
Abstract
The explosion of interest in CRISPR-Cas systems over recent years has delivered rapid advances in precision genome engineering technologies that hold enormous potential for application across the biotechnology sector. Within the cell therapy space, adoptive immunotherapies continue to lead the translation of increasingly sophisticated genetic engineering strategies toward clinical application. Although early focus was placed on engineering primary immune cells collected from patient blood, the democratization of these therapeutics will require the development of off-the-shelf allogeneic cell therapy products that can be manufactured at scale. Pluripotent stem cells (PSCs) represent an attractive substrate for such products, and many of the challenges that must be overcome to realize the application of PSCs in adoptive cell therapies are common with those holding back regenerative cell-replacement therapies. This article discusses how genome editing can overcome these challenges and highlights the potential of rapidly advancing second-generation technologies in the development of novel cell-based therapeutics.
Introduction
Pluripotent stem cells (PSCs) hold the potential to generate any cell type found in the body, presenting vast opportunities for the development of cell-based therapeutics. Embryonic stem cells (ESCs) derived from surplus early-stage embryos generated for the purpose of in vitro fertilization were the first PSCs to raise hopes for clinical application in cell therapies. 1 The paradigm was shifted in the mid 2000s by the pioneering work of Kazutoshi Takahashi and Shinya Yamanaka who demonstrated that PSCs can be “induced” from adult cells by reactivating embryonic gene expression programs with defined cocktails of transcription factors (Fig. 1).2,3
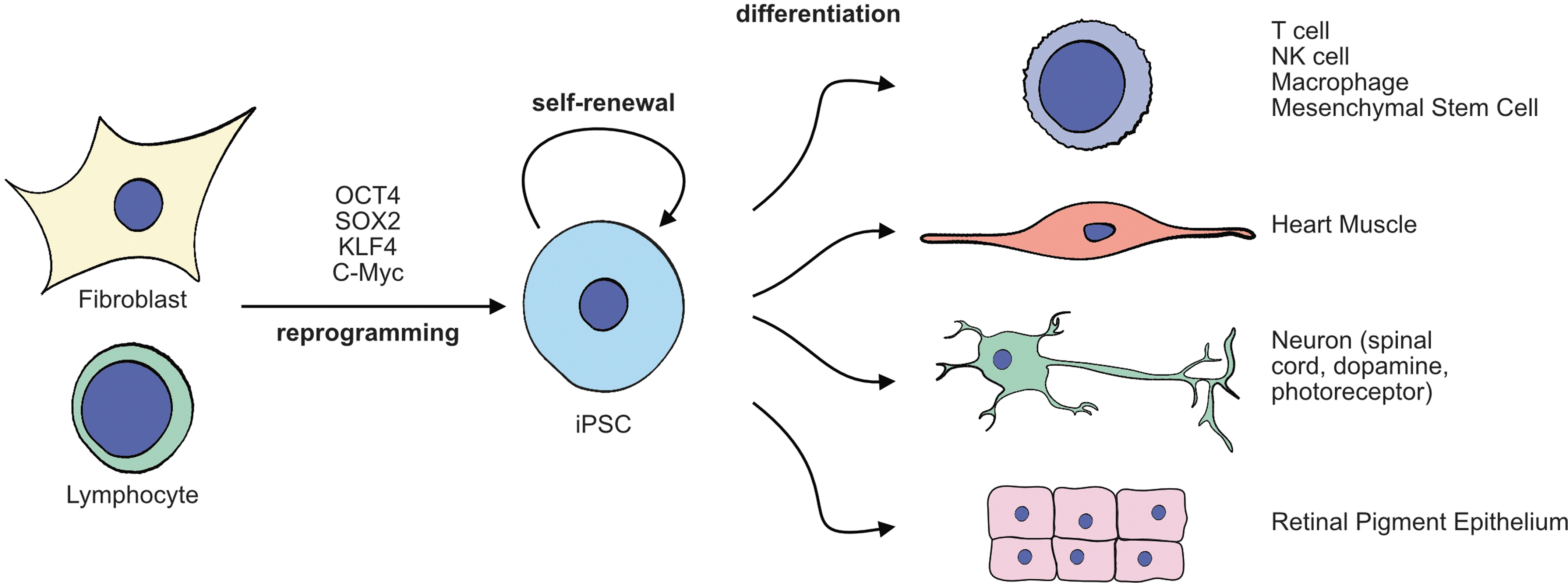
Such induced PSCs (iPSCs) can be derived either from prospective patients or adult donors, overcoming the practical and ethical considerations associated with the clinical translation of ESCs. Like ESCs, iPSCs can be stably propagated in culture, are amenable to transgenesis, gene editing, and clonal expansion, and can be banked for future use, circumventing several technical challenges inherent to the genetic manipulation of differentiated cells in ex vivo culture or intact tissues. Advances in the fields of developmental biology and bioengineering have refined capabilities to control the differentiation of iPSCs into functional immune cells for use in adoptive therapy and into an array of other cells and tissues with the potential to repair and replace diseased and damaged organs.
iPSCs, therefore, present a highly scalable platform for manufacturing advanced cellular therapeutics, and despite lagging behind ESCs, a number of trials are underway in patients to evaluate their utility in a range of clinical applications. 4 Owing to specific regulatory frameworks put in place to facilitate the translation of iPSC technology to the clinic, many first-in-patient trials with iPSCs were conducted in Japan. 5 However promising results from these studies have paved the way for a global shift toward iPSC-based therapeutics. 5
Application of iPSC Technology in Adoptive Cell Therapy
T cells expressing antigen-specific T cell receptors (TCRs) can be derived from patient tumors and have tremendous value in adoptive cell therapy. However, difficulties associated with consistently expanding a high enough yield of clinically effective T cells have hindered progress or wider deployment.6,7 Reprogramming technology has, therefore, been harnessed to derive iPSCs from T cells (T-iPSCs) with beneficial TCR configurations. 8
Knowledge of the developmental processes that lead to T cell differentiation has underpinned the refinement of in vitro differentiation protocols9–11 such that it is now possible to expand T-iPSC indefinitely in culture and then redifferentiate them to functional “rejuvenated” tumor antigen-specific T cells. These have great potential as effective adoptive cell therapies for the treatment of cancer. Similarly, T cells that have encountered HIV antigens can be expanded through this route as a potential therapeutic for HIV infection. 12
Transgenic expression of engineered chimeric antigen receptor (CAR) is an alternative approach to direct immune cells to therapeutically relevant target cells expressing a specific antigen that bypasses the requirement to collect them from the site of disease. CAR T cells are capable of identifying cancerous cells circulating in the blood and targeting them for elimination. 13 Similarly, CAR natural killer (NK) cells 14 and more recently CAR macrophages15,16 are showing promise in the treatment of solid tumors. In addition to their potential for treating cancer, CAR-expressing cells also show promise as immunomodulators for the treatment of autoimmune diseases.17,18
As with patient-derived antigen-specific T cells, the preparation of sufficient quantities of CAR-engineered immune cells is a major challenge to overcome. Moreover, CAR-engineered cells present additional technical challenges associated with ensuring the safe efficient genomic integration of the CAR transgene. Functional CAR-engineered T cells, NK cells, and macrophages have been generated from iPSCs,19–21 potentially providing a “once-and-done” approach to engineering and validating banks of clonal lines of CAR-expressing iPSCs for subsequent expansion and differentiation into therapeutic products.
The scalability and reproducibility of iPSC-based manufacturing processes thus have the potential to overcome the challenge of consistently generating sufficient quantities of safe therapeutically effective CAR cell therapies currently holding back their broad application and have stimulated the initiation of a number of clinical trials (Table 1). US-based Fate Therapeutics is a leading presence in the development of iPSC-based immune cell therapies, with an extensive pipeline of engineered NK and T cell candidates. 22 U.S. industry leading cell engineering company BlueRock Therapeutics 23 and iPSC technology pioneers Healios K.K 24 also list immune cell therapies in their development pipelines.
Clinical studies with induced PSC-derived cellular therapeutics
AML, acute myeloid leukemia; CAR, chimeric antigen receptor; CiRA, Center for iPS Cell Research and Application; IND, investigational new drug; MM, multiple myeloma; NIH, National Institutes of Health; NK, natural killer.
Application of iPSC Technology in Regenerative Therapy
Much of the therapeutic application of iPSC technology has focused on developing regenerative cell therapies to repair or replace diseased or damaged tissues. Similar to earlier studies in the gene therapy field, the eye has also served as a testbed for pioneering work evaluating iPSC-based regenerative therapies. The first-in-human trial of an iPSC-derived cell therapy was run at Kyoto University under the direction of Masayo Takahashi, 25 which demonstrated positive outcomes from grafting a sheet of retinal pigmented epithelial (RPE) cells differentiated from iPSCs into the eye of a patient with macular degeneration. The promising results from this study have led on to the initiation of clinical trials run by the National Institutes of Health (NIH) National Eye Institute,26,27 the first trial to investigate an iPSC-derived cell therapeutic in the United States, and a collaborative trial run by Healios K.K. and Sumitomo Dainippon Pharma 24 to further develop iPSC-based therapies for macular degeneration cells (see Table 1 for a nonexhaustive summary of ongoing trials using iPSCs). Researchers at Kobe University Hospital have also initiated a clinical trial to investigate the outcomes of transplanting iPSC-derived photoreceptors into the retinas of retinitis pigmentosa patients. 28
Clinical trials have also been initiated to evaluate safety and efficacy of transplanting iPSC- and ESC-derived dopaminergic neural progenitors into the brain of Parkinson's disease (PD) patients as part of an international consortium (GForce-PD) 29 that includes BlueRock Therapeutics and the world-leading Kyoto University Centre for iPS Cell Research and Application (CiRA) under the directorship of iPSC inventor, Shinya Yamanaka. Other clinical trials in Japan that have received approval include evaluating the potential of grafting iPSC-derived spinal cord neurons for the repair of spinal cord injury at Keio University30,31 and iPSC-derived heart muscle into the hearts of patients with severe ischemic myopathy at Osaka University. 32
U.S.-based Allele Pharmaceuticals in collaboration with South Korea's SCM Lifescience has announced plans to trial treatments for diabetes using iPSC-derived pancreatic beta cells, 33 while an ongoing trial run by U.S.-based Vertex Pharmaceuticals aims to treat type 1 diabetes using ESC-derived pancreatic islet cells. 34 In these early trials, focus will be placed primarily on determining whether the safety profiles of iPSC-derived cell grafts are comparable with those derived from ESCs. Should this prove to be the case, the stage will be set for a broad scale shift toward iPSCs as the primary source of cell-based regenerative therapies for the treatment of injury and degenerative diseases.
Histocompatibility
Therapeutic application of iPSC-derived cells in both adoptive and regenerative cell therapies is constrained by similar considerations applied to conventional cell and tissue donation. Decades of experience with hematopoietic stem cells and bone marrow transplantation have shown that patient-derived autologous grafts present fewer risks than donor-derived allogeneic grafts35–37 as they avoid alloreactivity that would lead to both graft rejection due to attack by the patient's immune system (host-vs.-graft disease) and the attack of patient tissue by donor immune cells (graft-vs.-host disease [GvHD]). 38 (See Box 1 for definitions of key terms used in this section.)
One iPSC-based approach to solving the challenge of GvHD is under development by Cynata Therapeutics in partnership with FujiFilm Cellular Dynamics in the form of iPSC-derived “Cymerus” mesenchymal stem cell therapy. 39 Cynata's phase 1 Cymerus trial was the first to employ allogeneic iPSCs in a clinical setting and demonstrated positive outcomes for the control of adverse inflammatory response invoked by the transplantation of donor bone marrow. 40 Cynata's Cymerus platform is also in trials to treat arthritis 41 and plans have been announced to investigate its potential in treating other autoimmune inflammatory diseases, such as diabetic ulcers.
Box 1. Key Terms
Histocompatible: a cell or tissue graft expressing alleles of the human leukocyte antigens that are sufficiently similar to those of the recipient to avoid transplant rejection
Histoincompatible: a cell or tissue graft expressing alleles of the human leukocyte antigens that are different to those of the recipient, leading to transplant rejection
Autologous: a cell or tissue graft derived from the same individual
Allogeneic: a cell or tissue graft derived from a donor
Alloreactivity: a T cell response mounted against transplanted cells or tissue expressing “nonself” human leukocyte antigens
Beneficial match: a cell or tissue graft with a mismatch of a single allele of either HLA-A or HLA-B
Hypoimmunogenic: a cell or tissue that evades immune rejection due to loss of HLA protein expression
To minimize the risk of rejection of allogeneic cell grafts by the host immune system, histocompatible donors can be identified that are sufficiently genetically matched with the patient for alleles of the human leukocyte antigen (HLA) genes.42,43 HLA proteins are antigen-presenting receptors present on the cell membrane that interact with the TCR to mediate immunosurveillance by the adaptive immune system to identify and clear virus-infected or cancerous cells. Class 1 HLA proteins encoded at the major histocompatibility 1 (MHC-1) locus (HLA-A/-B/-C/-E/-F/-G) form heterodimeric receptors with beta2-microglobulin (B2M) that present intracellular antigens at the surface of all cells.
Intracellular antigens that are recognized as foreign by the CD8/TCR complex activate CD8+ cytotoxic T cell responses leading to the direct attack of the antigen-presenting cell (Fig. 2A). In addition to recognizing antigens presented by HLA receptors, TCRs also directly engage and recognize HLA receptors themselves, identifying them as either “self” or “nonself” (Fig. 2B). 44
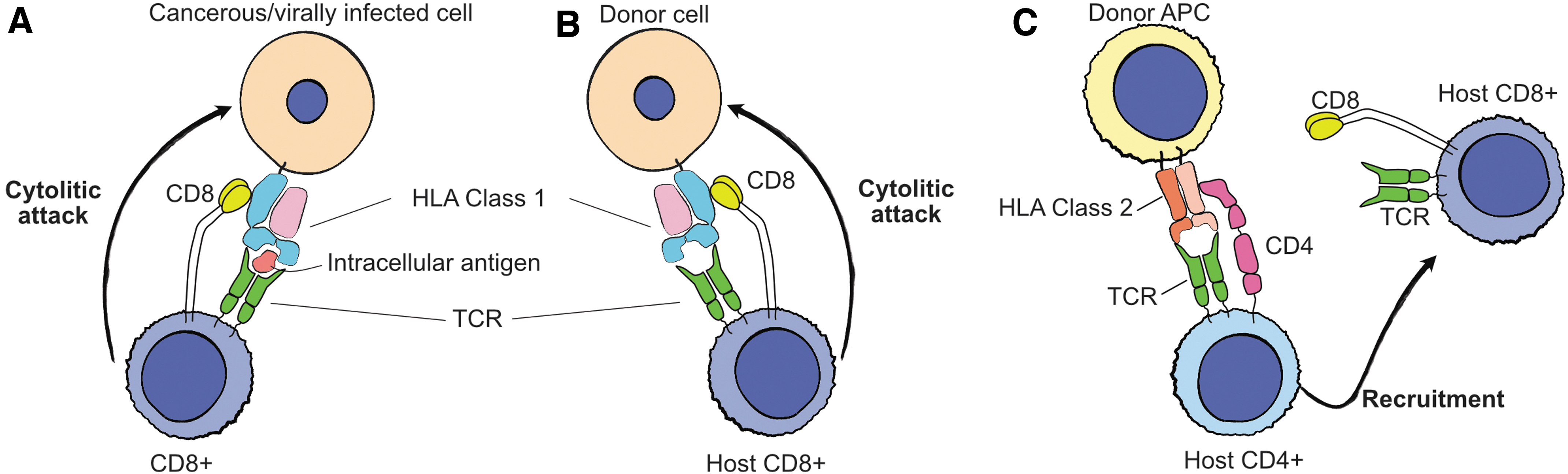
Class 2 HLAs encoded at the MHC-2 locus (HLA-DR/-DQ/-DP) form heterodimers composed of alpha and beta chains that present extracellular antigens and are constitutively expressed by specialized antigen-presenting cells such as macrophages and dendritic cells, and by microglia, endothelial, and epithelial cells in response to inflammatory cytokines.45–47 Foreign extracellular antigens presented by class 2 HLAs activate the CD4/TCR-mediated response of CD4+ helper T cells that recruit cytotoxic T cells and NK cells through secreted chemokines (Fig. 2C).
Recognition of allogeneic grafts as nonself, either through mismatched HLA proteins or through foreign antigen presentation, leads to host T cell alloreactivity and consequent rejection of the graft through inflammation and cytolytic attack. 43 HLA matching is, therefore, essential for successful cell and tissue grafting. However, the genes encoding HLA-A, HLA-B, HLA-C, and HLA-DQ are some of the most highly polymorphic coding loci in the human population, 48 likely due to selective pressure to diversify the antigen-binding potential of HLA receptors. 49 Owing to this enormous diversity of HLA genotypes, HLA matching of allografts is a major challenge to overcome for both conventional cell and tissue donation and for the application of iPSC-derived cellular therapeutics.
Autologous and Allogeneic iPSCs
To avoid graft rejection due to histoincompatibility, a number of early clinical trials opted to reprogram iPSCs from cells from the patients who would subsequently receive the derived autologous cell graft.25,26 Such approaches avoid the requirement for prolonged immunosuppression associated with imperfectly matched allogeneic donor tissue, and may result in better engraftment. Although the central nervous system (CNS) is an immune-privileged organ, postmortem analysis has shown that human dopaminergic neurons express MHC-1 proteins and may, therefore, be susceptible to T cell-mediated rejection. 50 Moreover, grafting experiments in nonhuman primates indicate that autologous iPSC-derived dopaminergic neuron grafts are better tolerated than allogeneic grafts,51–53 indicating that rejection is also an obstacle in the CNS. Similarly, immune rejection of allogeneic iPSC-derived RPE cells has been observed in preclinical animal models.54,55 However, despite the advantages of autologous donor material, in the case of heritable disease, patient-derived iPSCs will continue to harbor the underlying disease-causing genetic lesion and may, therefore, be unsuitable for therapy. Also, the cost of such a personalized approach to manufacturing iPSC-derived cell therapies may outweigh the reduced medical costs associated with continued immunosuppression and may not fit within the time constraints of disease progression.
Owing to the time and financial costs associated with patient-derived iPSCs, a lot of attention has turned to the prospect of manufacturing and banking allogeneic iPSCs derived from healthy donors as an off-the-shelf solution. As many of the known HLA variants are extremely rare, modeling of the allelic distribution in the U.K. population indicates that a collection of ∼150 randomly selected donor iPSC lines could provide a full match for ∼20% of recipients at HLA-A, HLA-B, and HLA-DR, which have been found to be most predictive of the success of HLA matching. Such a collection could also provide a beneficial match for 38%, with diminishing returns as the size of the collection is expanded. With access to sufficiently large number of prospective donors (>100,000), it has been calculated that as few as 10 selected ideal homozygous donors could provide a complete match for 38% of recipients and a beneficial match for 67%. 56 A similar exercise in the Japanese population suggested that due to lower ethnic diversity, a bank of 170 lines from unselected donors could provide beneficial or complete matches for 80% of patients. 57
Despite the impressive coverage of patients likely to benefit from feasibly small collections of banked donor iPSCs, coverage through such exercises is likely to remain incomplete. Also, although partially matched donor cells have the potential to provide benefit when combined with immunosuppression, the risk of graft rejection remains. The reduced engraftment efficacy and increased health and financial costs associated with prolonged immunosuppression continue to make a case for perfectly HLA-matched iPSC therapies. It is also well established that certain iPSC lines are more suitable for differentiation toward certain product cell types,58,59 further complicating exercises to bank comprehensive collections of iPSCs from donors with universal therapeutic potential.
Genome-Edited iPSCs to Enhance Therapeutic Application
With the advent of efficient genome-editing technologies, the prospect of engineering and manufacturing universal allogeneic iPSC stocks has become a reality. This would allow existing validated iPSC stocks with well-characterized differentiation potential to be engineered to evade the immune system, thereby enabling allogeneic iPSC-derived cell therapeutics to be grafted without the need for immunosuppression. Such hypoimmunogenic allogeneic iPSCs with deletions of multiple HLA molecules have been generated through a number of strategies.60–62
Owing to the dependence of HLA class 1 proteins on dimerization with B2M for presentation on the cell surface, 63 genetic modification of the B2M locus has the potential to render allogeneic cells invisible to cytotoxic CD8+ T cells and evade elimination by the patient's immune system (Fig. 3A). Similarly, disrupting the function of the class 2 transactivator protein (CIITA) switches off the expression of HLA class 2 proteins, 46 rendering cells invisible to CD4+ helper T cells (Fig. 3B). However, although such approaches evade T cell-mediated immunosurveillance, complete erasure of HLA expression elicits an NK cell-mediated “missing-self” response. 64 Moreover, by ablating all HLA class 1 expression, grafted cells are no longer able to present intracellular antigens, potentially creating a vulnerability to infection or oncogenesis by disrupting immunosurveillance.
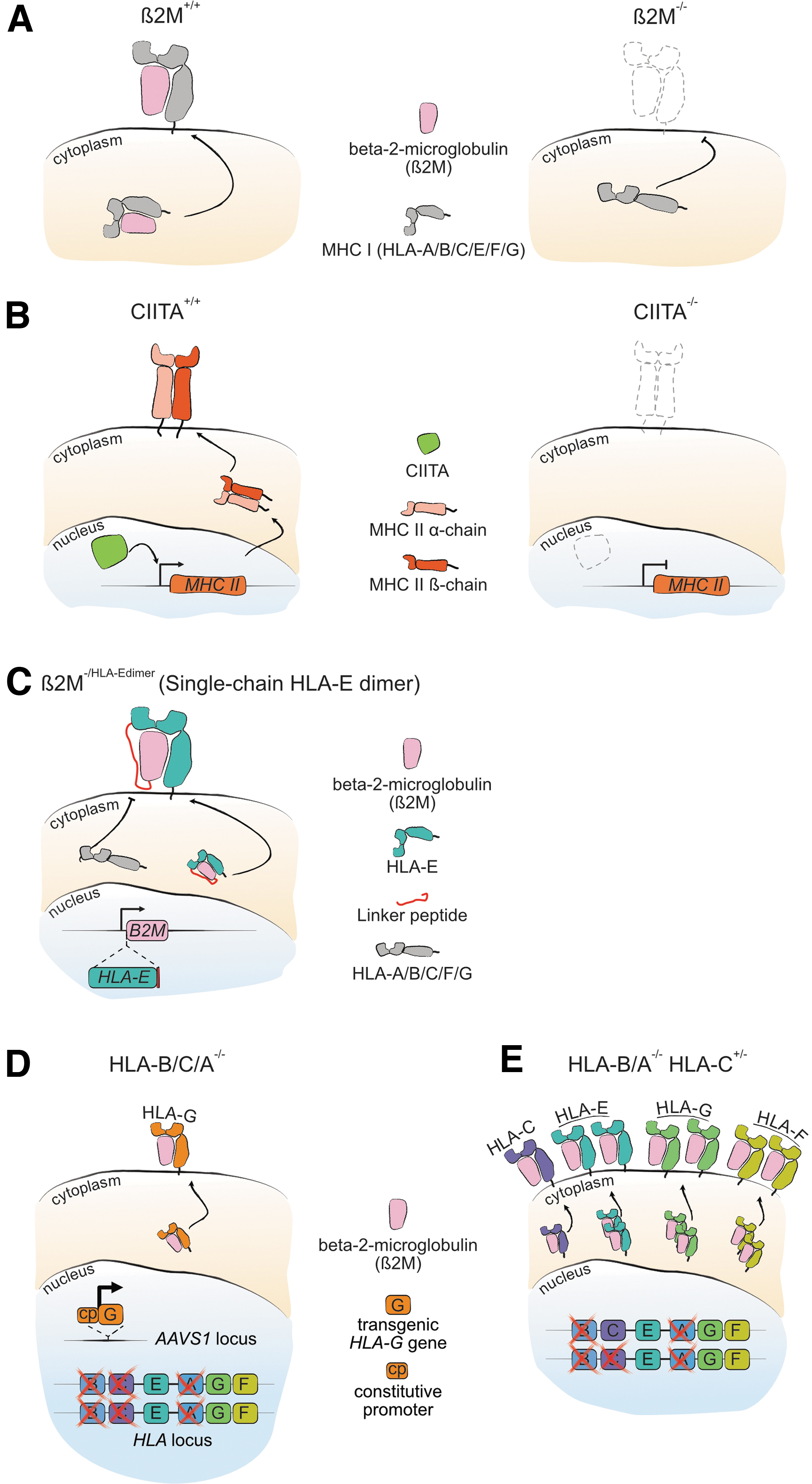
Several strategies have been employed to overcome the obstacles posed by complete removal of HLA classes I and II proteins. U.S.-based Universal Cells has developed adeno-associated virus (AAV)-based homologous recombination technology to modify the B2M locus in PSCs to create a single chain fusion protein comprising B2M and nonpolymorphic HLA-E that disrupts surface expression of immunogenic HLA-A/HLA-B/HLA-C proteins (Fig. 3C).62,65 As HLA-E can present classical peptides derived from intracellular proteins, such an approach maintains intact CD8+ T cell immune surveillance while also suppressing the NK cell missing-self response.66,67 Universal Cells was acquired in 2018 by the Japanese multinational Astellas Pharma that has been building a portfolio within the cell and gene therapy space with a strong focus on PSC technology, and lists partnerships with BlueRock Therapeutics and Adaptimmune Therapeutics, a forerunner in the T cell immunotherapy space.
An alternative to the AAV-based approach employed by Astellas/Universal cells produced universal donor iPSCs using CRISPR-Cas9 by ablating all MHC-1 class HLA proteins except for nonimmunogenic HLA-E and HLA-G. 61 However, expression and cell-surface localization of HLA-E were shown to depend on interferon gamma signaling in this system and would thus be limited to conditions of inflammation. In addition, expression of HLA-G, which mediates fetal–maternal tolerance during gestation, is restricted to a limited number of cell types. The investigators, therefore, knocked-in a constitutively expressed HLA-G transgene cassette into the AAVS1 locus to ensure its stable expression (Fig. 3D). A similar approach employing CRISPR-Cas9 editing of the MHC-1 locus deleted HLA-A and HLA-B while retaining expression of HLA-E/HLA-F/HLA-G, and a single copy of HLA-C (Fig. 3E). 60 Although this approach does not yield a truly universal iPSC donor cell, the requirement to match only a single HLA-C allele dramatically reduces the number of iPSC lines that would be required to cover the world population.
Complex engineering of the MHC-1 locus using Cas9-mediated double-strand break (DSB) technology, such as in the mentioned examples, required multiple rounds of editing and clonal selection to avoid deleting the entire MHC-1 locus. The subsequent introduction of cell-specific edits or transgenes (Table 2) into hypoimmunogenic iPSCs before differentiation to the final therapeutic product also demands further rounds of clonal selection. Such a sequential lengthy approach to genetic engineering is not only labor and resource intensive but also increases the risk of selecting for aberrant clones. The expansion of iPSCs tends to select for mutations in p53 due to the growth advantage conferred, 68 and Cas9-mediated DNA damage tends to select for cells defective in the p53-mediated growth-arrest response.69,70
Cell-type specific gene knockout and transgene knockin
GvHD, graft-versus-host disease; HLA, human leukocyte antigen; TCR, T cell receptor.
As clonal selection processes have the potential to generate iPSC lines at increased risk of oncogenic transformation, genetic screening is required to ensure that the p53 system remains intact in the final product. However, given that basic research in the CRISPR field continues to discover new unforeseen issues associated with gene editing through the generation of DSBs,71–75 new gene-editing technologies that minimize DNA damage and/or the number of rounds of clonal selection are much needed, as they would reduce both the extra costs associated with screening for anticipated side effects inherent to Cas9 technology, and reduce the risk posed by other as-of-yet unknown and potentially detrimental processes.
Second-Generation Genome Editors
Base editors and prime editors are emerging second-generation genome-editing platforms that utilize Cas protein/guide RNA (gRNA) complexes to localize editing enzymes to specific loci in the genome and directly modify target DNA sequences. Whereas CRISPR-Cas editing either creates semirandom insertions or deletions (indels) by error-prone nonhomologous end joining (NHEJ), 76 or introduces specific sequences by recombination through homology directed repair (HDR) of DSBs (Fig. 4A), 77 base editors and prime editors generate only a single-stranded DNA break during the process of DNA editing.
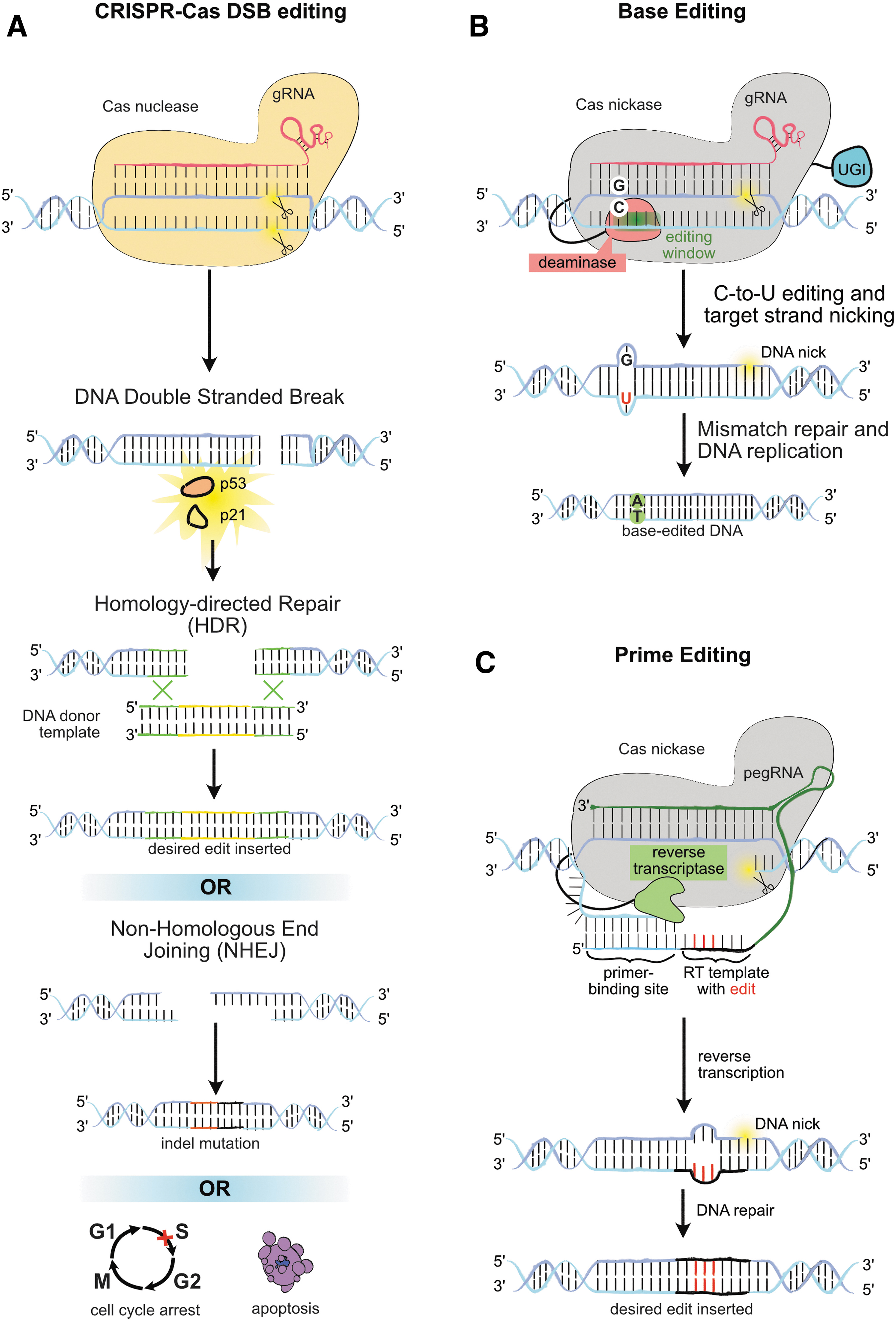
In the case of base editors, deaminase enzymes chemically modify target bases to convert them to the required nucleotide (Fig. 4B), 78 whereas prime editors utilize a reverse-transcriptase (RT) enzyme to synthesize edited DNA sequence from a template contained within the gRNA that is subsequently incorporated into the genome (Fig. 4C). 79 As with CRISPR-Cas DSB editing, the targets of base editors and prime editors are ultimately constrained by the protospacer adjacent motif (PAM) recognition sequence of the Cas protein employed in the editor.
The most widely used and characterized BE and PE employ SpCas9 that requires an NGG PAM sequence immediately adjacent to the guide sequence. Nonetheless, ongoing efforts continue to expand the scope of base editors and prime editors by employing alternative Cas proteins with different or more frequently occurring PAM motifs than that of SpCas9.80–86
The first base editors described were constructed in the laboratories of David Liu at the Broad Institute of MIT and Harvard, 87 and Akihiko Kondo at Kobe University 88 by coupling enzymatically inactive Cas9 to the cytosine deaminases APOBEC1 or AID, both of which convert cytosine (C) bases to thymidine (T). Initial developments focused on improving the efficiency and specificity of cytosine base editors (CBEs) by additional modifications aimed at tuning the DNA damage response and the position of C-to-T conversion within the target DNA sequence.
By employing a Cas9 mutant that nicks a single DNA strand, the DNA damage response could be harnessed to propagate editing to the nontargeted strand. Addition of a uracil glycosylase inhibitor domain to base editors was shown to improve both the efficiency of editing and the “purity” of the editing product by inhibiting the substitution of converted T nucleotides to other undesired bases.
Base editors edit target nucleotides positioned within a base editor-defined “editing window” located within the sequence targeted by the gRNA. 78 As current base editors have the potential to install additional undesired “bystander” edits at nontarget substrate nucleotides located within the editing window, a number of machine-learning exercises have been conducted to define the rules that govern the specificity and efficiency of base editing, which depend upon both the position of a target base within the editing window and the neighboring sequence context.89–91 Although still incomplete, there is now substantial capability to predict the editing outcome at a given target sequence and design editing strategies that avoid or minimize bystander editing.
Complementary to these computational efforts, alternative base editors have been engineered by introducing point mutations or modifying the linker between the Cas protein and deaminase components to reposition the editing window or increase the specificity of base-editing outcomes.92–94 Similarly, alternative cytosine deaminases such as AID and APOBEC family members from other species have been shown to exhibit distinct editing activities to those originally employed in early base editors, further expanding the repertoire of precision editing capabilities.92,95–98
After the demonstration of base editing with cytosine deaminases, the Liu laboratory developed Cas9 fusion base editors capable of converting adenine (A) DNA nucleotides to guanine (G) 99 through directed evolution of the tRNA-specific adenine deaminase. The addition of this second class of adenine base editors (ABEs) has greatly expanding the potential of base editing for gene correction applications, placing ∼60% of clinically relevant single nucleotide variants (SNVs) within the combined scope of CBEs and ABEs. 99 Nonetheless, CBEs and ABEs remain limited to catalyzing “transition” conversions between chemically similar pyrimidine or purine nucleotides.
A recent report has demonstrated base editors are capable of “transversion” edits from C (pyrimidine) to G (purine). 100 Although the editing purity of these C-to-G base editors (CGBEs) is less than that of transition base editors, the activity is not understood as well as that of CBEs and ABEs and depends significantly on the surrounding sequence context of the target cytosine. Nonetheless, CGBEs demonstrate the potential for progress toward base editors capable of catalyzing all conceivable nucleotide conversions.
The success of base editing has led to rapid advances toward their commercialization and clinical application. Beam Therapeutics was founded by David Liu and Feng Zhang, a pioneer of CRISPR-Cas technology, to commercialize the base-editing technologies invented in the Liu laboratory, and they have continued to develop the underlying platform 101 in parallel with advancing the technology into clinical trials.102–104 The laboratory of Shengkan Jin at Rutgers University has developed a modular base-editing system 105 in partnership with Horizon, commercialized as Pin-point™. In the Pin-point system, independent deaminase and Cas units are tethered through a protein recruiting aptamer encoded in the secondary structure of the gRNA. This provides the capability to flexibly pair deaminases with distinct editing activities with the ever-expanding range of Cas proteins, allowing the design and construction of application-specific base editors.
Prime editing was first described by the laboratory of David Liu in 2019 and holds the potential to overcome a number of the limitations of base editors. 106 Where base editors function through Cas coupled deaminase enzymes that catalyze nucleotide transversions from either C to T or A to G, prime editors employ a RT enzyme to install any of the 12 possible point mutations or generate short insertions or deletions encoded in the sequence of an extended prime editing guide RNA (pegRNA). As the edit is copied from the pegRNA rather than enzymatically catalyzed, prime editing is not prone to bystander editing associated with base editing.
Prime editing is initiated by the nicking of DNA sequence in the proximity of the intended edit leading to hybridization of the free 3′ DNA end with complementary sequence at the 5′ end of the pegRNA. This DNA–RNA duplex is then extended using modified sequence in the pegRNA as a template, thereby copying it onto the end of the free DNA strand creating an additional modified DNA “flap.” To date, it remains unclear precisely how the flap becomes incorporated into the DNA duplex, however, a recent study has highlighted an inhibitory role of the DNA mismatch repair pathway in the process. 107
As with base editors, prime editors modify only one of the strands of the DNA duplex themselves, and depend on an additional nick in the proximity of the edited sequence targeted by a second sgRNA to propagate the edit to the complementary strand after DNA repair. Thus whereas base editing requires only a single strand break within the complementary region of the gRNA for efficient editing, efficient prime editing requires a pegRNA and a gRNA and nicking of both DNA strands, which may explain the comparatively higher rate of indels observed with prime editors.79,106
Design of pegRNAs remains a largely empirical endeavor and is open to a range of additional considerations than must be accounted for when designing base-editing sgRNAs, including the position of the two nick sites relative to the intended edit, the sequence of homology between the pegRNA and the free 3′ end of the nicked DNA strand, and the sequence of the RNA template flanking the desired edit. An additional consideration is that degradation of the 3′ end of the pegRNA leads to inhibition of prime editing, and a recent study has sought to overcome this obstacle by incorporating structured RNA motifs into the 3′ sequence of engineered “epegRNAs.”
Although the position of prime editing targets is in principle less constrained by their exact proximity relative to an available PAM compared with base-editing targets, 106 a study comparing base editing and prime editing in iPSCs demonstrated a comparatively low efficiency of prime editing, 108 consistent with the generally lower efficiency of prime editing reported in other cell types.79,106 Nonetheless, the versatility offered by prime editing compared with base editing has inspired investment in Prime Medicine, Inc., and if the rapid translation of base editing is a fair indicator, it seems likely only a matter of time before the first trials with prime editors enter the clinic.
Engineering iPSCs Without Double-Strand Breaks
Whereas first-generation gene-knockout approaches with CRISPR-Cas rely on reading frame disruption after imperfect repair of DSBs at target sequences, gene knockout with base editing, and also in principle prime editing, is achieved either by directly installing premature stop codons in protein-coding regions, or by disrupting essential splice donor/acceptor sites.109,110
Single gene knockout through error-prone NHEJ DSB repair with first-generation CRISPR-Cas technology has the potential to generate large deletions in the genome,71,72 whereas the generation of multiple DSBs in the genome stimulate the formation of interchromosomal translocations after DNA repair. 111 Although iPSC clones with abnormal karyotypes can be identified by various approaches, base editing has been shown to reduce such undesirable editing outcomes to negligible levels 111 and, therefore, has the potential to minimize the resources deployed during safety screening. Also, CRISPR-Cas-induced DSBs are not well tolerated by PSCs and lead to cell cycle arrest and apoptosis, 112 reducing the efficiency of editing outcomes. 70 Although editing efficiency can be enhanced by blocking cell cycle arrest and apoptosis through transient inhibition of p53 activity, such approaches may increase the likelihood of genome instability in edited clones. 113
In addition to their enhanced safety profile in gene knockout applications, base editors and prime editors are also proving to be advantageous in correcting disease-causing SNVs and small insertions or deletions in preclinical models.104,106,114–120 A number of studies leveraging CRISPR-Cas-mediated DSB formation to stimulate recombination between a donor DNA sequence and a target gene have demonstrated the replacement of disease-causing alleles with “healthy” genetic material.121–128 HDR-based editing strategies are intrinsically less efficient than NHEJ-based gene knockout, and as with NHEJ, HDR repair is inhibited in iPSCs by the DNA damage responses elicited by DSB formation, which tends to both reduce editing efficiency and select for targeted cells with aberrant p53/p21 pathway activity. 69
HDR repair of DSBs is also prone to unwanted indels at the engineered site. 129 The enhanced efficiency and safety profile of base conversion with base editors and prime editors compared with DSB-mediated HDR may, therefore, prove useful for correcting disease-causing mutations in cell therapies employing patient-derived iPSCs in a manner similar to their application in gene therapy. If successful, this would enable the production of autologous iPSCs for transplant that are both disease free and perfectly matched to the recipient.
Although the drawbacks associated with first-generation genome-editing strategies through DSB formation have been overcome during the engineering of PSCs for use in preclinical studies and their subsequent therapeutic application in clinical trials, base editing and prime editing nonetheless offer a favorable safety profile by minimizing DNA damage and the associated risk of genome instability. When considering the complex multigene editing required for the engineering of both hypoimmunogenic allogeneic iPSCs and the installation of product cell-specific gene knockouts and knockins already discussed, base editing and prime editing offer complementary capabilities.
In highly multiplexed gene knockout applications, the high editing efficiency alongside negligible rates of indel formation of base editing compared with prime editing overcomes the need for multiple rounds of editing and associated risks associated with clonal selection. 130 In applications requiring the site-specific knock in of transgenes such as CARs during the generation of iPSC-derived adoptive cell therapies or HLA proteins in the various described strategies for the generation of hypoimmunogenic cells, the recently described twin prime editing, which employs two complementary pegRNAs, is capable of installing “landing sites” for subsequent site-specific integration of large DNA sequences. 131 Similarly, the demonstration of Cas-directed transposition of large DNA sequences in bacteria raises further prospects for targeted transgene insertion without the introduction of DNA DSBs.132–135
Future Prospects for Precision Genome Engineering
The advent of CRISPR-Cas technology has brought about a paradigm shift in genome engineering. The ability to efficiently edit multiple genes simultaneously in any desired cell type has dramatically expanded the scope for developing advanced cell therapies, and the enhanced safety profile of emerging second-generation base editor and prime editor technologies looks likely to further accelerate this trend. By combining gene edits that enhance therapeutic function with those that allow allogeneic cells to evade a patient's immune system, the scene is set to propel engineered iPSC-derived cell therapies into clinical trials. This raises the prospect of affordable off-the-shelf treatments for a broad spectrum of diseases, from leukemia to neurodegeneration.
Although much of the focus of gene editing has been placed on gene knockout and gene correction applications, as the technology continues to develop it can be envisioned that second-generation editing technologies could be applied to enhance or modulate protein function by installing known polymorphisms or rationally designed modifications into the genes encoding them. For example, the substrate preference of metabolic enzymes or the rate of flux through specific metabolic pathways could be engineered to modify the preference for available energy sources, enhance the production of anabolic precursors, or reduce the accumulation of toxic by-products to overcome challenges such as T cell exhaustion.136,137 Similarly, the affinity or specificity of receptor–ligand interactions, or the affinity of transcription factors for their target sequences, could be modified to rewire the signaling pathways and gene-regulatory networks that coordinate cellular signaling processes.
Beyond their application in modifying the protein-coding sequence of genes, base editors have proven effective at modifying gene expression by editing nucleotides in cis-regulatory elements in the noncoding genome.103,138 Such approaches raise the possibility of generating iPSC-derived cell therapies in which endogenous gene regulatory networks are “rewired” such that genes are either silenced or activated to alter cellular function in a context-dependent manner. 139 For example, the release of cytokines in response to specific stimuli by iPSC-derived graft cells in regenerating tissues, or by iPSC-derived adoptive immunotherapies, could be modified to enhance therapeutic efficacy or improve safety. Similarly, it can be envisioned that such approaches could be employed to enhance the efficiency of differentiation of iPSCs to challenging therapeutic cell types or to enhance the stability of the differentiated product by modifying the cis-regulatory elements of developmental gene regulatory networks. 140
As the fields of synthetic biology and developmental biology continue to develop, it seems likely that precision genome-editing technologies such as base editing and prime editing will play important roles in enabling increasingly complex iPSC-derived engineered cell therapies with truly novel functions.
Footnotes
Acknowledgments
The author thanks Andrea Frapporti for creating all figures and Jennifer Harbottle, Maren Mayer Gross, and Andrea Frapporti for providing critical feedback during article preparation.
Author Disclosure Statement
R.B. is an employee of Horizon Discovery, a PerkinElmer company. Horizon Discovery holds an exclusive license from Rutgers University to commercialize Pin-point™ base editing technology for therapeutic, diagnostics and service applications. The views expressed in this article are those of the author and do not reflect those of the company.
Funding Information
R.B. is an employee of Horizon Discovery. No additional source of funding supported this work.