprehensive coverage of algorithms for image analysis we point readers to reviews by Jeckel and Drescher 84 and Moen et al. 85
Optimal experimental design
Data labeling for synthetic biology problems is often very expensive compared with other fields, requiring specialist knowledge of the problem and sometimes full laboratory-based data collection pipelines. This cost is a particular problem for deep learning models, which require significant training data. This motivates interest in ensuring that practitioners do not waste time and resources labeling data that do not provide much additional information to a model. The selection of specific data to label, or experiments to perform, is a form of optimal experimental design referred to in the machine learning community as active learning. Taking such an approach with deep learning problems can significantly reduce the cost of data set creation.86,87 We provide an introduction to key ideas in optimal experimental design and active learning in the Supplementary Information S1.
Deep learning methods for optimal experimental design are not yet widely used in engineering biology; however, the potential of laboratory automation and preliminary results based on simulation suggests that this is a fertile area for future research. Treloar et al 88 used deep reinforcement learning to control a simulated chemostat model of a microbial coculture growing in a continuous bioreactor. The authors demonstrated that a satisfactory control policy can be learned in a single 24 h experiment by running five bioreactors in parallel and that deep reinforcement learning can be used to decide on the best sequence of inputs and control actions to apply to a continuous chemostat so as to maximize the product output of a microbial coculture bioprocess.
This constitutes a computational example of deep learning-driven optimal experimental design where reinforcement learning is used to infer near-optimal sequences of inputs to a bioreactor to control a complex system. Future efforts in optimal experimental design can build upon existing approaches from machine learning, such as those that have been deployed for metabolic engineering applications.13,89–93
Biomolecular implementations of deep learning networks
Although deep learning models are typically implemented using computers, several recent studies have demonstrated that ANN mimics can be constructed using biomolecular components. These designs engineer biochemical systems and living cells that can perform computations and “learn” to solve simple benchmark optimization problems. One of the key reasons why this is possible comes from the fact that inducible gene responses to chemical inducers typically resemble a sigmoidal function of the concentration of the inducers, and can thus serve as the nonlinear function in the neuron model.
Based on this, Moorman et al 94 presented the theoretical architecture of a biomolecular neural network, that is, a dynamical chemical reaction network that faithfully implements ANN computations, and demonstrated its use for classification tasks. The authors emphasized the usefulness of molecular sequestration for achieving negative weight values and of the sigmoidal activation function in its elemental unit called a biomolecular perceptron. Following up on this, Samaniego et al 95 theoretically demonstrated that interconnected phosphorylation/dephosphorylation cycles can operate as multilayer biomolecular neural networks.
As an application, they designed signaling networks that theoretically behave as linear and nonlinear classifiers. In a study by Sarkar et al, 96 a single-layer ANN was experimentally implemented in Escherichia coli cells, demonstrating the use of engineered bacteria as ANN-enabled wetware that can perform complex computing functions such as multiplexing, de-multiplexing, encoding, decoding, majority functions, or Feynman and Fredkin gates. In Li et al, 97 ANNs were implemented in consortia of bacteria communicating through quorum-sensing molecules.
These engineered consortia were then used to recognize 3 × 3 binary patterns. Prakash et al 98 developed memregulons, a new class of genetic switches acting as both memory systems and logic gates, and engineered them in E. coli cells to implement a reinforcement learning algorithm that allows engineered bacteria to play the tic-tac-toe game. Learning was achieved by persistently modifying the relative expression of memregulons through the application of external chemicals after each training game was won or lost. Bacteria learn by playing against other players or other bacteria in an unsupervised manner.
In the study by Sarkar et al, 99 simple genetic circuits distributed among various bacterial populations were used to solve chemically generated 2 × 2 maze problems by selectively expressing four different fluorescent proteins, demonstrating the possibility of using engineered bacteria to perform distributed cellular computations and optimizations. In van der Linden et al, 100 the authors genetically implemented a perceptron capable of binary classification. This was achieved through the use of toehold switch riboregulators to construct a synthetic in vitro transcription and translation-based weighted sum operation circuit coupled to a thresholding function. This synthetic genetic circuit was then used for binary classification, that is, the expression of a single output protein only when the desired minimum number of inputs is exceeded.
Using metabolic components, Pandi et al 101 presented an approach for biological computation through metabolic circuits implemented in both whole-cell and cell-free systems. The implementation relies on metabolic transducers used to build metabolic perceptrons, which are analog adders that implement a linear combination of the concentrations of multiple input metabolites with adjustable weights. Based on this, the authors built two four-input metabolic perceptrons that were used for binary classification of metabolite combinations, thereby laying the groundwork for rapid and scalable multiplex sensing through metabolic perceptron networks.
Following up on this, Faure et al 102 recently showed that artificial metabolic networks can be used to implement RNNs that can be trained to predict growth rates or the consensual metabolic behavior of an organism in response to its environment. As the proposed artificial metabolic networks can optimize various objective functions, they could be used to obtain optimal solutions in various industrial applications, such as searching for the optimum media for the bioproduction of compounds of interest or to engineer microorganism-based decision-making devices for the multiplexed detection of metabolic biomarker or environmental pollutants.
Such biological manifestations of ANNs and machine learning paradigms implemented at the biomolecular level open avenues for new research into the engineering of living cells for solving complex computing, decision-making, and optimization problems.
Opportunities for Reducing Experimental Data Set Requirements
Deep learning models can be notoriously data-hungry, which poses a major challenge for domains of synthetic biology where it is challenging to generate large data sets. However, there are several potential paths around this through methods such as transfer learning or with the use of simulated data, data augmentation strategies, or approaches that incorporate constraints based on physical models. We introduce these ideas and discuss their potential role in synthetic biology applications in the Supplementary Information S1.
Conclusions
Research at the intersection of synthetic biology and deep learning holds great promise for the design of novel sequences and constructs, the automation of data analysis, optimal experimental design, and many other applications. Although we have focused this review on deep learning methods, it is worth noting that simpler models can have distinct advantages. Deep learning models can effectively be black boxes due to the number or parameters and complexity of the architectures involved, reducing the interpretability of the model.
Before turning to deep learning models, it is often advisable to try more straightforward machine learning approaches to understand their performance first. For example, Sample et al 41 tested a linear regression model on their 5′ UTR data set, which served as a useful point of comparison with their CNN-based results. It will also be helpful to understand the trade-offs between performance and complexity for specific applications, and studies aimed at exploring this are likely to be valuable. For instance, Nikolados et al 103 compared models of increasing complexity to compare their ability to predict protein expression from DNA sequence. Finally, the amount of data available also places important constraints on whether deep learning approaches are appropriate, as deep models require large training sets.
Overall, deep learning methods have already had a substantial impact on the field of synthetic biology and we anticipate significant advances in this area moving forward. In this review, we have aimed to provide an overview of approaches and applications for deep learning in synthetic biology. We have also highlighted challenges and opportunities that exist for working with biological data sets, with the goal of helping researchers in engineering biology incorporate deep learning methods and insights into their toolset.
Footnotes
Authors' Contributions
All authors contributed substantially to the writing and editing of the article, with overall project management by M.J.D. with help from G.-B.S.
Acknowledgments
We thank Owen O'Connor and Caroline Blassick for their helpful comments.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Science Foundation grant MCB-2143289 to M.J.D. and the Royal Academy of Engineering Chair in Emerging Technologies for Engineering Biology to G.-B.S. (RAE CiET 1819\5).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.