
Abstract
To evaluate the in vitro anticancer activity and to investigate the mechanism of action of a thiophene heterocyclic compound, [3-Amino-5-[(2,6-dimethylphenyl)amino]-4-(phenylsulfonyl)-2-thienyl](4-fluorophenyl)methanone (APTM) against human colon cancer HCT116 cells. Sulforhodamine B assay and colony formation assay for cell proliferation assay; propidium iodide (PI) staining for cell cycle profile analysis; Hoechst staining; annexin V-FITC/PI double staining and Western blotting for apoptosis assay. APTM inhibits the growth of HCT116 cells dose and time dependently. The growth inhibitory effect of APTM on HCT116 cells was associated with induction of apoptosis but not cell cycle arrest. Also, the isogenic cell depletion of p53 was resistant to APTM-induced apoptosis and thus grows relatively better than the wild-type cells. The anticancer effect of APTM resulted from p53-dependent induction of apoptosis. Also, APTM is a promising lead compound for the treatment of human colon cancer.
Introduction
C
Chemotherapy is needed for stage III and more advanced diseases, including 5-fluorouracil (5-FU), capecitabine, irinotecan, oxaliplatin, and aflibercept (Van Cutsem et al., 2012), either used alone or in combination. However, 5-FU, the first-line therapy for colon cancer, only produces a response rate of 11% and a median survival time of 0.97 years when used alone (Advanced-Colorectal-Cancer-Meta-analysis-Project, 1992). Combination therapies can only increase the median survival time to 15–20 months (Giacchetti et al., 2000; Bouzid et al., 2003; Seymour et al., 2007). The adverse effects and treatment resistance further limit the benefits that patients could receive from the current therapeutic strategies (Crea et al., 2011). Thus, colon cancer remains one of the leading causes of cancer deaths, and the development of cancer-preventive and cancer therapeutic agents continues to be an area of considerable interest.
To screen for compounds with antiproliferative activities against colon cancer cells, we built up a library containing ∼2000 compounds, which were either synthesized or naturally isolated. The growth effect of these compounds on human colon cancer cell line HCT116 was measured by sulforhodamine B (SRB) colorimetric assay. A thiophene heterocyclic compound, [3-Amino-5-[(2,6-dimethylphenyl)amino]-4-(phenylsulfonyl)-2-thienyl](4-fluorophenyl)methanone (APTM, CAS No. 890817-75-1, Fig. 1A), drew our attention to its relatively high activity and structural rarity. The compound was first synthesized by Vlasenko et al. (2005), but has not been reported with any bioactivities yet. Similar compounds containing this thiophene heterocyclic structure have been shown to display diverse pharmacological activities, such as antibacterial (Hamama et al., 2012), hypolipidemic (Grashchenkova et al., 2007), antiprotozoan (Vlasenko et al., 2005), and antioxidant (Fadda et al., 2012; Mabkhot et al., 2013).
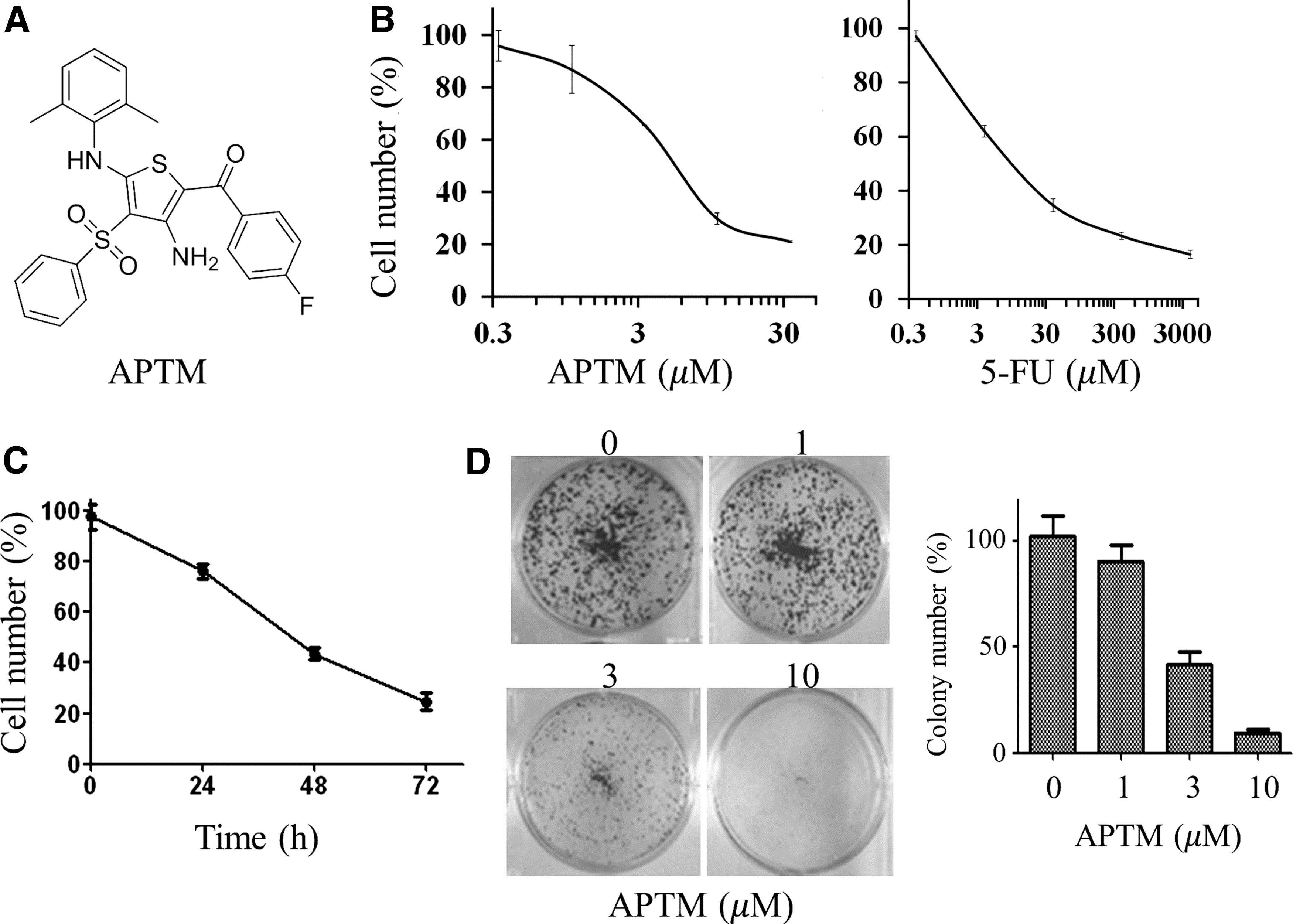
APTM inhibits proliferation of HCT116 cells.
In this study, we systematically evaluated the in vitro effect of APTM on the proliferation of human colon cancer HCT116 cells and proposed that the growth inhibition effect of APTM comes from its ability to induce apoptosis. By using the p53-deficient HCT116 cells, we found that induction of apoptosis by APTM is p53 dependent. Our work suggests that APTM is a promising lead compound against colon cancer, which deserves further study.
Materials and Methods
Chemicals and drugs
APTM was customer synthesized by Topscience Limited Liability Company, and was dissolved in dimethylsulfoxide and stored at −40°C until use. SRB, trichloroacetic acid (TCA), 5-FU, crystal violet, and Hoechst 33258 were obtained from Sigma Aldrich. McCoy's 5A (modified) medium, fetal bovine serum (FBS), TrypLE™ Express enzyme, and penicillin/streptomycin (10,000 U/mL) were purchased from Gibco. The Annexin V: FITC Apoptosis Detection Kit I was purchased from BD Biosciences, and the cell cycle detection kit was purchased from Nanjing KeyGen Biotech. The primary antibodies for cleaved nuclear poly (ADP-ribose) polymerase (cPARP), p53, Bcl-2, and Bax were purchased from Cell Signaling, Akt, pAkt, ERK, and pERK were from Cell Signaling Technology, and p73, Bid, and Bim were purchased from Abcam. β-Actin was purchased from Sigma. Horseradish peroxidase-conjugated secondary antibodies were purchased from Jackson ImmunoResearch, Inc.
Cell lines and culture
Human colon cancer cell line HCT116 (p53 +/+) was purchased from ATCC and the p53 −/− HCT116 cell line was provided by Bert Vogelstein, Johns Hopkins University. Cells were cultured in McCoy's 5A medium supplemented with 10% FBS, 100 U/mL penicillin G, and 100 μg/mL streptomycin in humidified atmosphere with 5% CO2. Cells were passaged twice weekly to maintain logarithmic growth.
In vitro cell proliferation assay (SRB assay)
The antiproliferative effects of APTM on cancer cell lines were assessed by SRB colorimetric assay as previously described (Lin et al., 2017). Briefly, cells were seeded in 96-well plates at densities of 5000 cells per well and cultured overnight. Then, cells were susceptible to APTM at indicated concentrations and cultured for indicated times. After incubation, attached cells were fixed with 50 μL cold 50% (w/v) TCA for 1 h at 4°C, washed five times with slow-running tap water, and stained with 100 μL 0.4% (w/v) SRB. Optical density at 515 nm (OD515) was measured using a microplate reader (Molecular Devices) after mixing the protein-bound dye with 200 μL of 10 mM Tris base solution (pH 10.5). The relative cell growth rate was determined with the following equation: Relative Growth (%) = OD (treated)/OD (control). The IC50 value was defined as the concentration required for a 50% reduction in cell growth.
Colony formation assay
HCT116 cells were plated at 3000 cells per well in six-well plates and cultured with indicated concentrations of APTM for 8 days. Cells were stained with 0.2% (w/v) crystal violet in buffered formalin for 20 min, and colonies were then photographed and quantified as previously described (Li et al., 2016).
Analysis of cell cycle by flow cytometry
Cell cycle distribution was determined by staining DNA with propidium iodide (PI). Briefly, 1.0 × 106 cells were incubated with or without APTM for 48 h. Cells were washed with cold phosphate-buffered saline (PBS) and fixed in 70% ethanol at −20°C for 2 h. After washing with PBS, cells were stained with cold PI solution (20 μg/mL PI and 200 μg/mL RNase in PBS) for 30 min at room temperature in the dark. The percentage of cells in different phases of the cell cycle was determined by a flow cytometer (BD Bioscience) and analyzed using FlowJo software.
Analysis of apoptosis by flow cytometry
The annexin V-FITC/PI double staining method was used for the apoptosis assay in HCT116 p53 +/+ and HCT116 p53 −/− cells (1 × 106/well, six-well plate). After 48 h of treatment with APTM, cells were harvested in 15-mL centrifuge tubes by gentle scraping followed by centrifugation at 300 g for 5 min at room temperature. Cell suspension was washed two times with cold PBS by centrifugation at 300 g for 5 min at room temperature. Then, cells were harvested, washed twice with cold PBS, and resuspended in 1× binding buffer (100 μL). Cells were transferred into 1.5-mL microcentrifuge tubes and stained with PI (5 μL) and FITC-annexin V (5 μL). Cells were briefly vortexed after incubation for 15 min in the dark at room temperature. Then, cells were filtered and analyzed by flow cytometry. Total apoptotic cells (FITC-annexin V positive) were counted.
Assessment of cell morphological changes
Cells were plated in six-well plates (200,000 cells/well) and treated with indicated concentrations of APTM. After incubation for 48 h, cells were collected, washed with PBS, and stained with Hoechst 33258 (11.1 g/mL) in buffered formalin solution containing 5.6% NP-40. Apoptotic and living cells were viewed through DAPI filter of fluorescence inverted microscope (Leica DM2500 Fluorescence Microscope) at 400 × magnifications.
Western blotting
HCT116 cells were treated with APTM at indicated concentrations for 48 h and harvested via trypsinization. Protein samples were prepared by scratching cells in RIPA buffer containing protease inhibitor cocktail (Roche) and diluted in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) protein sample buffer. Samples were heated for 5 min at 100°C. Protein concentrations were measured using the Direct Detect® Infrared Spectrometer (Millipore) according to the manufacturer's instructions. Equal amount of proteins were loaded on 4–20% SDS-PAGE gel. After electrophoresis, gels were transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore) and incubated with primary antibodies overnight at 4°C. The membranes were then washed with tris buffered saline with tween (TBST) and incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibodies (1:10,000; Santa Cruz, CA) for 45 min at room temperature. Proteins were visualized with SuperSignal West Dura Extended Duration Substrate or SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) using the Amersham Imager 600 Western blotting system. Densitometry analysis of proteins of interest was done by Image Studio Lite software (LiCor Biosciences).
Results
APTM inhibits proliferation of HCT116 cells
The proliferative effect of APTM on human colon cancer cell line HCT116 was examined using SRB assay. HCT116 cells were treated with a series of APTM concentrations from 0.33 to 33.3 μM for 48 h. As depicted in Figure 1B, APTM reduced cell viability of HCT116 cells in a concentration-dependent manner, with IC50 value of 6.57 μM, which is equivalent to 5-FU (IC50 = 9.12 μM), the first-line therapy for colon cancer treatment.
HCT116 cells were then treated with 10 μM of APTM and cytotoxicity was assessed at 24, 48, and 72 h after treatment. Consistent with the dose effect of APTM, the growth of HCT116 cells was also inhibited in a time-dependent manner (Fig. 1C). After treatment for 24 h, the relative growth rate of HCT116 cells was 76.36% and declined to 24.57% after 72 h. Thus, these results showed that APTM inhibited the growth of human colon cancer cells HCT116 both dose and time dependently.
This was further proved a long time by colony formation assay (Fig. 1D). The colony formation ability of HCT116 cells was significantly decreased after treatment with APTM at 3.0 μM and almost vanished at 10.0 μM.
These results suggest that APTM is potent in inhibiting the proliferation of HCT116 cells in vitro.
APTM does not inhibit cell cycle progression in HCT116 cells
Cell cycle arrest is the key intracellular event contributing to reduced cell proliferation, so we first checked whether APTM could alter cell cycle progression on colon cancer cells. Cell cycle distribution of HCT116 cells treated with APTM was analyzed by flow cytometry. As shown in Figure 2, the proportions of cells in G0/G1, S, and G2/M phases were almost unchanged after APTM treatment at different concentrations for 48 h. Therefore, our results suggest that the cause of reduced cell proliferation with APTM treatment is not due to the capture of cell cycle.
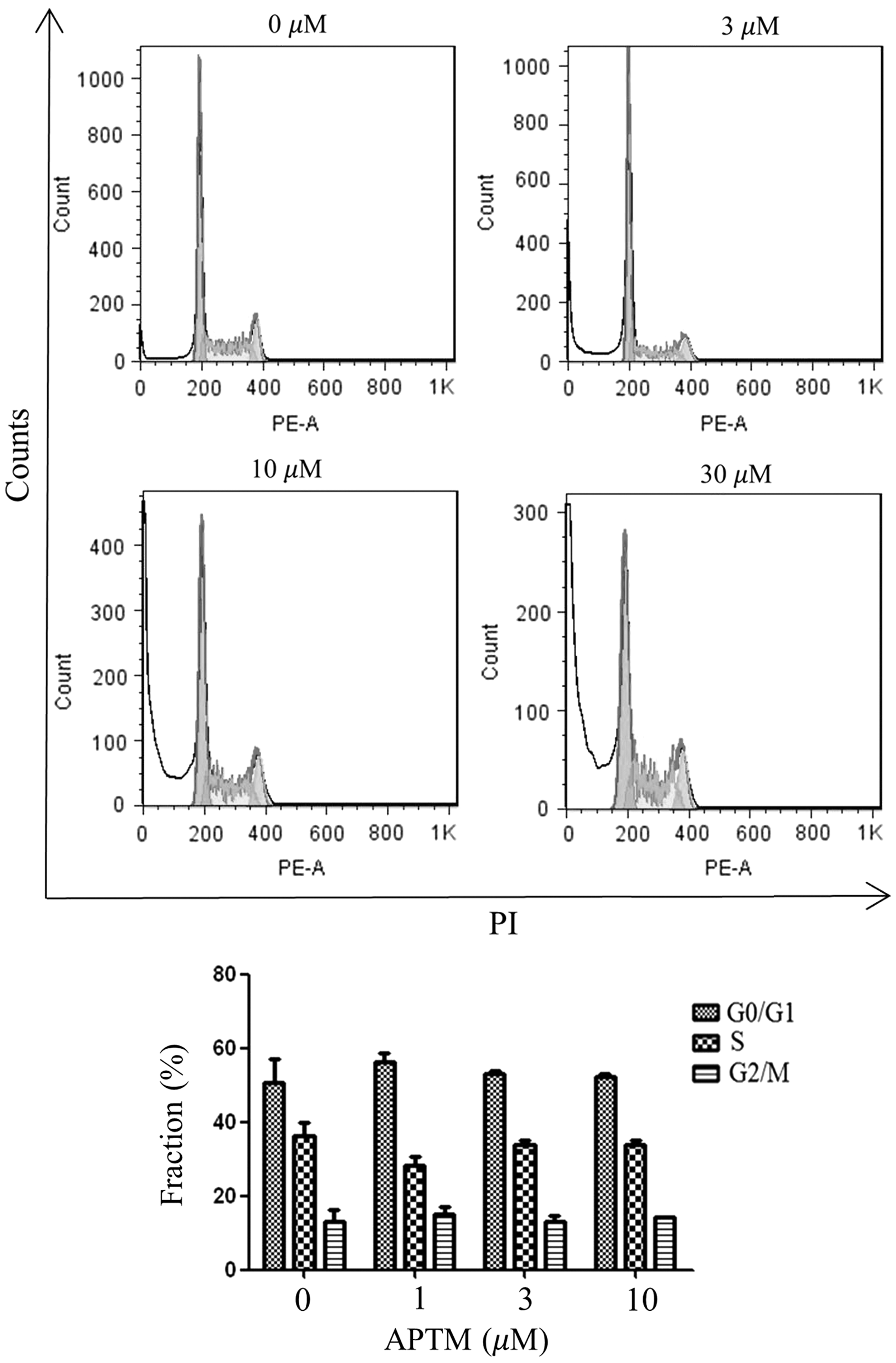
APTM does not induce cell cycle arrest in HCT116 cells. After treatment with APTM at indicated concentrations for 48 h, cell cycle distributions of HCT116 cells were analyzed by flow cytometry. Average values are from three independent experiments (n = 3). Data are shown as mean ± SD.
On the contrary, it is noticeably shown that the sub-G0/G1 fraction of cells was increased with APTM treatment, which is also dose dependent (Fig. 2). As cells in the sub-G0/G1 fraction represent apoptotic cells, we reasoned that APTM inhibits cell proliferation through induction of apoptosis.
APTM induces apoptosis in HCT116 cells
To further elucidate whether the growth inhibitory activity of APTM resulted from induction of apoptosis, HCT116 cells treated with 0, 3, 10, and 30 μM of APTM for 48 h were stained with Hoechst and visualized under a fluorescent microscope. As shown in Figure 3A, cells treated with APTM exhibit typical morphological characteristics of apoptosis, such as nuclear fragmentation and chromatin condensation.
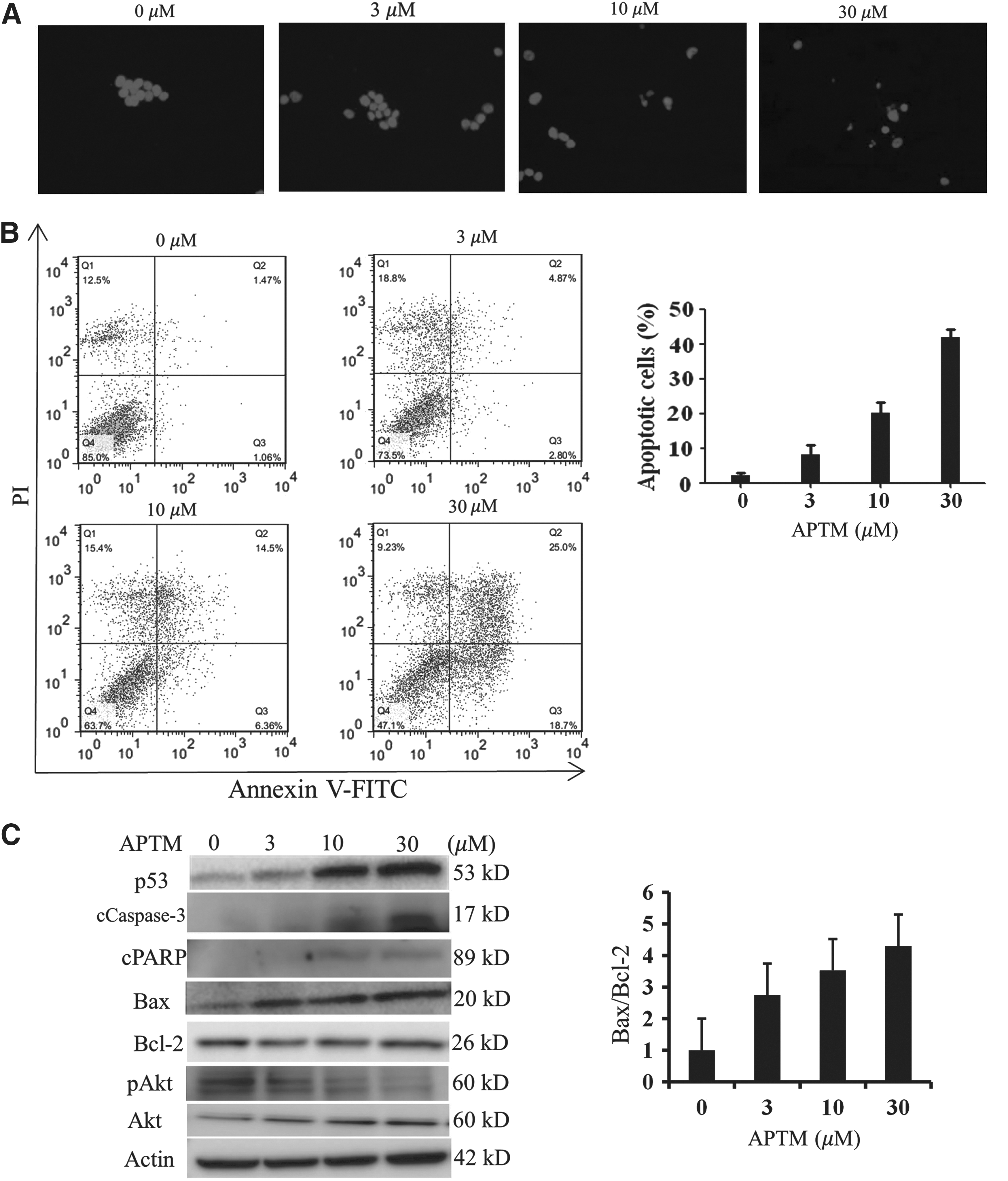
APTM induces apoptosis in HCT116 cells. HCT116 cells were treated with APTM at indicated concentrations for 48 h.
This was further confirmed by flow cytometry analysis of cells stained with annexin V-FITC/PI. APTM induced apoptosis of HCT116 cells in a dose-dependent manner. The percentages of apoptotic cells (annexin V positive) with APTM treatment for 48 h were 7.67% at 3 μM, 20.86% at 10 μM, and 43.7% at 30 μM, in comparison with 2.53% of the control (Fig. 3B).
Consistent with the induction of apoptosis, APTM treatment resulted in inhibition of phosphorylated Akt (Fig. 3C), one of the major pathways to promote growth and block apoptosis (Song et al., 2005).
The results were further demonstrated by the dose-dependent induction of cleaved caspase-3 (cCaspase-3) and cPARP with APTM treatment (Fig. 3C). Both proteins are well-known apoptosis markers (Porter and Janicke, 1999; Soldani and Scovassi, 2002).
We also found that Bax, the proapoptotic Bcl-2 family member, was induced by APTM treatment, while the antiapoptotic protein Bcl-2 remained unchanged (Fig. 3C). The ratio of Bax/Bcl-2 was dose dependently increased with APTM treatment (Fig. 3C), which is consistent with the notion that it is the ratio between pro- and antiapoptotic proteins rather than the level of any single Bcl-2 family member determining apoptosis (Vander Heiden and Thompson, 1999). On apoptotic stimuli, Bax undergoes conformational changes and oligomerizes at the mitochondrial outer membrane to promote its permeabilization and the releasing of cytochrome C and consequently activates caspase cascade (Pena-Blanco and Garcia-Saez, 2017).
Because Bax is a known target of p53 (Hemann and Lowe, 2006), a tumor suppressor mediating a variety of stress responses, we reasoned that APTM induces apoptosis through upregulation of p53 protein. Indeed, p53 protein was substantially induced by APTM treatment (Fig. 3C). p73, a member of the p53 family, is transcriptionally activated by p53 and is critical for p53-mediated apoptosis (Chen et al., 2001; Wang et al., 2007). However, p73 was not obviously induced on APTM treatment (data not shown).
Taken together, our data indicate that APTM induces apoptosis in human colon cancer HCT116 cells through activation of the p53/Bax/cPARP signaling axis.
Depletion of p53 attenuates APTM effect on apoptosis and cell proliferation
To test whether the effect of APTM on apoptosis induction and cell proliferation inhibition is dependent on p53, we used the isogenic HCT116 cell line lacking p53 (HCT116 p53 −/−) for the rest of our studies. Comparing with the parental wild-type cells (HCT116 p53 +/+), which showed apoptotic morphological changes with APTM treatment (Fig. 3A), HCT116 p53 −/− cells reserved normal nuclear staining even at the highest concentration (30 μM) tested in the study (Fig. 4A). Consistent with these results, HCT116 p53 −/− cells were resistant to APTM-induced apoptosis as analyzed by flow cytometry with annexin V-FITC/PI staining (Fig. 4B). Thus, we can conclude that p53 protein is essential for APTM-induced apoptosis in human colon cancer HCT116 cells.
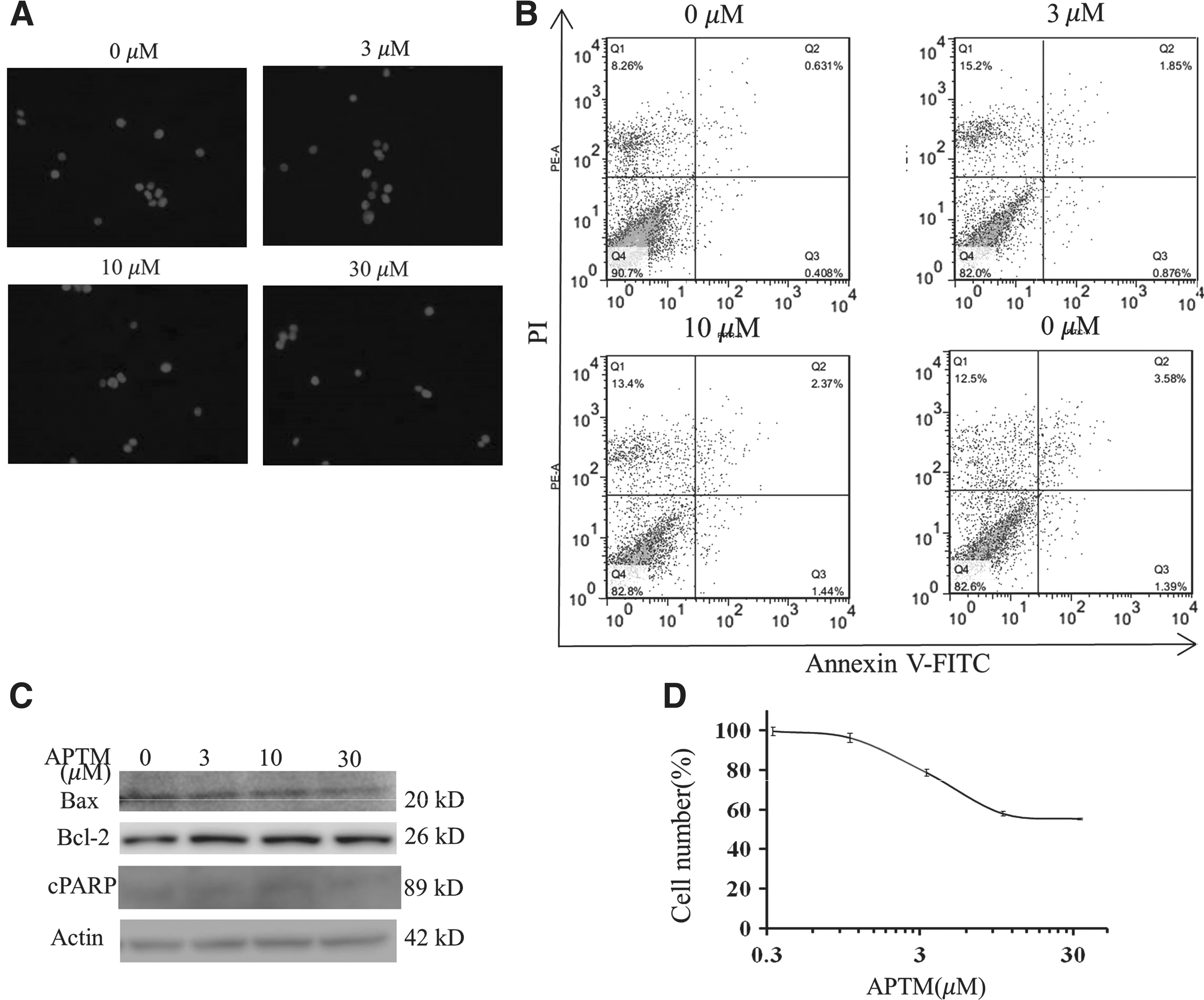
Depletion of p53 attenuates APTM effect on apoptosis and cell proliferation. HCT116 p53
−/− cells were treated with APTM at indicated concentrations for 48 h.
This was subsequently verified by Western blotting analysis, as loss of p53 blunted Bax expression and kept the Bax/Bcl-2 ratio constant at different APTM concentrations (Fig. 4C). cPARP was not induced and cCaspase-3 could not be detected with APTM treatment, indicating loss of apoptotic response with loss of p53 protein.
Finally, we tested the antiproliferative activity of APTM on HCT116 p53 −/− cells using SRB assay. As shown in Figure 4D, the IC50 value of APTM on HCT116 p53 −/− cells was greater than 33.3 μM, the highest concentration tested in this study. Comparing with the value of 6.57 μM on HCT116 p53 +/+ cells (Fig. 1B), there was more than fivefold resistance to APTM from the p53-deficient cells.
These results indicate that the apoptosis induction and growth inhibition activity of APTM are p53 dependent.
Discussion
APTM is a thiophene heterocyclic compound that has not been reported with any biological activity yet. The thiophene skeleton of APTM is widely used as a building block in many drugs with various pharmacological activities (Mishra et al., 2011; Gramec et al., 2014). Thiophene derivatives exhibit a broad spectrum of anticancer activities (Akatsuka et al., 2016; Abdallah et al., 2017; Mohareb et al., 2017; Unver and Canturk, 2017).
In this study, for the first time, we reported the antiproliferative effect of APTM on colon cancer HCT116 cells with an IC50 value of 6.57 μM. The growth inhibitory effect of APTM on colon cancer cells was confirmed by time course experiment and colony formation assay. Our data indicate that APTM is a promising lead compound for colon cancer treatment. However, before reaching this goal, more work is still needed, such as the in vitro activities tested in more (colon) cancer cell lines, evaluation of the in vivo anticancer activity in xenograft animal models or genetic colon cancer animal models, as well as the structure–activity relationship (SAR) study to improve the anticancer activity of APTM.
Under physiologic conditions, p53 is negatively regulated by E3 ubiquitin ligase MDM2 and maintained at low levels (Haupt et al., 1997). While in response to a wide range of stress stimuli, such as DNA damage, oxidative stress, nutrient deprivation, oncogene expression, and hypoxia, the interaction between p53 and MDM2 is perturbed and p53 protein is stabilized (Kruse and Gu, 2009). Once stabilized, p53 stands as the “guardian of the genome” through induction of apoptosis, cell cycle arrest, DNA repair, and senescence (Kruiswijk et al., 2015; Meek David, 2015; Laptenko and Prives, 2017). Tremendous efforts have been devoted to developing small molecules to reactive wild-type p53 for cancer treatment (Nguyen et al., 2017).
In this study, we have shown that APTM could induce p53 protein dose dependently in HCT116 p53 +/+ cells (Fig. 3C). However, how p53 proteins get stabilized on APTM treatment remains elusive. It should be noted that we could not rule out the possibility that APTM affects cofactors or other proteins that can cooperate with p53 and alter its functions (Hoe et al., 2014). Future studies toward this question will find the direct target and clarify the mechanism of action of APTM.
One of the most remarkable functions of p53 is the induction of apoptosis. This occurs through the extrinsic death receptor pathway (Bennett et al., 1998), transcription-dependent intrinsic mitochondrial pathway (Amaral et al., 2010), or transcription-independent cytosolic pathway (Chipuk et al., 2004). The proapoptotic protein Bax could be activated by BH3-only proteins, such as Bid and Bim (Westphal et al., 2011). It could also be activated by cytosolic p53 directly, independent of its transcriptional activity (Chipuk et al., 2004). Since we could not observe obvious induction of Bid and Bim (data not shown), it is possible that Bax was activated by cytosolic p53 with APTM treatment.
Furthermore, Bax was induced with increased p53 protein following APTM treatment (Fig. 3C), and the induction of Bax was diminished in HCT116 p53 −/− cells (Fig. 4C), which is consistent with the fact that Bax is a transcriptional target of p53 (Hemann and Lowe, 2006).
Thus, it is reasoned that APTM promotes apoptosis through both transcription-dependent and transcription-independent mitochondrial pathway of p53. That is to say, p53 protein stabilized on APTM treatment, increases Bax expression transcriptionally, and promotes its oligomerization and mitochondrial translocation. Bax oligomers insert into the mitochondrial outer membrane and promote the releasing of cytochrome C, leading to caspase activation and apoptosis.
However, there is a complex interaction between p53 and Bax in response to different anticancer agent-induced apoptosis (Zhang et al., 2000). To find a more precise mechanism of action of APTM, it is vital to compare apoptosis in HCT116 wild-type cells, p53 −/− cells, and Bax −/− cells.
On the contrary, p53 induction by APTM did not markedly change cell cycle distribution of HCT116 p53+/+ cells (Fig. 2). This selective induction of apoptosis but not cell cycle arrest may be because of high levels of p53 following APTM treatment. It was reported that higher levels of p53 promote apoptosis, whereas lower levels lead to cell cycle arrest (Chen et al., 1996; Kracikova et al., 2013).
Conclusions
In summary, we have shown that APTM, a thiophene heterocyclic compound, inhibits human colon cancer cell proliferation in vitro. Therefore, APTM is a promising lead compound for the development of new anticancer reagents for colon cancer. We have further shown that the growth inhibitory effect of APTM was due to the induction of apoptosis but not cell cycle arrest. Furthermore, the apoptosis induction activity of APTM is p53 dependent. Future studies to evaluate the in vivo anticancer activities, to explore the SAR, and to investigate the direct target of APTM will speed up the development of APTM as a novel anticancer reagent.
Footnotes
Acknowledgments
The p53 disrupted HCT116 cell line was a generous gift from Dr. Bert Vogelstein, Johns Hopkins University. This work was supported by grants from the Science and Technology Development Fund (FDCT) of Macau (Project codes: 034/2015/A1 and 005/2014/AMJ).
Disclosure Statement
No competing financial interests exist.