
Abstract
Background:
Ultra rapid lispro (URLi) is a new insulin lispro formulation that has accelerated absorption and improved postprandial glucose control compared with insulin lispro (Humalog®). The compatibility and safety of URLi versus lispro were evaluated in patients with type 1 diabetes using continuous subcutaneous insulin infusion (insulin pump).
Methods:
In this phase 3, double-blind, crossover study, 49 patients were randomized to two 6-week treatment periods, after a 2-week lead-in period on lispro. The primary endpoint was the rate of infusion set failures due to a pump occlusion alarm, or unexplained hyperglycemia with blood glucose >13.9 mmol/L (250 mg/dL) that did not decrease within 1 h after a correction bolus.
Results:
There was no significant difference in the rate of infusion set failures between URLi and lispro (0.03 vs. 0.05 events/30 days, P = 0.375). A higher rate of premature infusion set changes was observed with URLi (1.13 vs. 0.78 events/30 days; P = 0.028), translating to one additional infusion set change approximately every 3 months. A trend toward improved glycemic control was observed with URLi treatment: Time in range 3.9–10.0 mmol/L (71–180 mg/dL) was 65.7% ± 1.3% versus 63.0% ± 1.3%. Treatment-emergent adverse events (TEAEs) were reported by 46.9% of patients on URLi treatment and 18.8% on lispro. This difference was driven by an increase in infusion site reactions—more than 90% were mild. Incidence of all other TEAEs and severe hypoglycemia was similar between treatments.
Conclusions:
URLi was compatible with insulin pump use with a safety profile similar to lispro.
Introduction
Continuous subcutaneous insulin infusion (CSII, insulin pump) offers patients with diabetes an alternative to multiple daily injections (MDI) of insulin. 1 CSII continuously delivers small doses of the rapid-acting insulin throughout the day (basal) and variable amounts at mealtimes (bolus). Evidence suggests that CSII use results in similar or improved glycemic control when compared with MDI therapy. 2,3 However, achieving optimal glycemic control, particularly in the postprandial period, remains a challenge for patients with diabetes.
Currently, most insulin pumps are intended for use with rapid-acting insulin analogues such as insulin lispro (Humalog® or Admelog®), insulin aspart (Novolog®), and insulin glulisine (Apidra®). However, the time-action profile of these analogues may not be rapid enough to match carbohydrate absorption, therefore potentially compromising overall glycemic control. An ultra-rapid-acting insulin with pharmacokinetic and pharmacodynamic (PK/PD) profiles of faster onset and shorter duration of action would more closely match endogenous prandial insulin secretion and may allow better postprandial glucose (PPG) control. 4
Ultra rapid lispro (URLi; LY900014) is a new ultra-rapid formulation of insulin lispro that utilizes two locally acting excipients, citrate and treprostinil, to accelerate insulin lispro absorption and the insulin time-action profile. Citrate increases vascular permeability at the site of injection or infusion whereas treprostinil causes increased local vasodilation, with no measurable systemic exposure. In PK/PD studies, URLi has consistently demonstrated accelerated absorption and faster onset of action with reduced PPG levels when compared with lispro in patients with type 1 or type 2 diabetes. 5 –8 With euglycemic clamps, URLi showed faster absorption, more rapid onset, and shorter duration of action compared with lispro. 5,6 The onset of appearance of insulin lispro in serum was at least 5 min faster with URLi, leading to a 6- to 7-fold higher insulin exposure in the first 15 min after injection. 5,6 In addition, URLi demonstrated improved PPG control when administered as a single subcutaneous dose immediately before a 5-h mixed meal tolerance test: The PPG excursion was reduced by 1.5 mmol/L (27.0 mg/dL) at 1 h (P < 0.001) and by 1.2 mmol/L (21.6 mg/dL) at 2 h (P = 0.042) with URLi compared with lispro. 7 The accelerated absorption and trend toward improved PPG control was also demonstrated when URLi was administered via insulin pump in adults with type 1 diabetes. 8 The area under the curve (AUC) for the first 15 min was >50% higher with URLi compared with lispro after bolus administration on days 1 and 3 after infusion set catheter insertion with a 45% and 47% reduction in the 1-h PPG excursion during standardized meals on days 1 and 3, respectively. 8
Given its potential for improved PPG control, URLi is an attractive candidate for insulin pump therapy. However, it is important to evaluate the physicochemical properties of a new insulin formulation that may affect its stability in an insulin pump, resulting in problems such as pump occlusions. 9 This could lead to hyperglycemia, diabetic ketoacidosis (DKA), and other complications. Therefore, the aim of this study was to evaluate the compatibility, safety, and short-term efficacy of URLi and lispro with CSII in adults with type 1 diabetes. The primary objective was to compare the rate of infusion set failures that led to premature infusion set changes, due to a pump occlusion alarm or due to unexplained hyperglycemia with blood glucose (self-monitored blood glucose [SMBG]) >13.9 mmol/L (250 mg/dL) that did not decrease within 1 h after a correction bolus delivered via the pump.
Research Design and Methods
Study design
This phase 3 trial was a randomized, double-blind, two-treatment, crossover, active-controlled study in adults with type 1 diabetes on CSII therapy. The study included a screening (1 week) and lead-in period (2 weeks) before randomization, followed by two 6-week treatment periods and a 4-week safety follow-up (Supplementary Fig. S1).
The study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonization guidelines for Good Clinical Practice, and applicable laws and regulations in the respective countries. All procedures were approved by an independent ethics committee and all patients provided written, informed consent before participating in the study.
Participants
Adults (≥18 years) diagnosed with type 1 diabetes (based on the World Health Organization classification) for ≥1 year and receiving insulin by CSII were eligible to participate in the study if they met the following criteria: body mass index (BMI) ≤35 kg/m2; hemoglobin A1c (HbA1c) ≤8.5% at screening; had been using CSII (MiniMed 530G, 630G or 640G) for ≥6 months and had been treated with the same rapid-acting insulin analogue ≥30 days before screening; and had been using continuous glucose monitoring (CGM) or flash glucose monitoring (FGM) for ≥60 days in the 12 months before screening.
Key exclusion criteria were: hypoglycemia unawareness; >1 episode of severe hypoglycemia within the preceding 6 months; >1 emergency room visit or hospitalization due to DKA within the preceding 6 months; significant lipohypertrophy, lipoatrophy, or scars within subcutaneous tissue in areas of infusion; an abscess at an infusion site within 90 days before screening; and regularly changing infusion sets more frequently than every 3 days in the preceding 6 months. Investigators at five study centers and two countries participated in the study.
Study procedures
All patients entering the study were switched from their previous rapid-acting insulin analogue to lispro (Humalog U100) at the beginning of the 2-week lead-in period. Patients used their personal insulin pump throughout the study but were required to use the reservoirs (MiniMed® Reservoir) and infusion sets (MiniMed® Mio®) provided by the study sponsor. They were not allowed to use personal CGM or FGM and the SmartGuard/Threshold Suspend feature of their pump, during the lead-in and treatment phases of the study as requested by the U.S. Food and Drug Administration (U.S. FDA).
After lead-in, patients were randomized in a 1:1 ratio to 1 of the 2 treatment sequences: Sequence A: 6 weeks of URLi and then 6 weeks of lispro Sequence B: 6 weeks of lispro and then 6 weeks of URLi
Assignment to treatment sequences was determined by a computer-generated random sequence using an interactive web-response system and randomization was stratified by country, historical use of SmartGuard/Threshold Suspend (yes/no), and HbA1c stratum (≤7.3%, >7.3% at screening). Patients filled their pump reservoirs from blinded vials with each infusion set change, and boluses were delivered at mealtime (0–2 min before meals) using a standard bolus delivery speed of 1.5 U/min. Patients, in consultation with the investigator, were encouraged to adjust basal rates and bolus calculator settings (insulin-to-carbohydrate ratio, insulin sensitivity factor, and active insulin time) during the first 4 weeks of each treatment period to achieve glycemic targets so that these settings were fairly stable during the last 2 weeks of each treatment period. Patients were instructed to use the bolus calculator (Bolus Wizard®) to determine all meal and correction bolus doses. Glycemic targets were as follows: pre-prandial, range 4.4–6.1 mmol/L (80 to <110 mg/dL); 1- to 2-h postprandial, <10.0 mmol/L (180 mg/dL); and bedtime, range 5.0 to 7.2 mmol/L (90 to <130 mg/dL) with no hypoglycemia (blood glucose ≤3.9 mmol/L [70 mg/dL]).
Infusion set changes
Patients were required to routinely change their infusion sets every 72 ± 4 h and record the date and time of all infusion set changes (routine or premature) and the reason for any premature infusion set changes. Premature infusion set changes could have been due to any of the following categories of reasons: infusion set leaks, kinks, or disconnection; an empty pump reservoir; pain or redness at the infusion site; pump occlusion alarm; unexplained hyperglycemia; and other reasons. Unexplained hyperglycemia was defined as high blood glucose that could not be explained by a missed prior bolus, dietary indiscretion, rebound or treatment of hypoglycemia, a pump failure, an empty pump reservoir, an infusion set complication (e.g., kinked, came out, leaking), or an infusion site complication (e.g., pain, redness). If the premature change was due to unexplained hyperglycemia, this was to be confirmed by SMBG using the study glucose meter (MyGlucoHealth® Blood Glucose meter; Entra Health Systems LLC, USA). Patients were to first check that the pump was in place, the prior bolus dose was delivered, and the infusion set was in place without any leaks or kinks in the system. If a correction bolus was administered, glucose response to the correction bolus was to be confirmed by SMBG within ∼1 h. All non-meal-related correction boluses and the respective date, time, and dose were to be recorded.
Continuous glucose monitoring
The DexCom Generation 5 Mobile Continuous Glucose Monitor (G5; DexCom, Inc., USA) was used by all patients in real-time mode beginning at the start of lead-in and continuing throughout the treatment phase of the study. Patients were instructed to change the G5 sensor every 7 days as per the product label. Patients received training on the use of the G5 system, including expectations for completion of the daily requirements such as: eating a similar breakfast each day to the extent possible; avoiding snacks within 2 h after breakfast unless necessary to treat hypoglycemia; recording the start of the breakfast meal and the time of the breakfast meal bolus into the G5 receiver; keeping the receiver within 6 m (20 feet) of the transmitter to minimize loss of transmitted data; calibrating the sensor every 12 h; and avoiding use of acetaminophen/paracetamol.
Self-monitored blood glucose
Patients were instructed to measure a minimum of four SMBG readings daily (fasting [pre-morning meal], pre-midday meal, pre-evening meal, and pre-bedtime), with additional readings as needed for glucose self-management. Site personnel could request additional SMBG from patients and/or assess SMBG values at other times for clinical decision making. SMBG values were required for mealtime bolus calculations, calibrating G5 (every 12 h), and for confirming episodes of hypoglycemia, unexplained hyperglycemia >13.9 mmol/L (250 mg/dL), and the glucose response to a correction bolus.
Hypoglycemic events were recorded throughout the clinical study along with date, time, blood glucose level, and hypoglycemia treatment and outcome data.
Safety
All adverse events (AEs) were recorded and assessed throughout the study. Patients were encouraged to perform SMBG whenever hypoglycemia was suspected and record the blood glucose value, any associated symptoms, and the treatment administered. Reports of hypoglycemia were classified by the investigator as “severe” or “not severe” based on data collected and in consultation with the patient. Severe hypoglycemia was defined as an episode requiring assistance of another person to actively administer treatment due to neurological impairment of the patient as confirmed by the investigator. All episodes of severe hypoglycemia were reported as serious adverse events (SAEs).
Assessments
The primary endpoint was the rate of infusion set failures that led to premature infusion set changes, due to a pump occlusion alarm or due to unexplained hyperglycemia with blood glucose (SMBG) >13.9 mmol/L (250 mg/dL) that did not decrease within 1 h after a correction bolus delivered via the pump. No specific criteria were used to define a minimum blood glucose decrease.
Secondary endpoints included the: incidence of infusion set failures as defined in the primary endpoint; rate and incidence of premature infusion set changes for any reason; time interval (overall and due to any suspected infusion set occlusion) until infusion set change during the treatment period; bolus, basal, and total insulin dose and bolus/total insulin ratio; interstitial glucose reduction from hyperglycemia (>10.0 mmol/L [180 mg/dL] and ≤13.9 mmol/L [250 mg/dL]) to recovery (<10.0 mmol/L [180 mg/dL]) after a non-meal-related correction bolus delivered via the pump; rate of severe hypoglycemia during the treatment period; duration of time that glucose values were within target range (3.9–10.0 mmol/L [71–180 mg/dL]), as well as below and above the target range based on CGM analysis; actual and change from baseline to week 6 in HbA1c; and actual and change from baseline to week 6 in 1,5-anhydroglucitol (1,5-AG).
Statistical analysis
The study was designed to assess the safety and compatibility of URLi and insulin lispro with CSII and included elements required by the U.S. FDA. This study was not powered to be a non-inferiority or superiority trial for any safety, CGM, or efficacy endpoints.
All analyses were conducted based on all the randomized patients. Analyses of AEs (including severe hypoglycemia) included all data collected during the entire 6-week treatment period for each treatment group regardless of investigational product (IP) use. Pump-related safety analyses (e.g., infusion set failures, interstitial glucose reduction rate) excluded data (if any) that were collected over the 6-week treatment period while patients were temporarily off pump or off IP.
The CGM-related analyses included all data collected while patients were on pump and on IP. For the interstitial glucose reduction rate calculated from CGM, treatment comparisons were based on the derived outcome variables for 0–6 weeks. For other CGM outcome variables, treatment comparisons were based on the derived outcome variables for 4–6 weeks, to minimize the potential carryover effect. Only CGM data collected on the days with at least 70% of the total CGM measures that were supposed to be obtained (i.e., 288 points for a 24-h period) were included in the derivation of the CGM outcome variables. Similarly, for the meal-related outcome variables such as the incremental AUC (iAUC), only meals with at least 70% of the total measures that were supposed to be obtained were included. Incremental AUC was calculated by applying the trapezoidal rule to both positive and negative glucose increments. The area of each small trapezoid was first calculated and then the iAUC was derived as the total sum of those individual areas. If any trapezoid fell below the starting glucose, a negative value was obtained for its area that was, in effect, subtracted from the area obtained from above the starting glucose (the average of the CGM values in the time window [−19, 0 min] relative to the start of the meal).
The interstitial glucose reduction rate within 4 h after a non-meal-related correction dose delivered via pump was calculated by dividing the interstitial glucose reduction by the time it took to recover (interstitial glucose ≤10.0 mmol/L [180 mg/dL]) from hyperglycemia after the correction dose. If within 4 h a second correction bolus was given before the recovery, only the first non-meal-related correction bolus time was used and the data after the second correction bolus were censored. If the glucose never recovered to ≤10.0 mmol/L (180 mg/dL) within 4 h, the glucose rate was calculated by dividing the greatest glucose reduction within 4 h by the time it took to reach this level. The interstitial glucose reduction rate (mmol/L/min) was analyzed by sub-intervals of interstitial glucose levels at the time of the non-meal-related correction bolus: >10.0 to ≤13.9, >13.9 to ≤16.7, and >16.7 mmol/L (>180 to ≤250, >250 to ≤300, and >300 mg/dL).
For event rate measures, the aggregated rate was calculated by using the total number of events divided by the cumulative days on treatment (and on pump, for pump-related events) from all patients within that treatment, then times 30 days (for rate per 30 days). Wilcoxon signed-rank test was used for analysis of event rate, and Prescott's exact test was used for event incidence. A restricted maximum likelihood-based, mixed-effect model repeated-measure analysis was used to analyze continuous longitudinal variables. The model included the fixed class effects of treatment, period, sequence, strata (region, historical use of SmartGuard/Threshold Suspend, HbA1c), and the continuous, fixed covariate of baseline value. For the analysis of variables that were measured at multiple designated visits within each randomized treatment period (e.g., weight), the model also included the fixed class effects of week and week-by-treatment interaction.
Treatment group comparisons were performed for the primary objective at the full significance level of 0.05. No multiplicity adjustment was made for secondary and exploratory objectives.
The glucose thresholds pre-specified for calculation of time in range parameters in this study were different from those recommended in newer consensus guidelines. 10 As a result, data were re-analyzed by using recent thresholds and these results are presented in Supplementary Table S1 where different.
Results
Forty-nine patients were randomized to the study, 24 on sequence A and 25 on sequence B. The mean age was 39.6 years, with BMI 26.9 kg/m2, baseline HbA1c 7.06%, and mean duration of CSII use 8.7 years (Table 1). Ninety-four percent (N = 46) of patients completed the study (Supplementary Table S2). Three patients discontinued the study during periods I and II: 2 during URLi treatment and 1 during lispro treatment (Supplementary Table S2). One patient discontinued after completing period II.
Baseline Characteristics
Data are mean (standard deviation) unless otherwise stated.
BMI, body mass index; CSII, continuous subcutaneous insulin infusion; HbA1c, hemoglobin A1c; n, number of patients in population.
Compatibility
Infusion set failures
There was no difference between URLi and lispro in the rate of infusion set failures, as defined in the primary endpoint during the treatment period (P = 0.375). The aggregated rate of infusion set failure was 0.03 events per 30 days with URLi and 0.05 events per 30 days with lispro treatment. Five events in five patients met the primary study endpoint, all due to pump occlusion alarm (Fig. 1) and occurring on day 1 (URLi treatment) or day 2 (lispro treatment) of infusion set wear. The incidence of infusion set failures was not significantly different between treatments: 2 (4.1%) patients on URLi treatment reported two events; 3 (6.3%) patients on lispro reported three events.
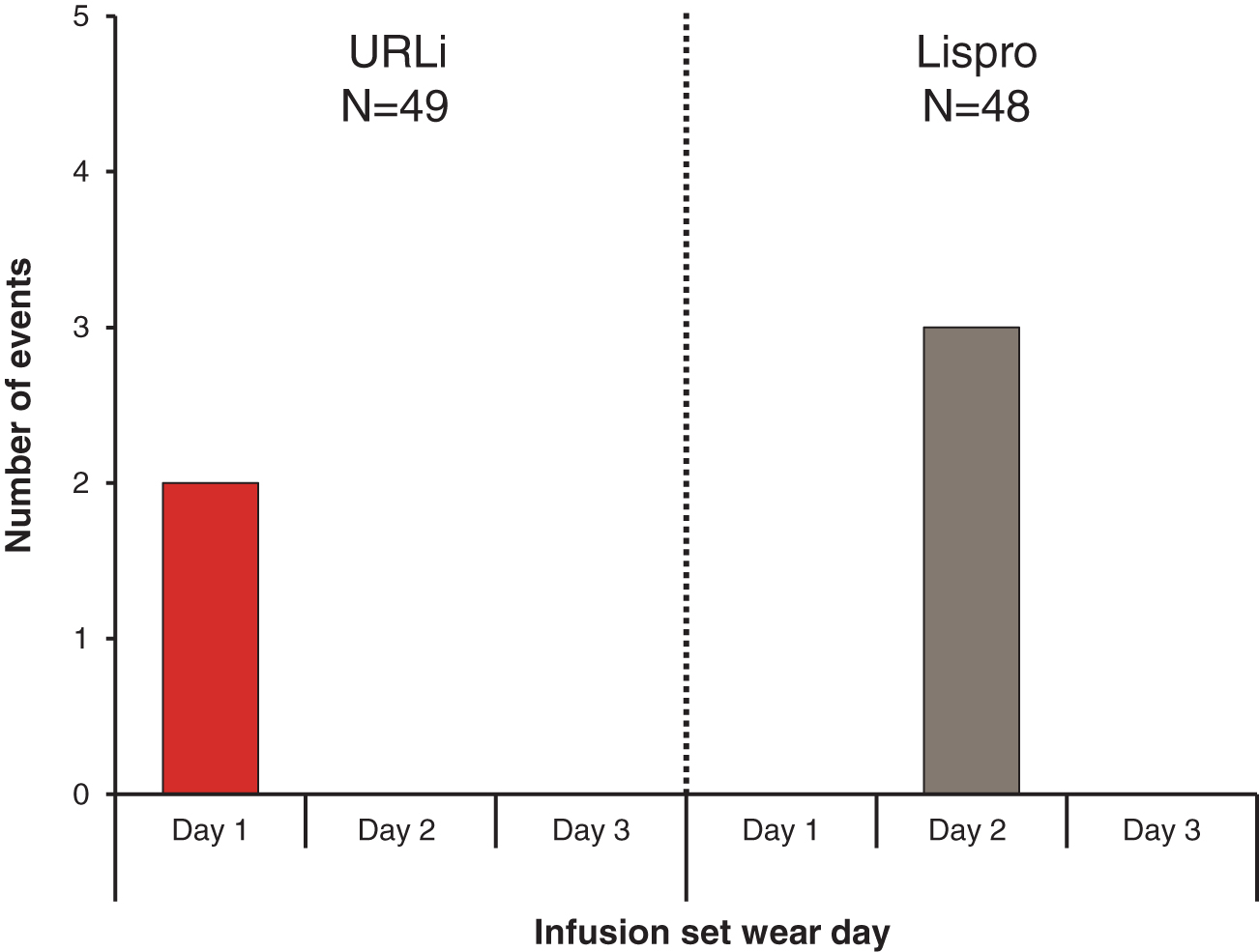
Number of infusion set failures by infusion set wear day. URLi, ultra-rapid lispro.
Premature infusion set changes: overall
The overall incidence of premature infusion set changes was not different between treatments: URLi, 32 (65.3%); lispro, 25 (52.1%); and P = 0.317. The rate of premature infusion set changes during the two treatment periods was higher for URLi (aggregated rate 1.13 vs. 0.78 events per 30 days; P = 0.028), but the difference was small, translating to 1 additional infusion set change with URLi approximately every 3 months, based on routine changes every 3 days. The mean time interval until infusion set change was similar between treatments: URLi, 74.3 h; lispro, 76.1 h.
Premature infusion set changes: by reason
The rate of premature infusion set changes categorized by reason was generally similar between URLi and lispro for all categories except for a higher rate of premature infusion set changes due to infusion site pain or redness with URLi treatment (0.32 vs. 0.02 events per 30 days; P = 0.001) (Supplementary Table S3).
The incidence of premature infusion set changes categorized by reason was not significantly different between treatments for all categories except for infusion site pain or redness (URLi, 12 [24.5%]; lispro, 1 [2.1%]; P = 0.003) and unexplained hyperglycemia (URLi, 15 [30.6%]; lispro, 8 [16.7%]; P = 0.026) (Supplementary Table S3). None of the events of unexplained hyperglycemia met the primary endpoint definition. There were no episodes of DKA. Similar rates of premature infusion set changes due to unexplained hyperglycemia were reported on days 1 and 3 of infusion set wear (0.10 and 0.12 events per 30 days) during URLi treatment. Overall, the rate of infusion set changes due to unexplained hyperglycemia was not different between treatments and was lower during the treatment period with both URLi (0.26 events per 30 days) and lispro (0.14 events per 30 days) compared with the open-label lead-in period on lispro (0.57 events per 30 days).
Efficacy
CGM assessments
A trend toward improved glycemic control was observed with URLi treatment during the daytime and 24-h period with increased time in target range (Fig. 2). The URLi treatment had significantly higher percent time in target range between 0–1 and 0–2 h after the start of breakfast (Fig. 2B). In addition, both the time spent in hypoglycemia (<2.8 mmol/L [50 mg/dL]) and time spent in hyperglycemia (>10.0 mmol/L [180 mg/dL]) were numerically lower with URLi treatment during daytime, nighttime, and the 24-h period, as well as at 0–1 and 0–2 h after the start of breakfast, with significantly less time in hyperglycemia at 0–1 and 0–2 h after breakfast (Figs. 3 and 4). Similar trends were observed with other glucose thresholds (time in hypoglycemia <3.3 mmol/L [60 mg/dL] and <3.9 mmol/L [70 mg/dL] and time in hyperglycemia >13.9 mmol/L [250 mg/dL] and >16.7 mmol/L [300 mg/dL]), although none reached statistical significance.
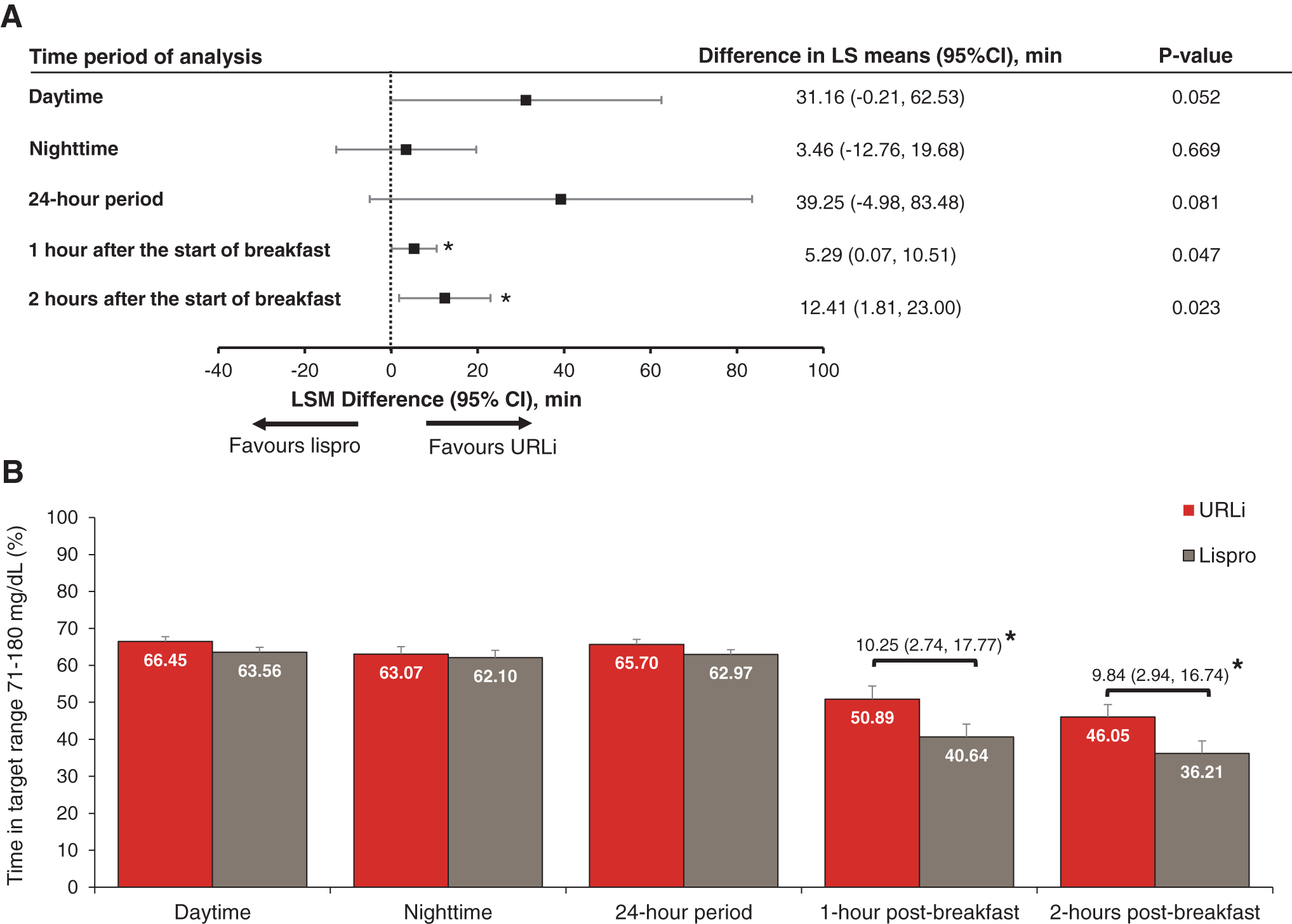
Forest plot of difference in mean time in target range 3.9–10.0 mmol/L (71–180 mg/dL) (URLi—lispro)
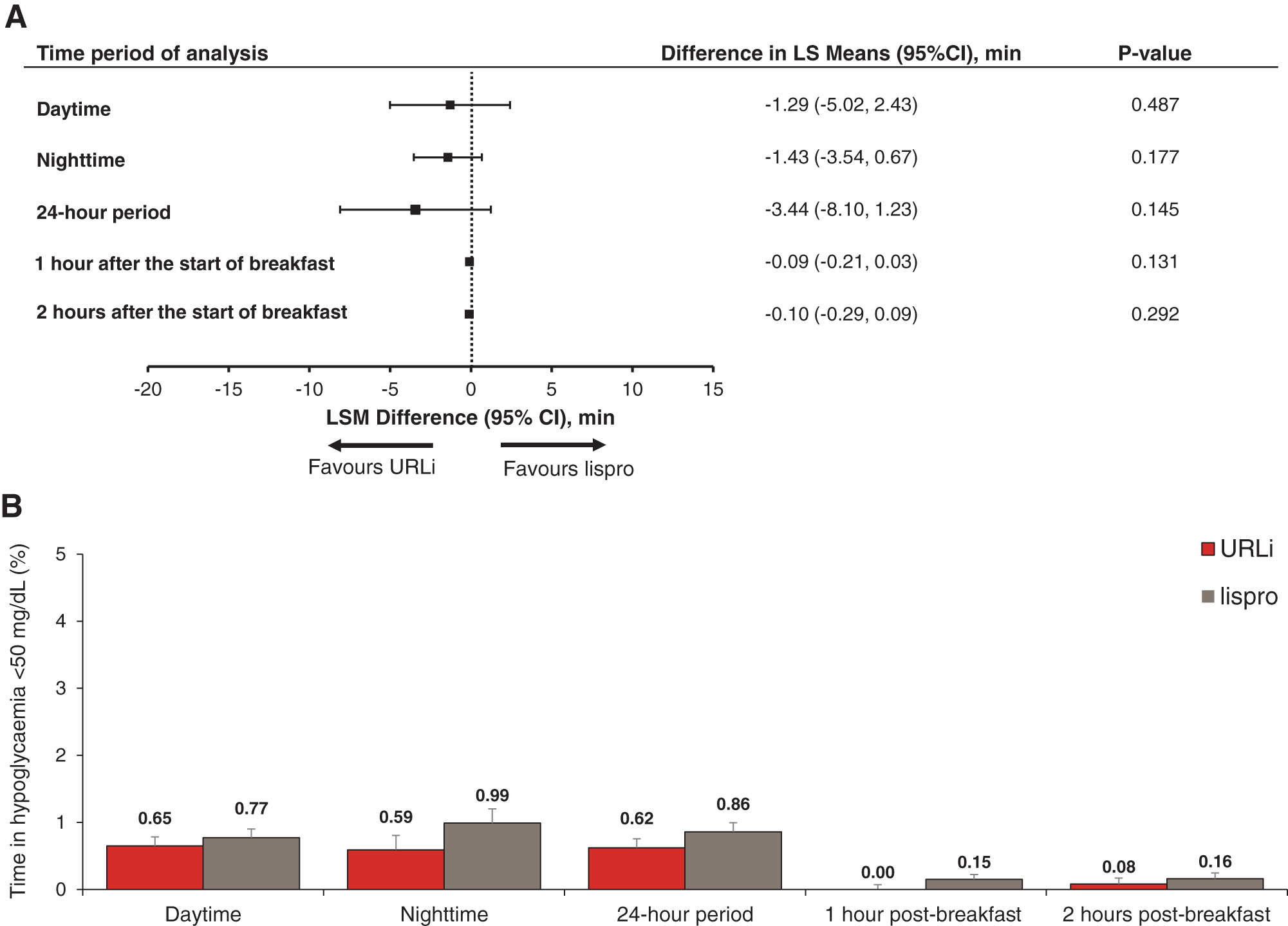
Forest plot of difference in mean time in hypoglycemia <2.8 mmol/L (50 mg/dL) (URLi—lispro)
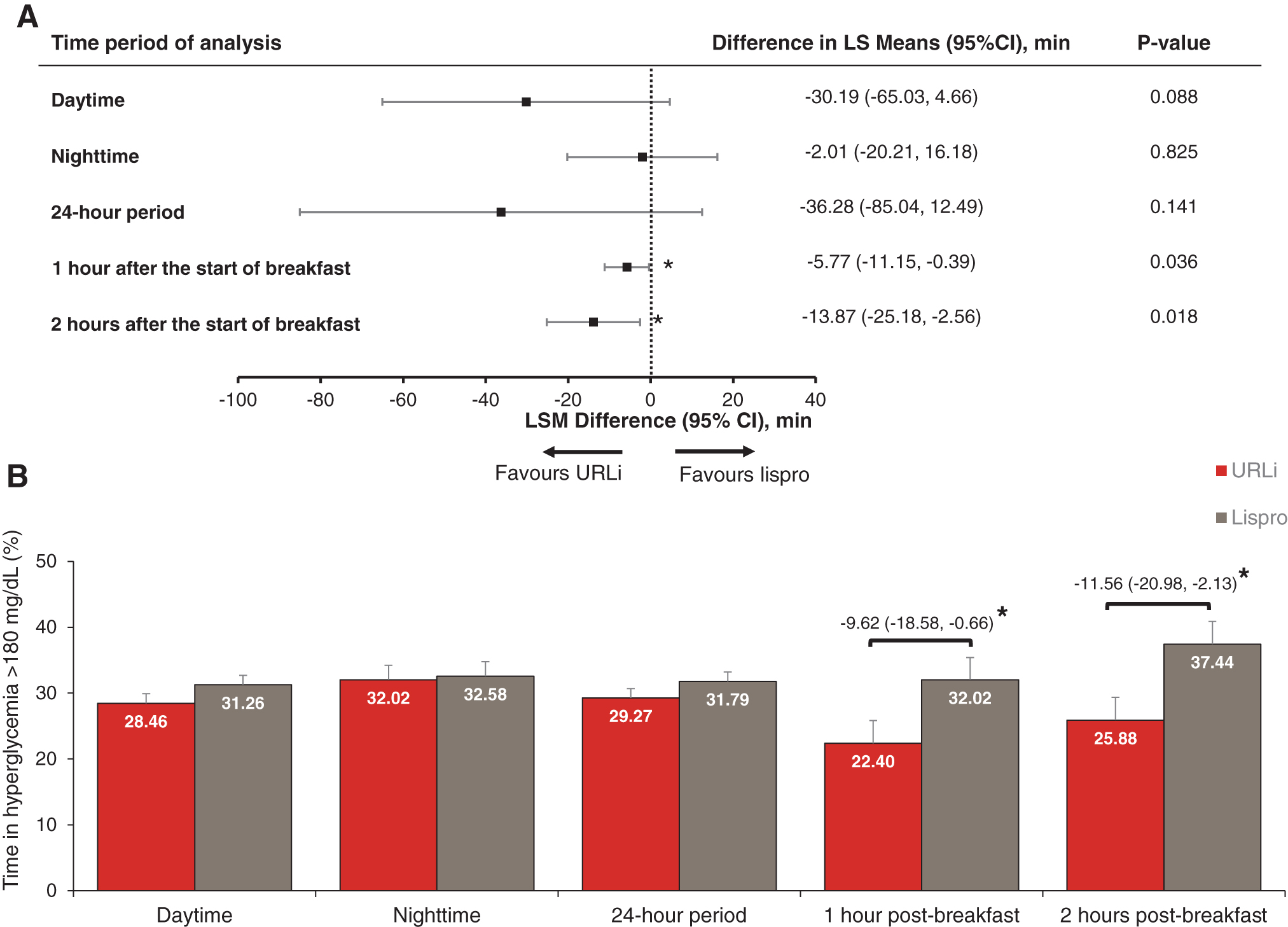
Forest plot of difference in mean time in hyperglycemia >10.0 mmol/L (180 mg/dL) (URLi—lispro)
Numerically lower iAUC after breakfast was observed with URLi treatment. However, this difference did not reach statistical significance at either 1 or 2 h after breakfast: least squares mean difference −5.19 mg·h/dL (−12.58, 2.19); P = 0.159 and −12.64 mg·h/dL (−33.43, 8.16); P = 0.222, respectively.
Overall, glucose control as measured by time above, below, and in range, and iAUC after breakfast was similar across all days (days 1, 2, and 3) of infusion set wear (Supplementary Fig. S2).
The interstitial glucose reduction rate from hyperglycemia (>10.0 mmol/L [180 mg/dL] and ≤13.9 mmol/L [250 mg/dL]) to recovery (≤10.0 mmol/L [180 mg/dL]) within 4 h after a non-meal-related correction bolus was significantly faster for URLi compared with lispro treatment: URLi, 0.03 mmol/L/min (0.62 mg/dL/min) vs. lispro 0.01 mmol/L/min (0.21 mg/dL/min); P = 0.049]. No treatment differences were seen for other hyperglycemic endpoints (>13.8 and ≤16.7 mmol/L [>250 and ≤300 mg/dL]; >16.7 mmol/L [300 mg/dL]).
Other efficacy endpoints
Both treatments showed significant reductions in HbA1c from baseline in period I (Fig. 5). However, the reduction was numerically greater for URLi compared with lispro (−0.39% [−4.2 mmol/mol] vs. −0.25% [−2.8 mmol/mol]). Minimal changes in HbA1c were observed for both treatments in period II.
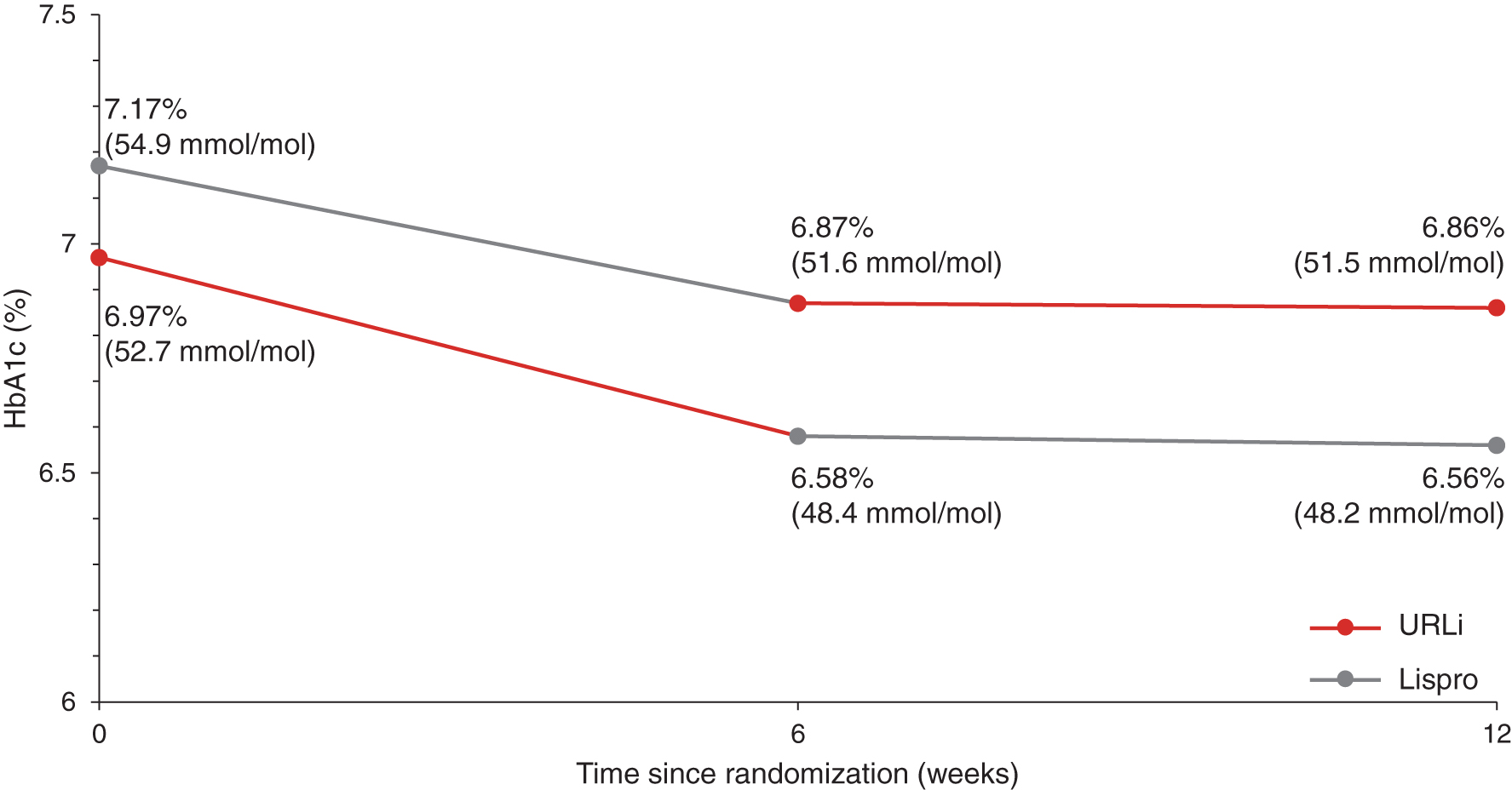
Mean HbA1c from baseline to week 12. HbA1c, hemoglobin A1c.
URLi showed a significant increase in 1,5-AG from baseline in period I (mean: 1.52 mg/L, 95% confidence interval: 0.69 mg/L, 2.35 mg/L) with non-significant change from baseline for lispro (mean: 0.46 mg/L [−0.02, 0.95]) (Fig. 6). Minimal changes in 1,5-AG were observed for both treatments in period II.
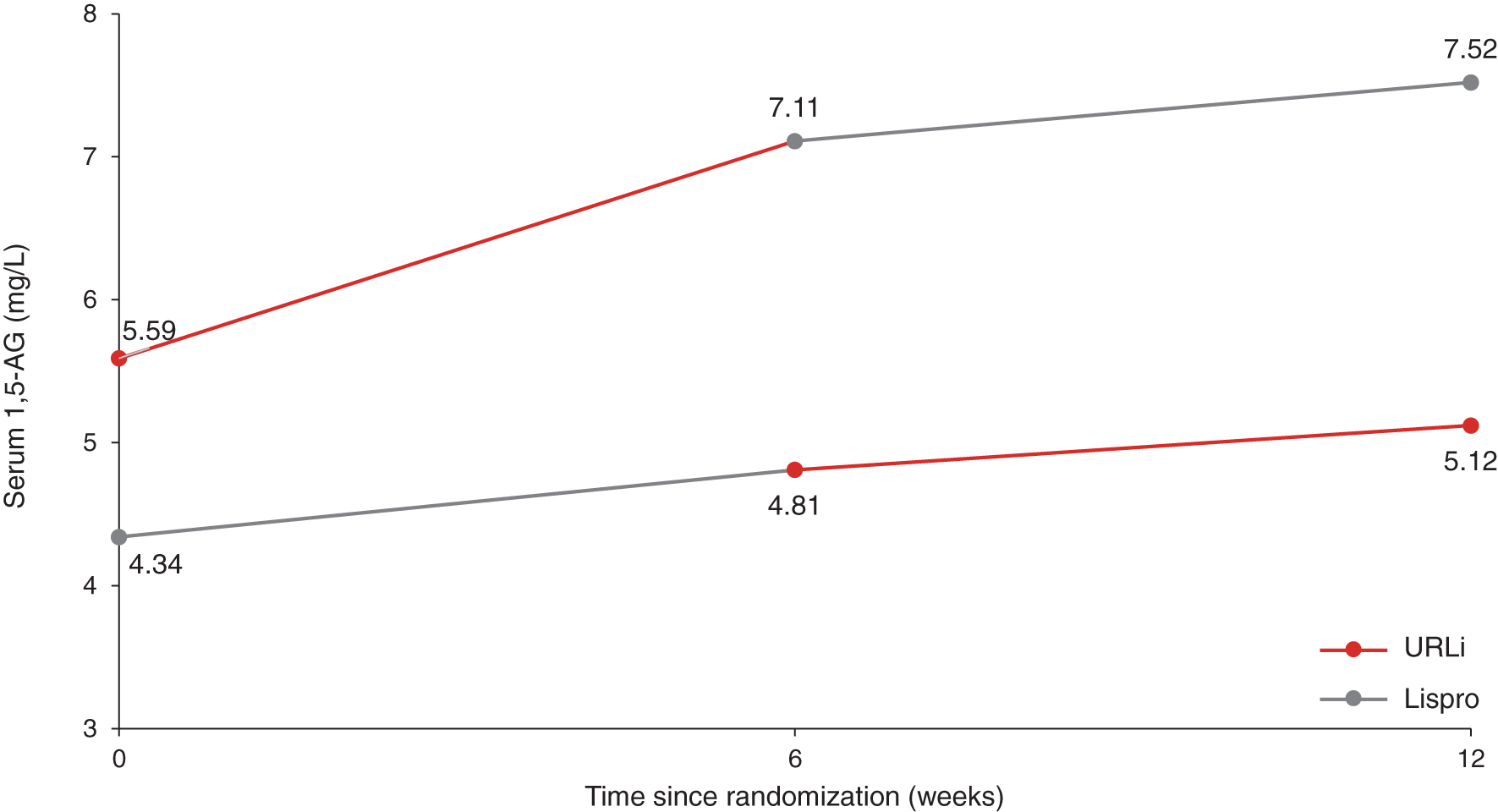
Mean 1,5-AG from baseline to week 12. 1,5-AG, 1,5-anhydroglucitol.
Pump basal rates and bolus calculator settings at week 6 were not significantly different between URLi and lispro treatments: Breakfast carbohydrate ratio was 9.1 versus 9.2 g/U; active insulin time, 3.6 versus 3.5 h; and insulin sensitivity factor, 50.0 versus 49.8 mg/dL/insulin unit, respectively. Mean insulin doses (basal, bolus, and total daily insulin dose, U/kg) were similar between treatments. The mean bolus-to-total insulin ratio at week 6 was also similar: URLi, 44.2%; lispro, 46.6%.
Safety
During the treatment period, the incidence and rate of severe hypoglycemia were similar between treatments with one patient experiencing one severe hypoglycemia event with each treatment.
No discontinuations due to an AE occurred in the study. Overall, 26 (53.1%) patients experienced a treatment-emergent adverse event (TEAE), with more patients on URLi treatment (46.9%) reporting TEAEs than those on lispro treatment (18.8%). This difference was driven by an increase in infusion site pain with URLi treatment (URLi, 18.4%; lispro, 4.2%) and infusion site reaction (URLi, 18.4%; lispro, 8.3%). Infusion-site-related events were mostly of mild or moderate severity (>90%), all were resolved during the study, and none resulted in discontinuation from study treatment.
A greater increase from baseline in diastolic blood pressure was observed for URLi compared with lispro (URLi, 2.5 mm Hg vs. lispro, 0.1 mm Hg; P = 0.038). However, no treatment differences were observed for changes from baseline in mean systolic blood pressure as well as pulse, weight, and BMI.
Discussion
This study was designed to assess the compatibility of URLi treatment with CSII use by evaluating infusion set failures when URLi or lispro was administered via insulin pump. The infusion set failures, as defined in the primary objective, were low and comparable between treatments, indicating that URLi was compatible with CSII use.
Although the overall incidence of premature infusion set changes was similar between treatments, a higher incidence of premature infusion set changes due to unexplained hyperglycemia was observed with URLi treatment. It is unlikely that these events were due to occlusions since no pump occlusion alarms were reported. Although it can be argued that not all occlusions will lead to an occlusion alarm and the time from the occurrence of an occlusion to an occlusion alarm may be longer than expected, 11,12 indicators of an occlusion such as prolonged hyperglycemia and ketosis were absent and hyperglycemia resolved quickly after infusion set change. Finally, although occlusions would be expected to increase with longer infusion set wear, 13 in this study the rate of premature infusion set changes due to unexplained hyperglycemia during treatment with URLi did not increase with time of infusion set wear. Also, CGM-based measures of glucose control were maintained from days 1 to 3 of infusion set wear.
A higher incidence of TEAEs was observed for URLi in this study, driven by infusion site pain and infusion site reaction. The exact cause of the increase in local infusion site symptoms is unknown but could be attributed to one or both enabling excipients in the URLi formulation (citrate and treprostinil). The majority of these events were mild, and none led to treatment discontinuation. However, infusion site reactions contributed to higher rates of premature infusion set changes with URLi treatment. In total, the rate of infusion set changes during URLi treatment corresponded to approximately one additional change every 3 months, assuming routine changes every 3 days. Injection site reactions were reported when URLi was administered via subcutaneous injection as part of MDI therapy; however, the incidence was lower than in the current study (URLi, 2.7%; lispro, 0.2%). 14 Most events were mild, and none led to treatment discontinuation.
Although this study was not powered to evaluate differences between treatments, safety and efficacy objectives were assessed. Similar lowering of HbA1c was observed for both treatments at the end of period 1. Mean HbA1c at the end of period 2 was 6.6% with sequence A (URLi/lispro) and 6.9% with sequence B (lispro/URLi), indicating good glycemic control over the course of the study.
The CGM assessments also showed that treatment with URLi resulted in a trend toward less time in hypoglycemia (<2.8 mmol/L [50 mg/dL]), and more time in the pre-specified target range (3.9–7.8 mmol/L [71–140 mg/dL] and 3.9–10.0 mmol/L [71–180 mg/dL]). Postprandial ambulatory glucose profiles and iAUCs after breakfast were numerically lower with URLi treatment, indicating a trend toward improved PPG control. These results are consistent with findings from the PRONTO-T1D CGM sub-study assessing CGM outcomes for patients using URLi or lispro in an MDI regimen. 15
The strengths of this study include the use of a crossover design allowing for within-subject comparison of compatibility and safety measures across the treatments. The analysis results are, however, limited by certain factors. In this study, the compatibility of URLi with insulin pump use was assessed in patients using the Medtronic MiniMed 530G, 630G, or 640G. Further investigation using alternative insulin pumps may provide additional insights into the behavior of the ultra-rapid insulin with other insulin pumps, as occlusion rates may differ. Patients in the study had generally good glycemic control. However, further investigation on the efficacy of URLi with CSII in a clinical trial with a larger sample size and of longer duration may be necessary to confirm these findings.
In conclusion, the compatibility and safety of URLi and lispro with CSII were comparable in patients with type 1 diabetes. URLi also demonstrated a trend toward improved glycemic control in this study. Although infusion site reactions were more common with URLi treatment, they were mostly mild and did not lead to treatment discontinuation. The results support that URLi can be administered safely by insulin pumps and that patients can be transitioned to URLi from a rapid-acting insulin analogue by using similar doses and pump settings without loss of glycemic control. As diabetes technologies continue to advance toward more fully automated systems, future studies will need to evaluate the use of URLi in sensor-augmented, hybrid closed-loop, and fully closed-loop insulin delivery systems.
Authors' Contributions
B.W.B., S.K.G., P.N., and C.M. participated as trial investigators and reviewed and edited the article. R.L. contributed to the study design, the statistical analyses, the interpretation of the research, and writing of the article. T.H. contributed to the study design, medical oversight, interpretation of the research, and writing of the article. D.I. was responsible for medical oversight during the trial and contributed to the study design, the data analysis and interpretation of the research, and writing of the article. All authors approved the final article to be published. D.I. is the guarantor of this work; as such, they had full access to all the data in the study; and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Acknowledgments
The authors would like to thank the study participants, as well as the investigators and study coordinators who supported them. The authors also thank Dr. Janet Tobian and Mary Anne Dellva (Eli Lilly and Company, Indianapolis, IN) for critically reviewing the article and Farai Chigutsa (Eli Lilly and Company) for medical writing and editorial assistance.
Author Disclosure Statement
This study was funded by Eli Lilly and Company. B.W.B., employed by Atlanta Diabetes Association, has received research support and fees for being a consultant for Eli Lilly and Company. S.K.G. has received consulting fees from Boehringer Ingelheim, Eli Lilly and Company, Lexicon Pharma, Mannkind, Medtronic, Merck, Novo Nordisk, Roche, Sanofi, Senseonics, and Zealand Pharma, and research grants from Animas, Dario, Dexcom, Eli Lilly and Company, The Juvenile Diabetes Research Foundation, Lexicon Pharma, Medtronic, Merck, National Cancer Institute, National Institute of Diabetes and Digestive and Kidney Diseases, Novo Nordisk, Sanofi, and T1D Exchange, Inc. All his research grants and honoraria were received through the University of Colorado. C.M. received research grants from Astra Zeneca, Boehringer Ingelheim, Eli Lilly and Company, FPS, Hanmi, Janssen, Lexicon Pharma, Merck, Novo Nordisk, Pfizer, Roche, Sanofi, Takeda, and Theracos; he received speaker fees from Abbott, Astra Zeneca, Boehringer Ingelheim, Eli Lilly and Company, Janssen, MSD, Novo Nordisk, Roche, and Sanofi; and he is on the advisory board for Abbott, Astra Zeneca, Boehringer Ingelheim, Eli Lilly and Company, MSD, Novo Nordisk, and Sanofi. T.H., R.L., and D.I. are employees and shareholders of Eli Lilly and Company. No other potential conflicts of interest relevant to this article were reported.
Funding Information
This study was funded by Eli Lilly and Company.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.