
Abstract
Salmonella serovar Kentucky is frequently isolated from chickens and dairy cattle, but recovery from humans is comparatively low based on the U.S. National Antimicrobial Resistance Monitoring System (NARMS) reports. We aimed to better describe the genetic diversity, antimicrobial resistance, and virulence determinants of Salmonella Kentucky isolates from humans, food animal ceca, retail meat and poultry products, imported foods and food products, and other samples. We analyzed the genomes of 774 Salmonella Kentucky isolates and found that 63% (54/86) of human isolates were sequence type (ST)198, 33% (29/86) were ST152, and 3.5% (3/86) were ST314. Ninety-one percent (570/629) of cecal isolates and retail meat and poultry isolates were ST152 or ST152-like (one allele difference), and 9.2% (58/629) were ST198. Isolates from imported food were mostly ST198 (60%, 22/37) and ST314 (29.7%, 11/37). ST198 isolates clustered into two main lineages. Clade ST198.2 comprised almost entirely isolates from humans and imported foods, all containing triple mutations in the quinolone resistance-determining region (QRDR) that confer resistance to fluoroquinolones. Clade ST198.1 contained isolates from humans, ceca, retail meat and poultry products, and imported foods that largely lacked QRDR mutations. ST152 isolates from cattle had a lineage (Clade 2) distinct from ST152 isolates from chicken (Clade 4), and half of ST152 human isolates clustered within two other clades (Clades 1 and 3), largely distinct from Clades 2 and 4. Although clinical illness associated with Salmonella Kentucky is low, ST198 appears to account for most human infections in the Unites States but is uncommon among ceca of domestic food animals and retail meat and poultry products. These findings, combined with human exposure data, suggest that fluoroquinolone-resistant ST198 infections may be linked to the consumption of food products that are imported or consumed while traveling. We also found unique differences in the composition of virulence genes and antimicrobial resistance genes among the clades, which may provide clues to the host specificity and pathogenicity of Salmonella Kentucky lineages.
Introduction
Data generated by the U.S. National Antimicrobial Resistance Monitoring System (NARMS) reveal that Salmonella enterica serovar Kentucky (Salmonella Kentucky) is frequently isolated from the ceca of chickens (29% of 1133 isolates) and dairy cattle (3.7% of 1467 isolates) and is the predominant serovar found in retail chicken products (28% of 3171 isolates) (U.S. Food and Drug Administration [FDA], 2022). Because an estimated 10% of foodborne Salmonella illnesses in the Unites States are attributed to chicken (Batz et al., 2021), one would expect the relative proportion of Salmonella Kentucky infections to be high. Salmonella Kentucky causes very few human clinical infections, however, amounting to only two cases per 1 million people per year (Centers for Disease Control and Prevention [CDC]). Outbreaks are rare, with only five reported through the National Outbreak Reporting System since 1998 (P. Sundararaman, pers. Comm., July 12 2021; CDC, 2021b).
It is generally thought that serovar Kentucky is less harmful to humans than other more virulent serovars (e.g., Salmonella Typhimurium) because it lacks many virulence genes (Dhanani et al., 2015; Tasmin et al., 2017; El Hage et al., 2020). However, the recent global emergence of multidrug-resistant Salmonella Kentucky strains in humans indicates evolutionary pathways for this serotype toward more virulent subtypes that can become common causes of human illness (Le Hello et al., 2013a).
Salmonella Kentucky is a polyphyletic serovar, meaning it consists of multiple sequence types (ST) that do not share a recent common ancestor (Timme et al., 2013). This has resulted in Salmonella Kentucky STs that are highly divergent phylogenomically, appearing as multiple clades (Haley et al., 2016). Some research points to these STs having different ecologies, host characteristics, and geographic dispersions (Haley et al., 2016). In the United States, most Salmonella Kentucky isolated from domestic agricultural sources are ST152 (Haley et al., 2016; Rauch et al., 2018). Salmonella Kentucky ST198 is also found in agricultural sources in the United States, primarily dairy cattle (Haley et al., 2016). ST198 can be resistant to multiple antibiotics, including ciprofloxacin, a first-line therapy for the treatment of complicated Salmonella infection in people. The most commonly detected clinical isolates from patients in the United States are ciprofloxacin-resistant (CipR) ST198 (Le Hello et al., 2011; Rickert-Hartman and Folster, 2014; Haley et al., 2019).
The CipR phenotype is conferred by triple mutations in the quinolone resistance-determining region (QRDR) (GyrA_Ser83/Asp87 and ParC_Ser80). In countries where CipR ST198 circulates, poultry is the major source of infection (Le Hello et al., 2011; Soltys et al., 2021). CipR ST198 can be found in high levels among poultry isolates in countries where there is believed to be uncontrolled administration of fluoroquinolones in poultry production (Collignon and Voss, 2015; Igomu, 2020; Xiong et al., 2020), so it stands to reason that extensive use of fluoroquinolones could provide the selective pressure necessary for spread and persistence of this strain. Although fluoroquinolones are not approved for use in poultry production in the United States, these strains may still reach consumers through other food sources, including imported foods (Le Hello et al., 2013a). International travel is a significant risk factor for CipR ST198 clinical infection (Le Hello et al., 2011, 2013b; Rickert-Hartman and Folster, 2014; Haley et al., 2019).
Since its establishment in 1996, NARMS has isolated over 11,000 Salmonella Kentucky from ill persons and domestic agricultural sources, offering one of the largest collections in the United States. Approximately 17% of those isolates have been sequenced and the genomes have been made publicly available at the National Center for Biotechnology Information (NCBI). In this study, we sought to examine the genomic profiles of many temporally and geographically distributed Salmonella Kentucky isolates from the NARMS collection and from imported and domestic product samples collected by the FDA Office of Regulatory Affairs (ORA). Previous work by NARMS has demonstrated high correlations between the presence of resistance genes and mutations and phenotypic resistance in Salmonella (McDermott et al., 2016).
Therefore, by using these data, we can further our understanding of the strain relatedness and predicted antimicrobial resistance, virulence, and plasmid profiles of Salmonella Kentucky derived from various sources and make inferences about possible sources of Salmonella Kentucky infection. Furthermore, by understanding the genetic differences in Salmonella Kentucky subtypes from different sources, we may be able to detect and respond to the emergence of strains with higher pathogenic potential earlier on.
Materials and Methods
Sampling
As part of routine NARMS surveillance, Salmonella Kentucky were isolated from retail meat and poultry (ground beef, pork chops, ground pork, ground turkey, and chicken parts) collected from 2002 through mid-2020, ceca of domestic food animals (beef cattle, dairy cattle, market hogs, sows, turkeys, and chickens) collected from 2014 through mid-2020, and humans between 1996 and 2019. NARMS sampling and testing protocols have been previously described (U.S. FDA, 2019). The isolates were sequenced, regardless of antimicrobial resistance profiles. Imported foods, animal feed and pet foods, and produce and swabs from production facilities were tested by FDA ORA laboratories for FDA microbiological surveillance sampling of targeted foods between 2005 and 2019.
Data mining
Genomic sequences were gathered as raw sequencing reads from NCBI Sequence Read ArchiveSRA database under Bioprojects PRJNA292661, PRJNA292666, PRJNA186035, PRJNA230403, and PRJNA287785. A total of 774 genomes of Salmonella Kentucky isolates recovered between 1996 and the first few months of 2020 were downloaded from NCBI, including human (93), turkey ceca (24), retail ground turkey (5), dairy cattle ceca (42), beef cattle ceca (27) retail ground beef (8), sow ceca (2), market swine ceca (10), and retail pork (1). Due to the relatively large number of Salmonella Kentucky collected from chicken sources during the time frame of the study, we selected sequences of only 149 chicken ceca isolates (from 435) and 375 retail chicken isolates (from 968).
Chicken isolates were randomly chosen within state, month, and year strata using simple random sampling without replacement (PROC SURVEYSELECT, SAS version 9.4 [SAS, Cary, NC]). State locations, where domestic food isolates were collected, were converted to U.S. Department of Health and Human Services (HHS) regions to be consistent with human metadata. We also downloaded sequences from 37 Salmonella Kentucky recovered from U.S. FDA ORA product sampling of fish, spices, animal feed, and pet food.
Bioinformatic analyses
All whole genome sequencing (WGS) data were assembled using SPAdes v3.13.0 (Bankevich et al., 2012). Contigs with coverage <10 or with length <200 bps were removed from the assemblies.
The analysis of classical multilocus sequence typing (MLST), which used seven different loci, was performed using the whole-genome sequencing data. Salmonella MLST allelic profiles and sequences were downloaded from the PubMLST database (
Antimicrobial resistance genes were identified using the Resfinder database (Bortolaia et al., 2020) (downloaded from
Virulence genes were extracted from the NCTR-database, transformed to binary data, and imported into BioNumerics (ver. 7.6; Applied Maths, Austin, TX) for phylogenetic analysis using dice coefficients and unweighted pair group method with arithmetic mean. Strains were color coded based on their ST and phylogenetically relevant virulence factors (i.e., those that were not present or absent in all isolates) were selected to generate a maximum parsimony tree using BioNumerics default settings. Of the 404 virulence genes identified among all isolates, only 25 differed among STs. The 25 virulence genes were used to build the heat map in Figure 5b.
Epidemiological data
We summarized exposure histories collected by CDC for a previous report on the emergence of ST198 in human cases (Rickert-Hartman and Folster, 2014), including five of the six ST198 cases presented in the report and seven additional cases that were not ST198. We then paired the results with the Salmonella Kentucky isolate IDs (Supplementary Table S1). This activity was reviewed by CDC and was conducted consistent with applicable federal law and CDC policy. Patient travel histories were obtained through interviews as part of a study reviewed and approved by the Centers for Disease Control and Prevention IRB, and all participants provided verbal consent.*
Results and Discussion
ST and plasmid distribution
The majority of the 774 isolates sequenced fell among four predominant STs: ST152 (52%), an ST152-like clade (similar to ST152 but with one allele difference; 26%), ST198 (17%), and ST314 (2%). STs varied for the remaining 22 isolates (3%), and included 1 ST80, 1 ST471, 1 ST138, 5 ST696, 1 ST3220, 1 ST6086, 1 ST33, 1 ST142, 3 ST15, 3 ST27, and 4 ST19 (Supplementary Fig. S1). Because there was no overlap in ST types among human and food samples, these 22 isolates were excluded from the remainder of the analysis, leaving 752 isolates. Like Haley et al. (2016), we found that the majority of NARMS chicken ceca and retail chicken isolates (98.6%, 507/514) were either ST152 or ST152-like, whereas just 1.4% (7/514) were ST198 (Fig. 1a). Similar distributions were seen in swine ceca isolates. When grouped together, cattle isolates were more evenly split with 50% (39/77) categorized as ST152 or ST152-like, but 48% (37/77) appearing as ST198. One cattle isolate was ST314. There was an apparent discrepancy in ST distributions between dairy and beef sources.
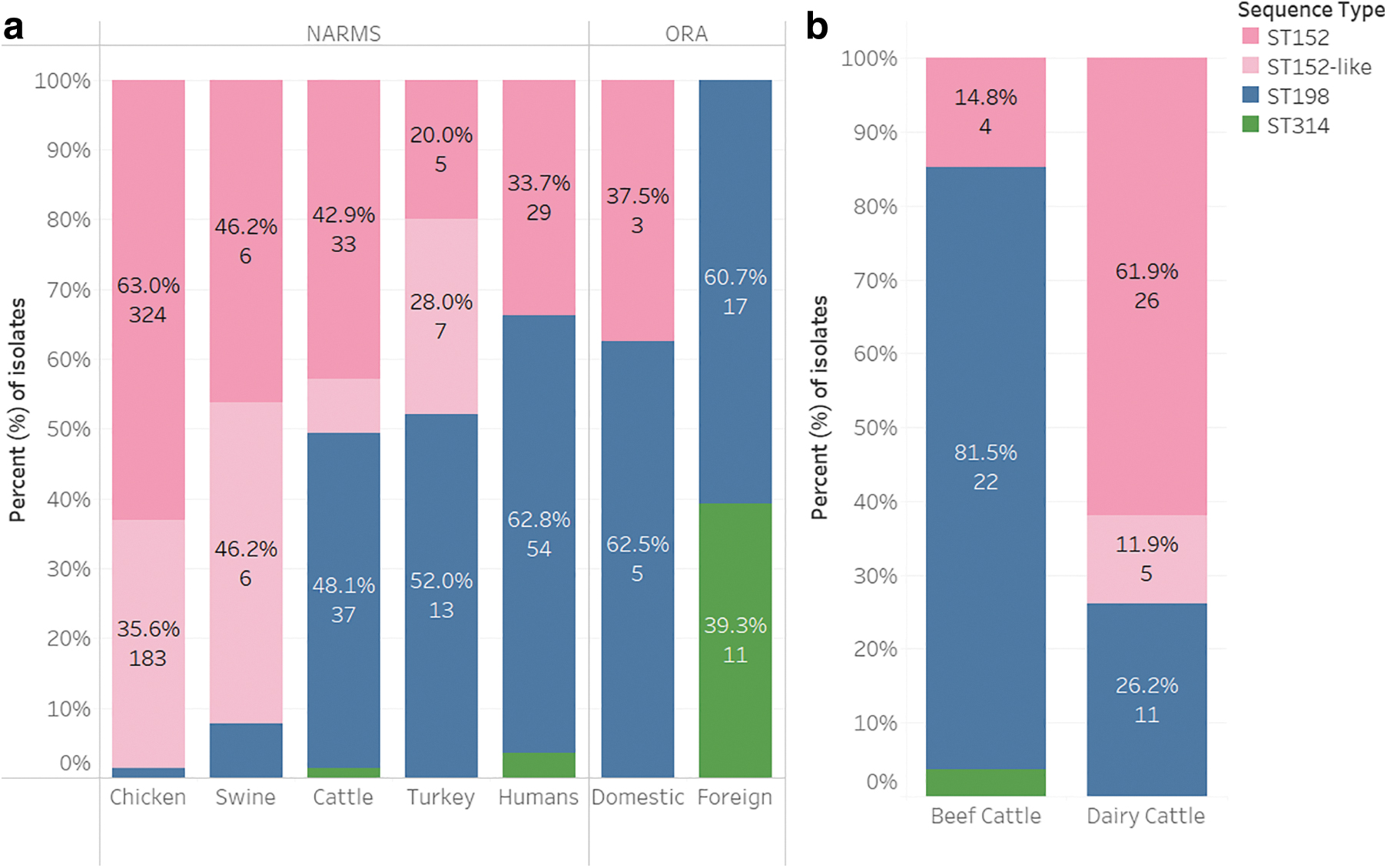
While beef cattle ceca isolates were more likely to be ST198 (81%, 22/27), dairy cattle ceca isolates were more likely to be ST152/152-like (74%, 31/42) (Fig. 1b). NARMS turkey ceca and food isolates were also evenly split between ST152/152-like and ST198. The majority (63%, 54/86) of human isolates were ST198, but 34% (29/86) were ST152 and three isolates were ST314 (Fig. 1a). There were no ST152-like human isolates. Twenty-eight of the 37 FDA-ORA isolates originated from imported samples. All foreign-derived FDA-ORA isolates (originating from Canada, China, Egypt, France, India, Malaysia, Mexico, Morocco, Pakistan, Syria, UAE, and Vietnam) were either ST198 (61%, 17/28) or ST314 (39%, 11/28). Eight FDA ORA isolates were labeled as derived from the United States. Five of these isolates were ST198 and three were ST152/152-like. One isolate (not shown in the figure) collected by FDA ORA was ST152-like but had no associated country of origin data.
Eighty-eight percent (660/752) of Salmonella Kentucky strains carried at least 1 plasmid replicon, covering 9 major incompatibility (Inc) replicon types, 8 Col plasmid replicon types, and 1 pKPC plasmid with no designated Inc type. Plasmid replicons were found in 97% of the ST152 and ST152-like group (Table 1), 54% of the ST198 isolates, and 2 of the 15 ST314 isolates. Inc and Col plasmid distribution was remarkably different between the STs.
Plasmids Found in Salmonella Kentucky Isolates
Many isolates had more than one plasmid. Those isolates are counted more than once in this table.
ST, sequence type.
ST152 and ST152-like
ST152 isolates separated into four distinct clades (Fig. 2). Half (15/29) of ST152 human isolates appeared to group within two geographically and temporally heterogeneous clades (1 and 3) that were largely absent of animal or food sources. Clade 1 isolates originated in five different regions and spanned 18 years and clade 3 isolates also originated in five different regions and spanned 15 years. The other half of human isolates were dispersed throughout clade 4, a large group containing isolates from varied source types but predominated by chicken sources. Five of the human ST152 isolates with associated epidemiological information (AM45701, AM45844, 2012AM-1440, AM44610, AM42793) fell into clade 4. All five patients had previous meat, poultry, or seafood exposures and one of the four patients for whom travel histories were available did travel internationally (Fig. 2, Supplementary Table S1). As observed elsewhere (Haley et al., 2016), ST152 isolates from chicken (clade 4) clustered in a lineage distinct from most ST152 isolates from dairy cattle (clade 2).
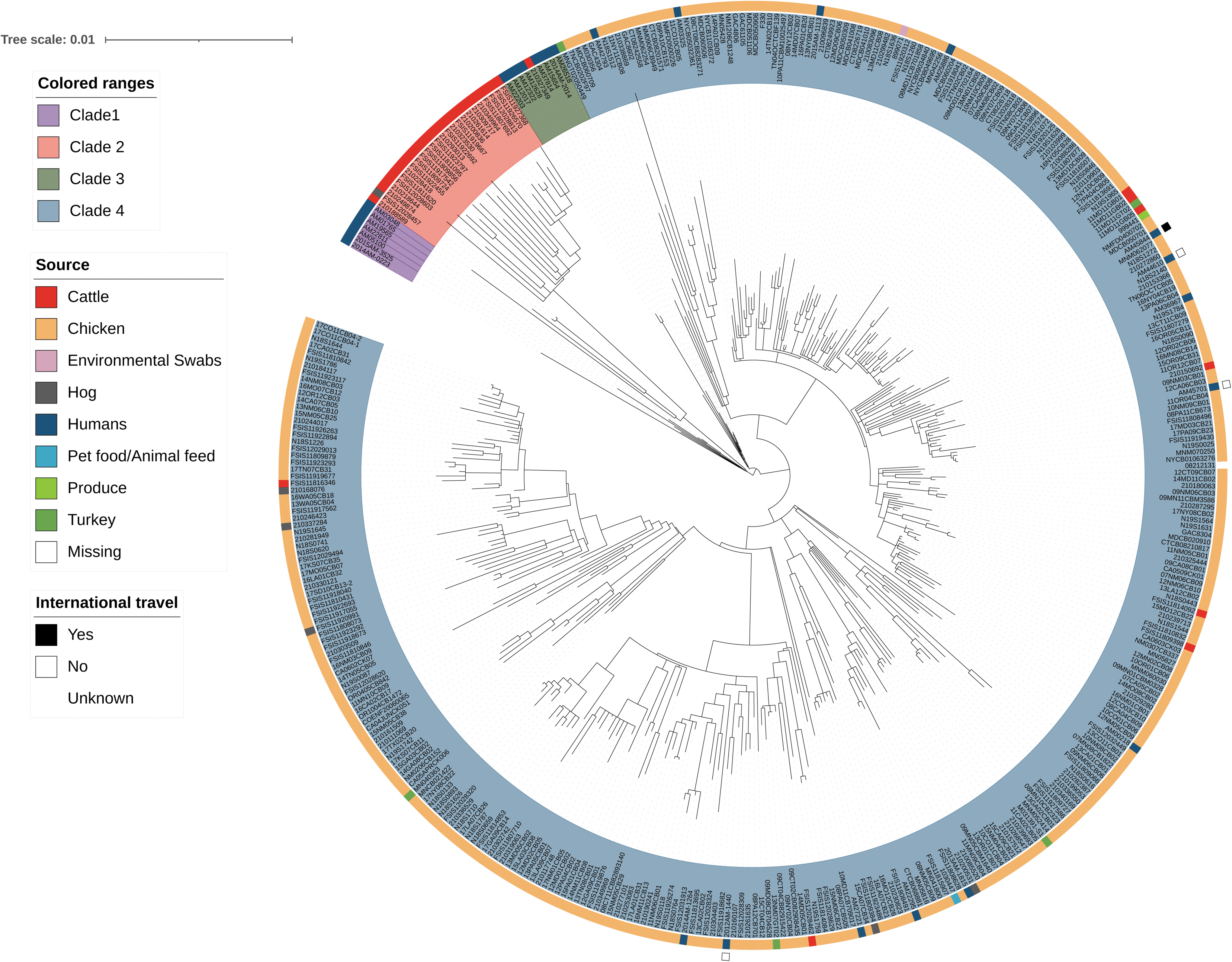
Maximum likelihood phylogeny of 400 Salmonella Kentucky ST152 isolates. Metadata in Supplementary Tables S1 and S2 were used to fill the outer rings of the tree and indicate international travel status (where known) and source. The colored ranges on the labels indicate strain sequences associated with clades 1 (purple), 2 (red), 3 (green), and 4 (blue). ST, sequence type.
Antimicrobial resistance genotypes were varied among ST152 clades (Supplementary Table S2). Notably, 95% of the 41 isolates within clades 1, 2, and 3 contained only the cryptic aac(6′)-Iaa gentamicin resistance gene, which is normally silent (Magnet et al., 1999). Two of the 41 isolates contained this gene and a tetracycline resistance gene (tetA). In contrast, 286 of the 359 isolates (79.7%) in clade 4 had predicted resistance to at least one antibiotic, as they carried aac(6′)-Iaa and at least one additional antimicrobial resistance gene. Tetracycline resistance genes were found in 213 of the clade 4 isolates. β-Lactam resistance-conferring bla genes were also identified in 70 of the clade 4 isolates, including bla CTX-M-1 (1), bla HERA -3 (1), bla CMY -150 (66), and bla TEM -1 (4). Other resistance genes (cmlA1, dfrA14, dfrA15, floR, sul1, sul2) were detected among clade 4, although to a lesser degree (<4% of isolates).
Ninety-seven percent (585/603) of the ST152/ST152-like isolates contained at least one Inc plasmid sequence and 8% (46/603) contained a Col plasmid sequence (Table 1). Plasmids were missing from all but one of the isolates in two human clades of ST152 (clades 1 and 3). However, all dairy cattle isolates in clade 2 displayed IncI1 and multiple Col plasmid replicons, including Col(pHAD28) and Col440I. The isolates lacked resistance genes (Supplementary Table S2), so the biological role of the IncI1 plasmid is unclear. Haley et al. (2016) suggest that IncI1 creates a selective advantage for these bovine-associated strains, particularly in cattle from the Mid-Atlantic region. These plasmids have been shown to be more common among pathogenic rather than commensal Escherichia coli (Johnson et al., 2007).
A majority of isolates in clade 4 carried at least two Inc replicon types, the most frequent being IncX1, followed by the IncF group and IncI1. Chicken sources, which comprised a large portion of clade 4, commonly carry Inc plasmids. Additional research is needed to elucidate the function of IncX1 plasmids in chicken; however, it is widely recognized that IncI1 is associated with chicken isolates in the United States, particularly those that are resistant to cephalosporins (Folster et al., 2011, 2017). Similarly, IncFIB plasmids can be associated with multidrug resistance as well as virulence factors that increase bacterial virulence (McMillan et al., 2020).
ST198
Previous work has shown that Salmonella Kentucky ST198 isolates recovered in the United States could be divided into two major clusters, a largely fluoroquinolone-susceptible clade (198.1), which comprised mostly domestic agricultural sources and a fluoroquinolone-resistant clade (198.2), composed of human clinical isolates (Haley et al., 2016). Consistent with those findings, we found that many isolates that grouped within clade 198.1 originated from domestically produced agricultural sources and foods (dairy and beef cattle, chicken, turkey, produce, pet food/feed) (Fig. 3). Sixty-five percent (35/54) of human clinical ST198 isolates also fell within clade 198.1. All isolates in clade 198.1 lacked the triple mutations in QRDR that confer high-level resistance to fluoroquinolones; however, subclade 198.1-1 contained three isolates with a point mutation in QRDR [gyrA (S83F/A) or gyrA (D87Y)] (Fig. 3). Two of these isolates were from spices imported from South Asia, and one isolate was from a human. We observed that 59% (48/81) of the isolates in subclade 198.1-2 had no functional resistance genes, consistent with reduced plasmid content.
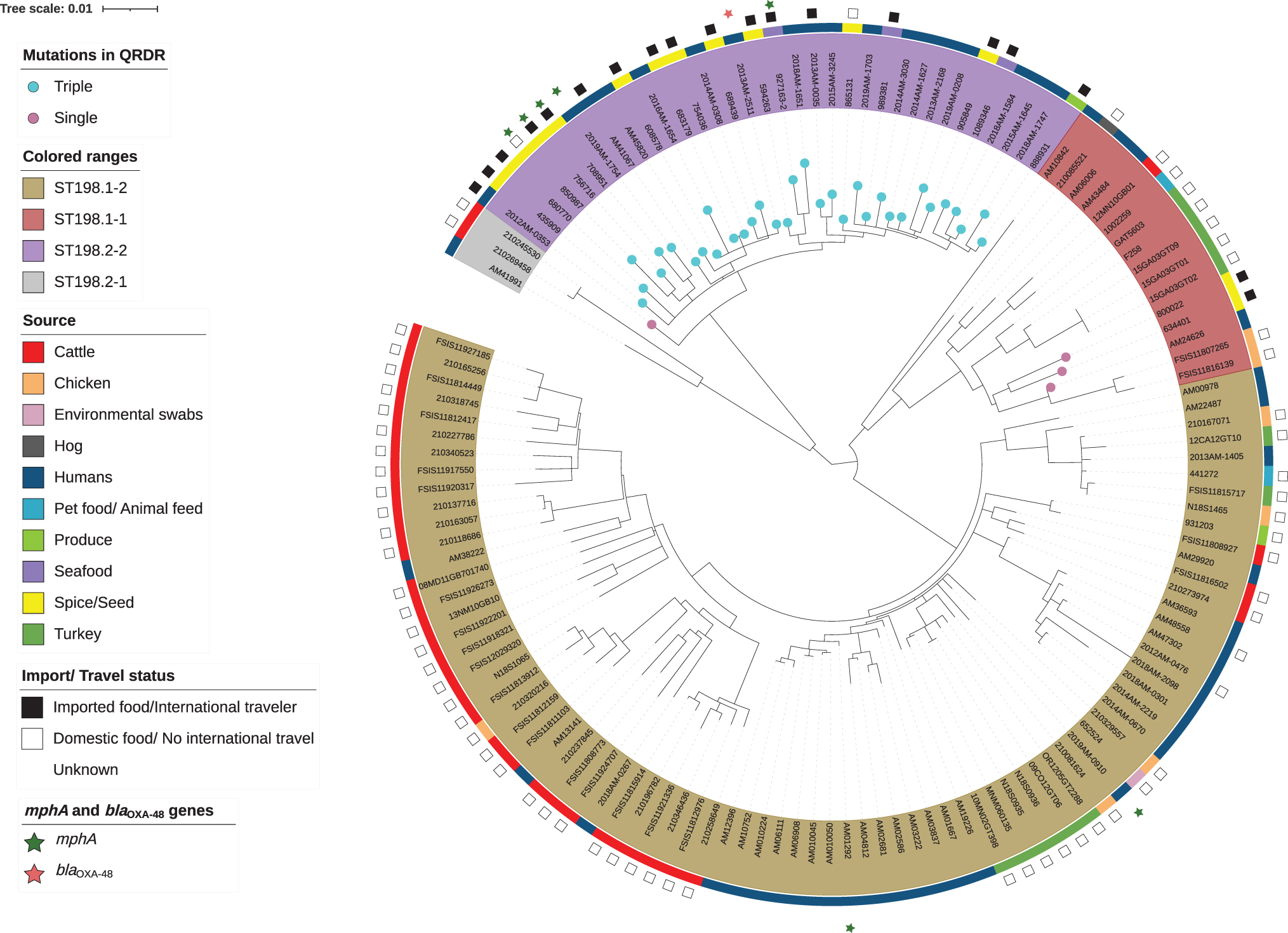
Maximum likelihood phylogeny of 134 Salmonella Kentucky ST198 isolates. Metadata in Supplementary Tables S1 and S2 were used to fill the outer rings of the tree and indicate the presence/absence of mphA and blaOXA-48 genes (stars), import (food) and (human) travel status (boxes), and source (colored strips). The colored ranges on the labels indicate strain sequences associated with ST198.1-2 (tan), ST198.1-1 (red), ST198.2-2 (purple), and ST198.2-1 (gray). Branch circles indicate the presence of single (purple) or triple (light blue) QRDR mutations. QRDR, quinolone resistance determining region; ST, sequence type.
Furthermore, turkey and human isolates were more likely than cattle isolates to contain functional resistance genes. We identified source-specific clusters within subclade 198.1-2, where turkey, human, and cattle isolates generally clustered separately, suggesting that the majority of these strains circulated independently from one another (Fig. 3). However, we did identify some cases where human isolates were embedded in the cattle cluster (Supplementary Fig. S2, cluster 3).
The geographic and temporal association in the human and cattle clusters of ST198.1-2 is worth noting. We found that two distinct clusters (separated by 70 SNPs) comprised human isolates predominately from HHS Region 2 (Supplementary Fig. S2, clusters 1 and 2). However, cluster 1 contained isolates collected between 2008 and 2014 and cluster 2 contained isolates collected between 1997 and 2003. This suggests that there was a potential common source for these strains. Three cases in cluster 1 (isolates AM48558, AM47302, 2012AM-0476) had exposures to dairy product (Supplementary Table S1). Similarly, 77% (24/31) of cattle isolates in cluster 3 were collected from slaughterhouses in Region 6 between 2014 and 2020. Additional investigation might lead to a common exposure.
Cluster 198.2 consisted of two sublineages. One subclade of three isolates (198.2-1) did not yield fluoroquinolone resistance genes, and contained only the silent aac(6′)-Iaa gene; yet, all but one isolate in the other subclade (198.2-2) contained double amino acid substitutions in gyrA (S83F and D87G,N,Y) and a single substitution in parC (S80I) (Fig. 3, Supplementary Table S2). Subclades 198.2-1 and 198.2-2 were distantly related, differing by 304 SNPs. Human isolates grouped with imported spices, seafood, and bread in subclade 198.2-2, differing by just 46 SNPs, suggesting a possible epidemiological link, although we know that three human isolates (2012AM-0353, AM41607, and 2013AM-0035) also had prior travel history (Rickert-Hartman and Folster, 2014) (Supplementary Table S1). Notably, we found that human clinical ST198 isolates displayed a temporal trend across the two main clades, with isolates appearing in clade 198.2 only after 2008.
This finding is in accordance with Le Hello et al. (2011), who determined that MDR ST198 emerged from Egypt in the early 2000s. Twenty-nine of the 34 isolates (85%) in subclade 198.2-2 carried genes conferring resistance to at least three classes of antibiotics. Previous research has shown that the multidrug resistance in these strains is likely due to integration of the Salmonella genomic island (SGI-1) in the chromosome (Le Hello et al., 2013b; Hawkey et al., 2019). The SGI-1 structure is highly mosaic, which may explain the differences in resistance profiles among the 198.2-2 isolates. In light of its multidrug resistance, this strain's more recent acquisition of cephalosporinases (extended spectrum beta-lactamase [ESBL] and AmpC) and carbapenemases (Le Hello et al., 2013b; El Hage et al., 2020) is especially concerning because third-generation cephalosporins are recommended as therapies for the treatment of complicated or severe Salmonella infections (Shane et al., 2017), and carbapenems are considered drugs of last resort.
None of the CipR isolates contained ESBLs or AmpC β-lactamases, but we did find one isolate (2013AM-2511) with the carbapenem resistance gene, bla OXA-48. Because azithromycin is also a recommended therapy (Committee on Infectious Diseases, 2021), the discovery of the macrolide resistance gene, mphA, in three spice/seed isolates, one seafood isolate, and one clinical isolate from subclade 198.2-2 was another significant finding. Isolates with mphA appeared sporadically between 2011 and 2019. The apparent chromosomal mediation of resistance attributes in the multidrug-resistant 198.2-2 subclade is consistent with the low level of plasmids in this group.
ST314
ST314 isolates were split into two clades with geographic interdependency (Fig. 4). One clade (314.1) contained isolates derived only from foods sampled for import product testing. The other clade (314.2) contained isolates from humans and one isolate from domestic cattle ceca. Discounting the silent aac(6′)-Iaa genes, all but one isolate had predicted pan-susceptibility. The isolate, from imported petfood (1018638), was only one of two that harbored a plasmid and contained multiple resistance genes, including those conferring resistance to fluoroquinolone (qnr) and macrolide drugs (mph). Others have observed ST314 in human clinical isolates from the United States (Haley et al., 2019), however, its global distribution is not well studied. While we found that ST314 is predominately pan-susceptible, others have seen multidrug-resistant ST314 in poultry-associated isolates from China (Gu et al., 2020).
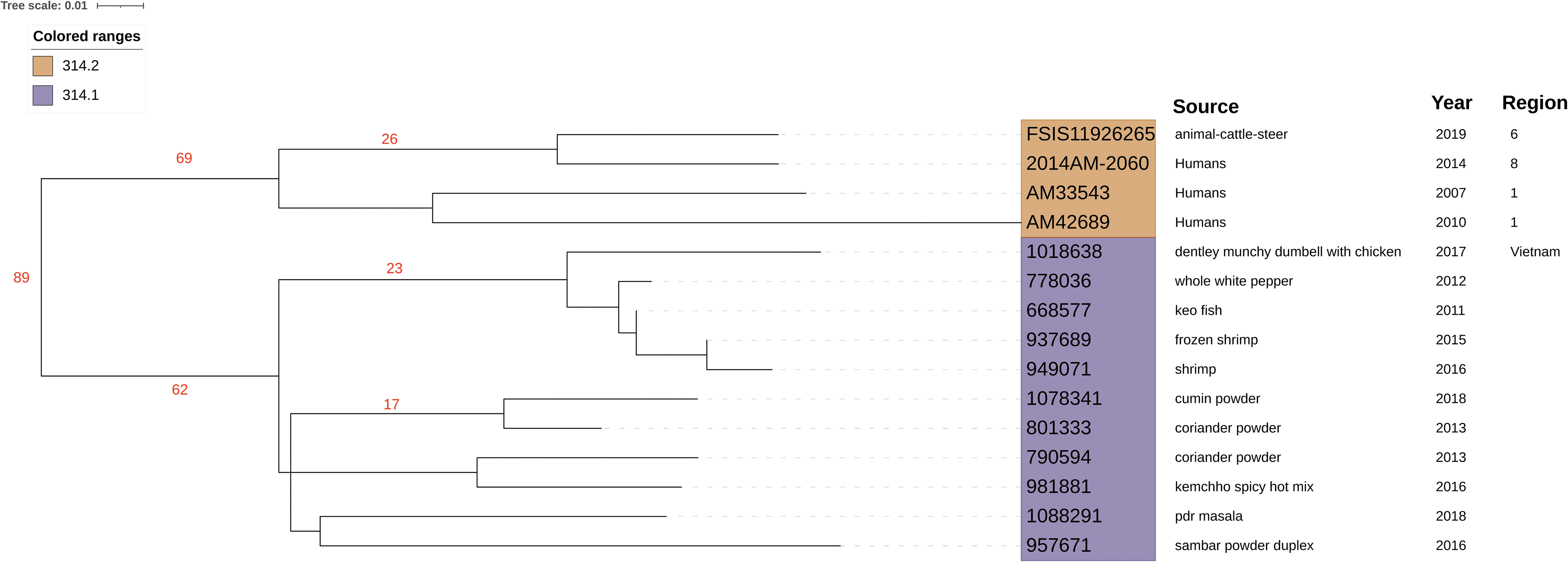
Phylogenetic tree of 15 Salmonella Kentucky ST314 isolates. The colored ranges indicate strain sequences associated with ST314.1 (purple) and ST314.2 (orange). The number of SNP differences between branches are shown in red. SNP, single nucleotide polymorphism; ST, sequence type.
Virulence genes
We created a maximum parsimony tree of all 774 Salmonella Kentucky isolates using only the 25 virulence genes that were differentially distributed among the STs (Fig. 5a), potentially signifying the importance of these genes in strain evolution. ST198 isolates hosted virulence genes not seen in ST152/152-like isolates, including determinants for a putative LysR family transcriptional regulator (STM0030), two putative metabolic proteins (STM0036 and STM4433), a fimbrial protein (lpfD), a type III secretion system effector (sseK2), and the entire fimbrial protein-associated tcf operon (Fig. 5b). All of the aforementioned genes appeared in 98–100% of ST198 isolates. It is possible that the absence of these genes from ST152 may contribute to the poor invasiveness (sseK and tcf) (Tasmin et al., 2017) of this ST, and may precipitate insufficient intracellular survival (STM0030) (Zhang et al., 2019), poor host cell adhesion (lpfD) (Weening et al., 2005), and an inability to colonize cells (STM0036 and STM4433) (Carnell et al., 2007).
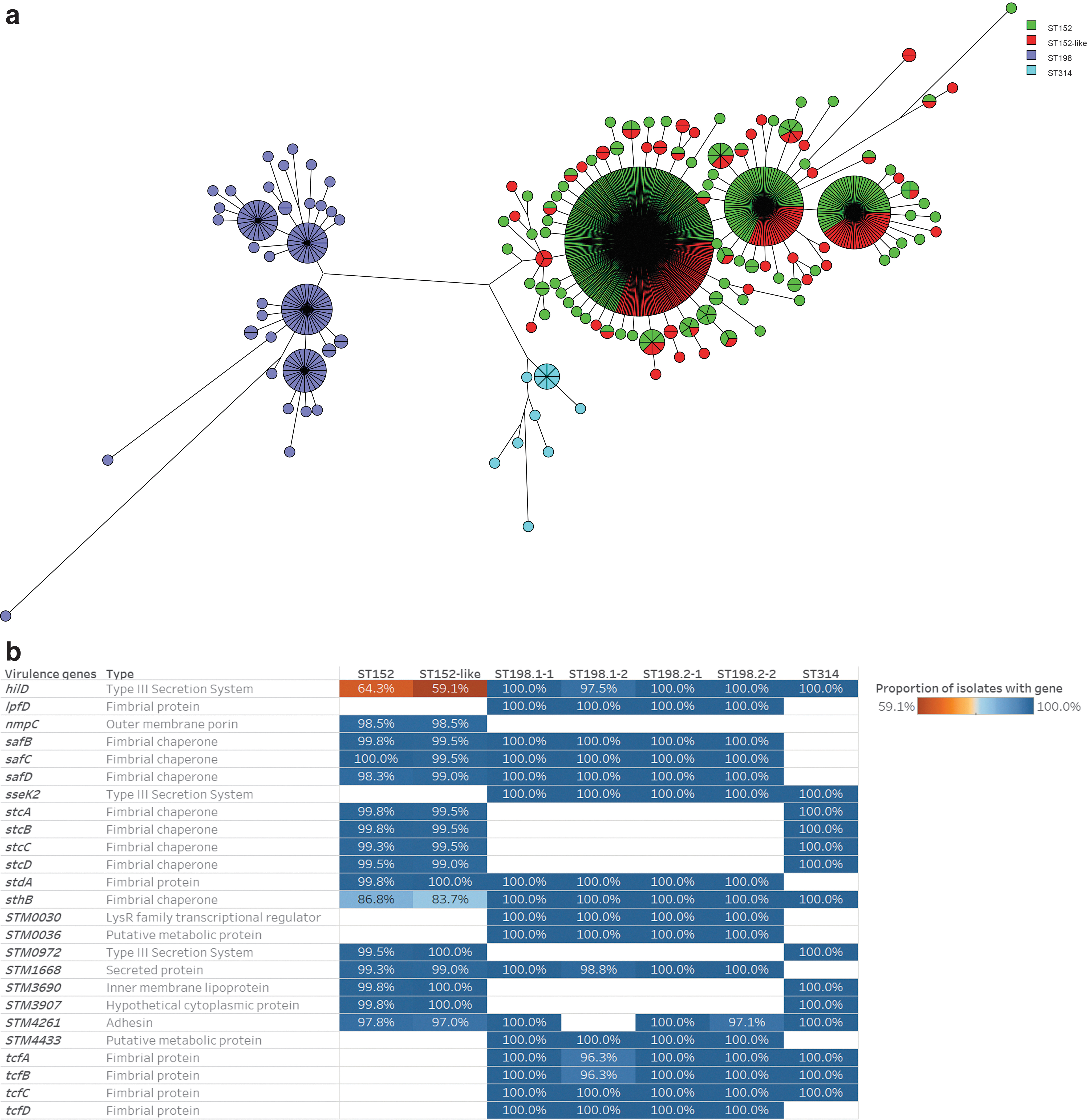
Genes found in ST152/152-like isolates but not ST198 isolates were STM3907, STM3690, the stc operon, nmpC, and STM0972 (Fig. 5b). STM3907 is a hypothetical cytoplasmic protein with no known function currently. STM3690 (also named SadB) is an inner membrane lipoprotein thought to perform a chaperone function for cell adhesion factors (Grin et al., 2014), and the stc operon is one of a number of fimbrial operons that may be involved in cell colonization. It is unclear why ST198 would lack genes that facilitate cell adhesion and colonization, but one possibility is that these genes could play a role in ST152 colonization of the chicken intestinal tract.
The nmpC gene (also named ompD) encodes an abundant outer-membrane porin that has been associated with antimicrobial uptake and susceptibility (Correia et al., 2016), and has been studied as a critical target for antibody response (Gil-Cruz et al., 2009; Domínguez-Medina et al., 2020). Future studies could investigate the immunomodulatory role ompD/nmpC might play in human clinical Salmonella Kentucky ST152 infection. Deletion of ompD/nmpC increases the ability of Salmonella Typhimurium to invade cultured macrophages and survive and replicate in target organs of a mouse model (Ipinza et al., 2014).
We hypothesize that the absence of ompD/nmpC from ST198 may also promote the pathogenicity of this ST. STM0972 (aka sopD2) has an antagonistic role in Salmonella virulence, inhibiting the formation of the Salmonella-containing vacuole required for Salmonella intracellular replication (Schroeder et al., 2010). This virulence-inhibiting function supports the observation that ST152 is found less frequently than ST198 in human clinical samples.
There were three genes present in both STs but to variable degrees (Fig. 5b). The hilD gene, a master regulator of genes that encode the type III secretion system 1 (T3SS1) invasion mechanism (Lou et al., 2019), was in 99% of ST198 (132/134) isolates but in only 64% (257/400) of ST152 and 59% (120/203) of ST152-like isolates. Among ST152, hilD was present in all isolates of clades 1, 2, and 3, and specific clusters nested within Clade 4. The absence of hilD can compromise infectivity in murine models (Chowdhury et al., 2021). Almost 97% of human isolates were hilD+ suggesting that this gene is linked to human infection. The sthB gene from the β-fimbriae cluster was also variable, appearing in all ST198 isolates but only 87% of ST152 isolates (specifically Clades 1, 2, and 3 and specific clusters in Clade 4), and 84% of ST152-like isolates.
Finally, STM4261 (siiE), a gene in Salmonella Pathogenicity Island-4 that encodes a large adhesin, was found in almost all ST152/152-like isolates but was present only in clades ST198.2 and ST198.1-1. Inactivation of this gene decreases infection in mice but does not change infectivity of calves (Morgan et al., 2004). The association between the absence of siiE and the presence of the point mutations in gyrA or parC may be related to genomic rearrangements that caused divergence of the CipR ST198 strain and should be investigated further.
ST314 appeared to be a hybrid of ST198 and ST152/152-like virulence profiles (Fig. 5a, b). Like ST198, almost all ST314 isolates contained hilD, sseK2, and tcfABC, but were missing nmpC. Similar to ST152, ST314 isolates contained the stcABCD operon, STM0972, STM3690, and STM3907, but were missing lpfD, nmpC, STM0030, STM0036, and STM4433. Unlike the other STs, ST314 was missing the gene for a hypothetical secreted protein (STM1668), tcfD, genes from the fimbrial chaperone saf operon and the fimbrial protein gene, stdA. The StdA protein plays a significant role in adhesion of Salmonella Enteritidis to the intestinal mucosa of poultry (Shippy et al., 2013). ST314 was not found in any domestic poultry sources in our study; however, other countries have found ST314 to be endemic in their poultry (Gu et al., 2020). A deeper investigation of the virulence profile of this ST is warranted.
It is worth noting that, although the ST152 group had isolates from humans, the ST152-like group did not. We did not see any differences in virulence gene distributions between these two related STs, suggesting that either the virulence divergency described in this study had no association with human pathogenicity, or more likely that other factors are involved. Pathogenicity of all nontyphoidal Salmonella is mediated by a cascade of virulence factors and the presence or absence of a few virulence genes might not impact disease. Because the activity of some factors has been shown to be host specific (Morgan et al., 2004), future studies could investigate whether some of these virulence genes function in colonization or invasion of the chicken gut but not the human gut and vice versa. Finally, it is important to understand that presence of the gene is not synonymous with expression.
The differential presence of virulence genes in the three STs may explain why there are so few human Salmonella Kentucky infections despite chickens being the predominant source of Salmonella Kentucky and a major cause of foodborne infection. Another possible explanation for the discrepancy in chicken and human isolates is that ST152 may cause low-grade human illness that quickly resolves and is less likely to be reported than ST198 infections, except when persons have pre-existing conditions or other immunodeficiency-related factors (such as age). Unfortunately, we did not have enough epidemiological data to investigate this.
One of the limitations of this study is that we did not include isolates from water, soil, or other animals such as companion or exotic animals. For instance, Salmonella Kentucky ST314 has been found in reptiles (Zając et al., 2013) and may be possible vectors for infection. Additionally, we did not include in our analysis sequences from environmental sources acquired outside of FDA or clinical sources outside of CDC NARMS. Comparing those sequences to the discarded STs that were found only in clinical or environmental sources may also provide clues to potential sources of human infection. Another thing to note is that the virulence and antimicrobial resistance determinants identified this study are reliant on the databases we used. Using another virulence or antimicrobial resistance database may have produced slightly different results. Finally, the limited patient data and exposure histories prevent us from making more firm conclusions on specific sources of ST198 and ST152 Salmonella Kentucky infections and the potential clinical impacts caused by the different virulence profiles.
Conclusion
Taken together, our findings indicate that most clinical infections of Salmonella Kentucky in the human cases we examined are ST198. The recent introduction of CipR ST198 strains in the human population in the United States likely results from consumption of foods that are imported or consumed while traveling. Almost 50% of human CipS ST198 isolates and 90% of human ST152 isolates clustered separately from ceca of domestic food animals and domestic meat and poultry products, which suggests that the majority of sporadic CipS ST198 and ST152 Salmonella Kentucky infections may have a source other than food products derived from domestic cattle, chicken, turkey, or swine. The presence of virulence genes in some STs and absence in others provides clues to the apparent pathogenicity of Salmonella Kentucky.
Footnotes
Acknowledgments
The authors would like to acknowledge the work of the state public health departments, NARMS retail meat surveillance sites, the FDA NARMS group, the CDC NARMS group, FSIS Public Health Veterinarians, FSIS NARMS group, and Regan Rickert for their work on the sample collection, isolation, characterization, and epidemiological investigation of the Salmonella Kentucky isolates.
Authors' Contributions
H.T.: conceptualization (lead), data curation, investigation, visualization (lead), original draft, review, and editing. C.-H.H.: conceptualization, data curation (lead), formal analysis, and software. J.C.: investigation. J.H.: visualization, review, and editing. S.F.: conceptualization, review, and editing. J.F.: conceptualization, data curation, review, and editing. L.K.F.W., J.R., G.E.T., and E.N.: conceptualization, resources, data curation, review, and editing. S.Z.: conceptualization, methodology, supervision, review, and editing.
Disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the official policy of the U.S. Department of Health and Human Services, the U.S. Food and Drug Administration, U.S. Department of Agriculture, or the U.S. government. Reference to any commercial materials, equipment, or process does not in any way constitute approval, endorsement, or recommendation by the Food and Drug Administration.
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.