
Abstract
Background:
The prevalence of type 2 diabetes (T2D) in youth is increasing. Treatment options beyond metformin and insulin are needed. The safety, tolerability, pharmacokinetics, and pharmacodynamics of liraglutide once daily in youth (10–17 years old) with T2D were investigated in a randomized, double-blind, placebo-controlled trial.
Subjects and Methods:
Youth treated with diet/exercise alone or with metformin and having a hemoglobin A1c (HbA1c) level of 6.5–11% were randomized to liraglutide (n=14) or placebo (n=7). Starting at 0.3 mg/day, doses were escalated weekly to 0.6, 0.9, 1.2, and 1.8 mg/day (or placebo equivalent) for 5 weeks.
Results:
Nineteen participants completed the trial. Baseline characteristics were similar between groups, with mean (SD) values for age of 14.8 (2.2) years, weight of 113.2 (35.6) kg (range, 57–214 kg), diabetes duration of 1.7 (1.4) years, and HbA1c level of 8.1% (1.2%). No serious adverse events (AEs), including severe hypoglycemia, occurred. Transient gastrointestinal AEs were most common at lower liraglutide doses during dose escalation. No significant changes in safety and tolerability parameters occurred. There was no evidence of pancreatitis or lipase elevations above three times the upper normal limit; calcitonin levels remained within the normal range. For liraglutide 1.8 mg, mean half-life was 12 h, and clearance was 1.7 L/h. After 5 weeks, the decline in HbA1c level was greater with liraglutide versus placebo (−0.86 vs. 0.04%, P=0.0007), whereas mean body weight remained stable (−0.50 vs. −0.54 kg, P=0.9703).
Conclusions:
Liraglutide was well tolerated in youth with T2D, with safety, tolerability, and pharmacokinetic profiles similar to profiles in adults.
Introduction
T
Liraglutide is approved for the treatment of adult T2D in the United States, as an adjunct to diet and exercise (although not recommended as first-line therapy for patients with inadequate glycemic control on diet and exercise), and in Europe, as add-on therapy for participants not achieving control with single (metformin or sulfonylurea) or multiple (metformin+sulfonylurea or metformin+thiazolidinedione) oral agents. 6,7 In adult Phase 3 clinical trials, 8 –15 liraglutide (1.2 and 1.8 mg) as monotherapy or in combination with oral antidiabetes therapies demonstrated efficacy in reducing the hemoglobin A1c (HbA1c) level by 0.8–1.5%, fasting plasma glucose (FPG) level by 0.8–2.4 mmol/L (15.0–44.0 mg/dL), systolic blood pressure by 0.55–6.7 mm Hg, and body weight by up to 3.4 kg, 9 with transient gastrointestinal (GI) reactions (primarily nausea) being the most common adverse event (AE). 16 The antihyperglycemic effect of liraglutide involves glucose-dependent stimulation of insulin secretion and inhibition of glucagon release, whereas the weight-loss effect involves appetite suppression, which lowers energy intake. 17,18
Liraglutide's pharmacokinetic profile in adults showed dose exposure proportionality within the 0.6–1.8 mg dose range, 19,20 with a time to maximum concentration (t max) of 8–12 h, an elimination half-life (t 1/2) of approximately 13 h, and a mean apparent clearance (CL/F) of 1.2 L/h. 19 –21 Pharmacokinetics (PK) were not influenced by older age (>65 years), race, or ethnicity. 20,22,23 Although female sex was associated with a lower weight-adjusted drug clearance, this did not impact clinical response to the 1.2 and 1.8 mg liraglutide doses. 24 Body weight significantly affected the PK of liraglutide. Although increasing body weight resulted in lower drug exposure (area under the concentration–time curve [AUC]), the clinical response was unaffected. 24
Current U.S. and European regulatory requirements for the development of therapies for T2D mandate their evaluation in youth with T2D 10 years of age or older. 25 Pediatric T2D studies are difficult to conduct because of the low prevalence of the disease (0.42 cases/1,000 youth 10–19 years of age) with 3,600 new cases annually (15–20% of total youth diagnosed with diabetes in 2002–2005). 25,26
Furthermore, recruitment and executional challenges are delaying pediatric trial results. The liraglutide pediatric investigation plan was developed under the guidance of the European Medicines Agency. 27,28 As the first stage of this strategy, this trial assessed liraglutide (monotherapy or combination therapy with metformin) safety, tolerability, PK, and pharmacodynamics (PD) in youth with T2D over a 5-week period.
Research Design and Methods
Participants
Eligible participants with T2D were 10–17 years old, with a body mass index of >85th percentile for age and gender, and were treated with diet and exercise alone or in combination with a stable dose of metformin for at least 4 weeks before screening. Initially, inclusion criteria of an HbA1c level of 7–10% at screening and of an FPG level of 7.2–12.2 mmol/L (130–220 mg/dL) at randomization were used. However, after the first four participants were enrolled (over approximately 8 months), these criteria were broadened to 6.5–11.0% for HbA1c and 6.1–13.3 mmol/L (110–240 mg/dL) for FPG to facilitate recruitment. Diabetes-related antibodies were not measured.
Trial design and conduct
This double-blind, randomized, placebo-controlled, parallel-group trial involved 16 sites in four countries (the United States, the United Kingdom, Slovenia, and Belgium). The study was conducted in accordance with the Declaration of Helsinki and adhered to Good Clinical Practice Guidelines issued by the International Conference of Harmonization. Both participants and their legal-age representatives provided written informed consent/assent. Eligible participants were randomized 2:1 to once-daily subcutaneous injections of either liraglutide or placebo (Fig. 1). Liraglutide treatment was initiated with 0.3 mg daily during the first week and increased weekly to 0.6 mg, 0.9 mg, 1.2 mg, and 1.8 mg for a total of 5 weeks of treatment. Drug dose was not increased if FPG (average of measurements on 3 consecutive days) was <6.1 mmol/L (110 mg/dL). Participants whose dose was not escalated to the next higher one continued on the highest dose attained for the remainder of the trial.
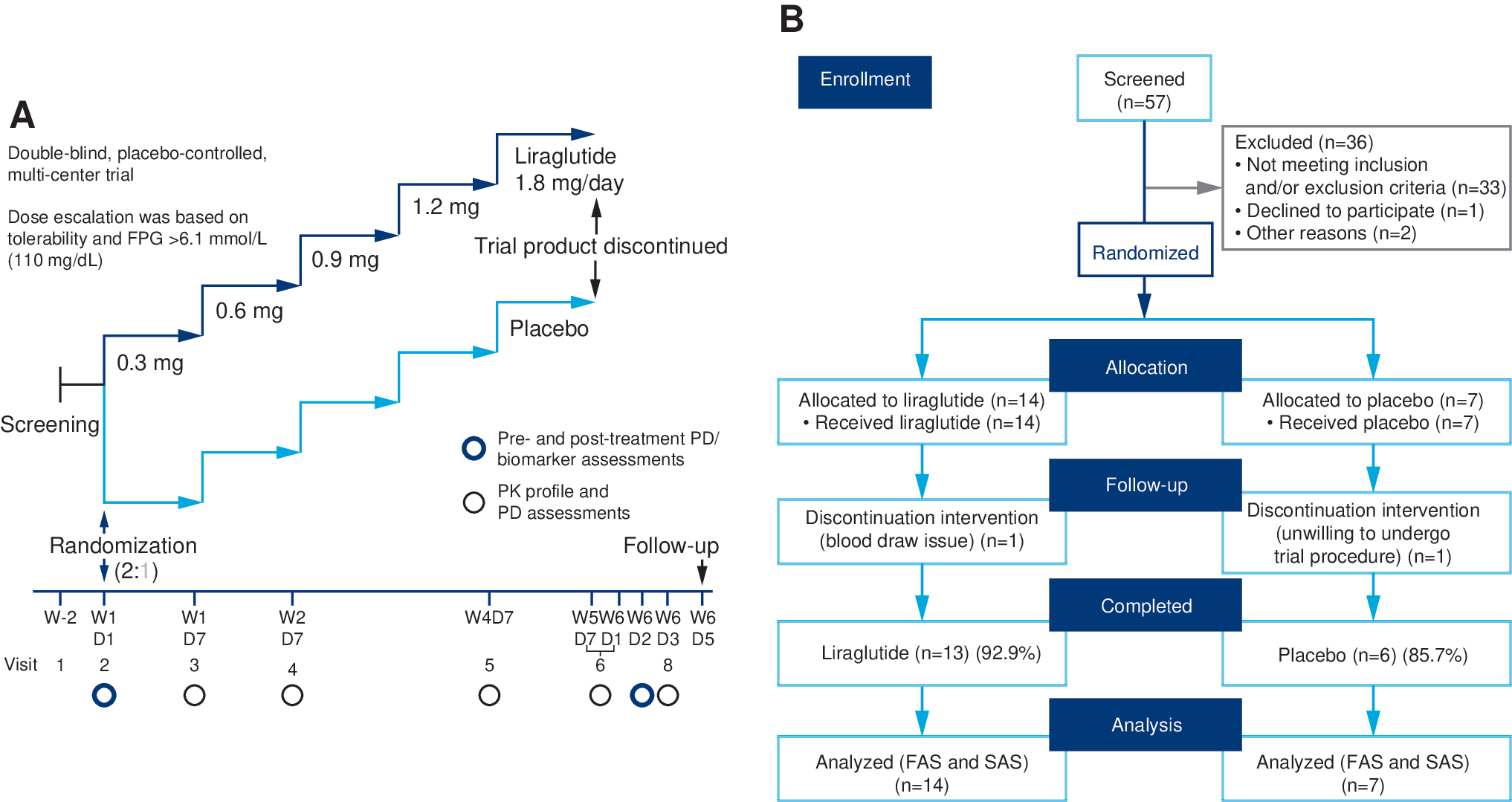
Trial design and participant flow.
Assessments and end points
Evaluation of safety and tolerability was the primary objective of the trial. Primary end points included AEs, laboratory tests (biochemistry, hematology, urinalysis, calcitonin, amylase, lipase, fasting lipids, vital signs, physical examination, electrocardiogram, fundoscopy, and anti-liraglutide antibodies), and a range of biomarkers and hormones. Anti-liraglutide antibody analysis was performed by a central laboratory using blood samples drawn before treatment and approximately 5 days after end of treatment. Hypoglycemic episodes were classified according to the definitions of Novo Nordisk (Cophenhagen, Denmark) for major (requiring third-party assistance) and minor (any symptomatic self-treated or asymptomatic episode, with confirmed plasma glucose <3.1 mmol/L [56 mg/dL]) and also according to the definitions of the American Diabetes Association (Supplementary Fig. S1; Supplementary Data are available online at
The secondary objectives of the trial were the determination of PK and PD parameters. PK end points, assessed at steady state, included maximum concentration (C max), t max, concentration before dosing (C trough), CL/F, t ½, and AUC 13 h post-dose administration and 24 h after final dose (1.8 mg) administration.
Samples for PK assessment were drawn at liraglutide doses of 0.3 (Week 1), 0.6 (Week 2), and 1.2 mg (Week 4) on the last day of each week before dose escalation (Day 7) and for liraglutide 1.8 mg during the 72-h period (3 days) following the last dose (given on Week 5, Day 7). The first four participants had a 24-h blood sampling schedule at the end of Weeks 1, 2, 4, and 5, with samples taken within 15 min before liraglutide administration and 2, 4, 8, 10, 11, 12, 14, and 24 h post-dosing. Following a protocol amendment that aimed to reduce the burden on remaining participants by eliminating the need for overnight clinic stays, the blood sampling period was changed from 24 h to 13 h, and samples were taken 15 min before liraglutide administration and 2, 5, 8, 10, and 13 h post-dosing. Blood samples were also taken at 24, 48, and 72 h after the final liraglutide dose. For participants whose dose was not escalated to 1.8 mg liraglutide, two types of pharmacokinetic profiles were obtained: a 13-h profile on Day 7 of treatment Weeks 1, 2, and 4 and an end-of-treatment profile (during the 72-h period after the last trial dose). Analysis of liraglutide in plasma samples was performed using a validated enzyme-linked immunosorbent assay method. 21 Exploratory PD end points included HbA1c (high-pressure liquid chromatography method), FPG, and body weight.
Statistical analysis
No statistical power calculation was performed. Safety, tolerability, and PK end points were summarized using descriptive statistics. Analyses of safety, tolerability, and PD analyses included all randomized participants. For C max, dose proportionality (i.e., slope and 95% confidence interval [CI]) was assessed using a random coefficient model with log C max at steady state (from the 0–13-h profile) as a dependent variable, a common intercept as the fixed factor, log dose as a fixed covariate effect, and a participant-specific intercept and slope (i.e., log dose) as random effects. The same model was used to assess C trough. Dose proportionality would be indicated for each of these analyses if, for a 95% CI, the point estimate was close to 1.0. Changes from baseline to end of trial in PD end points were analyzed using an analysis of covariance model, with treatment as a fixed effect and baseline value as a covariate (dose not included). A P value of <0.05 was considered significant.
Results
Participants and demographics
Of 57 youth screened, 19 did not meet the HbA1c inclusion criterion, and 17 were otherwise ineligible for enrollment. The remaining 21 participants were randomized to liraglutide (n=14) or placebo (n=7). Nineteen participants completed the study, and two withdrew: one from the liraglutide group after 7 days of treatment because of unsuccessful blood draw and one from the placebo group after 5 days of treatment because of unwillingness to undergo trial procedures (Fig. 1B). Nine of the 14 participants receiving liraglutide reached the highest dose (1.8 mg). Doses were not escalated for four participants who did not meet the FPG-based dose escalation criterion (see Subjects and Methods); these participants received either liraglutide 0.3 mg (n=1) or 0.6 mg (n=3) for the remainder of the trial. Liraglutide dose was not escalated in one other participant because of early withdrawal. All participants in the placebo group were escalated to the 1.8 mg dose equivalent.
Participants' average age was 15 years. Three youth were under 12 years of age: two (10 and 11 years old) were randomized to liraglutide, and one (11 years old) was randomized to placebo. Most participants were white, female, and post-pubertal. Mean baseline body weight and body mass index were high (113 kg and 40 kg/m2, respectively) and ranged widely (from 57 to 214 kg and from 29.2 to 71.6 kg/m2, respectively). Mean body mass index z scores for liraglutide (3.41) and placebo (3.38) reflected these values. Mean duration of T2D was 1.7 years; 76% of participants had been previously treated with metformin and continued the regimen unchanged during the trial. Demographic and baseline characteristics were generally similar between groups with the exception of baseline HbA1c, which was slightly higher (8.3%) in the liraglutide group compared with the placebo group (7.8%) (Table 1).
One subject in the placebo group was classified as “previously treated with metformin”; however, this subject stopped taking metformin before the study and is counted in the diet and exercise group.
BMI, body mass index; HbA1c, hemoglobin A1c; NA, not available.
Safety and tolerability
No serious AEs were reported during the trial. In the liraglutide group, 10 participants (71.4%) experienced 38 mild AEs. In the placebo group, three participants (42.9%) experienced 18 mostly mild AEs (12 of 18). Treatment-emergent AEs reported with a frequency greater than 10% are summarized in Supplementary Table S1. The most frequently reported AEs in both groups were GI related (particularly diarrhea, nausea, and vomiting). Although GI AEs were more common with liraglutide, their severity was mild, and they occurred mostly at the 0.3 and 0.6 mg doses (13 of 17 events) during dose escalation. GI AEs were body weight independent, and most lasted 1–4 days (data not shown).
No major hypoglycemic events occurred during the trial. Minor hypoglycemia (by the Novo Nordisk definition) occurred in three liraglutide-treated participants (four events) during 0.6 mg treatment (Table 2). One participant had two episodes with confirmed plasma glucose levels of 2.5 and 3.0 mmol/L (45 and 54 mg/dL); the other two participants each experienced single events with plasma glucose measurements of 2.5 and 2.1 mmol/L (46 and 38 mg/dL). Three of these minor episodes occurred 2–3 h after a meal. Of note is that two asymptomatic and one documented symptomatic episodes (American Diabetes Association definition) occurred after a prolonged fast (>9 h) (Supplementary Table S2).
Data are number of participants (%)/number of events, except as indicated.
The three classes with the highest number of events are presented.
ADA, American Diabetes Association.
Lipase levels (mean±SD) at screening and end of treatment were 25.5±7.1 and 37.5±18.4 U/L, respectively, in liraglutide-treated participants and 27.1±12.0 and 33.2±18.5 U/L, respectively, in participants receiving placebo (normal range: ≤13 years, ≤37 U/L; >13–18 years, ≤46 U/L) (Supplementary Fig. S2). Three liraglutide-treated participants with normal lipase levels at screening had elevated levels (53, 74, and 56 U/L) at a follow-up visit; one 10-year-old participant had elevated lipase levels at both screening (38 U/L) and follow up (60 U/L). One participant receiving placebo had elevated lipase levels at both screening (54 U/L) and end of trial (63 U/L). No participant experienced AEs associated with “abdominal pain,” and no lipase elevations reached more than three times the upper limit of normal (ULN). Therefore, no trial patient fulfilled the diagnostic criteria for pancreatitis.
Amylase levels (mean±SD) at screening and end of treatment were in the low-to-normal range in all participants in the liraglutide group (39.6±12.6 and 45.5±14.9 U/L), respectively) and in the placebo group (49.9±14.4 and 45.6±21.3 U/L, respectively) (normal range: 19–76 U/L for 10–16 years; 28–100 U/L for >16 years) (Supplementary Fig. S2).
During the trial, no clinically significant elevations in calcitonin levels occurred, with all participants remaining within gender-specific normal limits (Supplementary Fig. S3). No clinically significant changes in any other safety parameters (including the laboratory tests listed in Subjects and Methods and Supplementary Table S3) were noted at the end of the trial (data not shown); no anti-liraglutide antibodies were detected.
PK
PK end points for participants escalated to 1.8 mg liraglutide are summarized in Figure 2A: t ½ was 12.3 h, t max was reached in 8 h, and CL/F was 1.7 L/h. Liraglutide concentration profiles over a 13-h period are presented by dose in Figure 2B and show higher exposure with increasing drug dosage. The corresponding PK end points are shown in Supplementary Table S4. The point estimate of the slope for the best linear fit of steady-state liraglutide C max at 0.3 mg, 0.6 mg, 1.2 mg, and 1.8 mg was 0.97 (slope [95% CI 0.78–1.17]), indicating dose proportionality. The corresponding estimate for C trough was 0.68 [0.03, 1.32]. One participant receiving liraglutide at 1.8 mg was suspected of noncompliance because of low liraglutide concentrations; however, noncompliance was not confirmed, and the data for this subject are included in the end points.
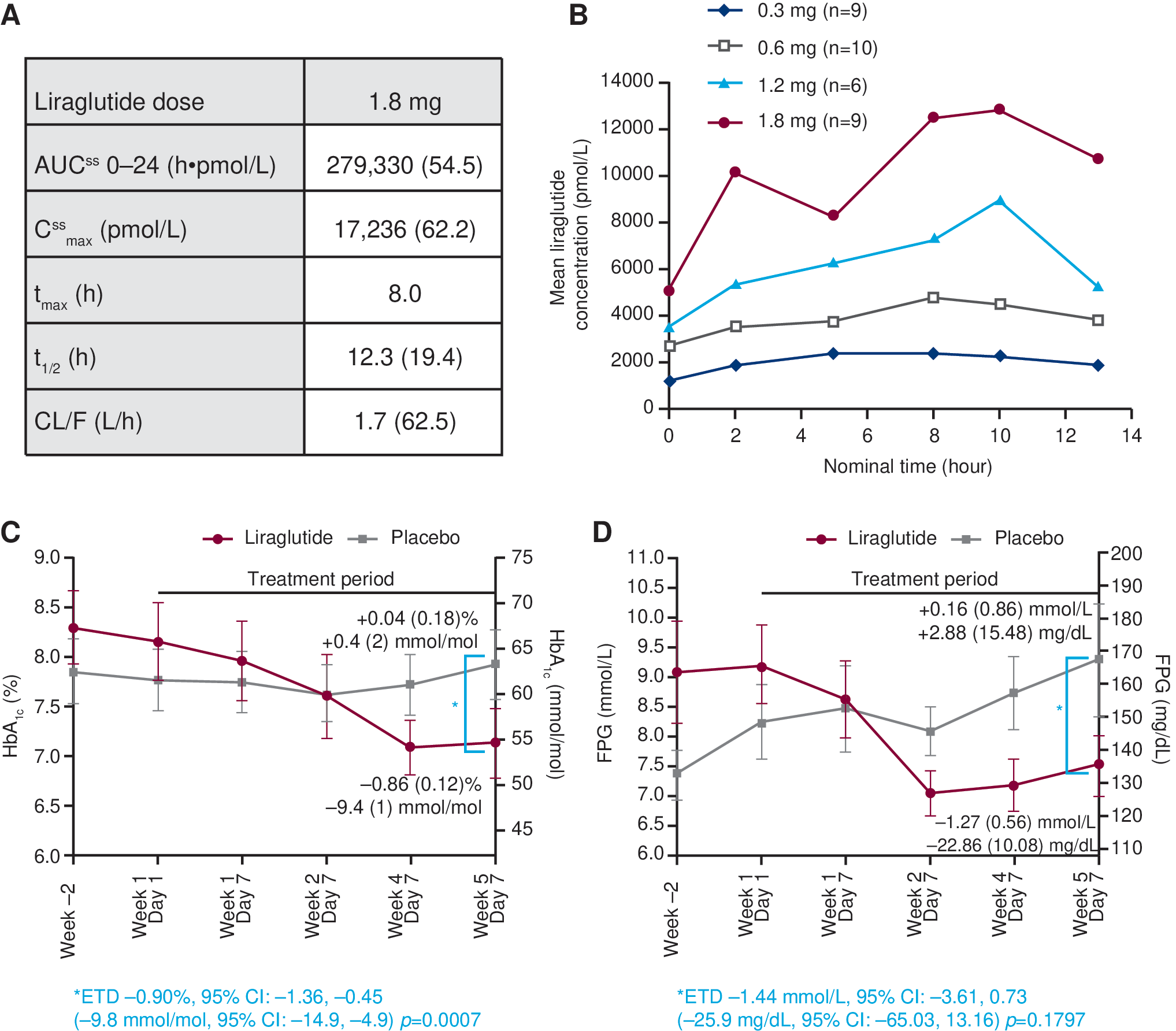
Pharmacokinetic and pharmacodynamic assessments during 5 weeks of treatment.
PD
After 5 weeks, the mean HbA1c level had decreased in liraglutide-treated participants and remained stable in placebo-treated participants (−0.86% vs. 0.04% [−9.4 vs. 0.4 mmol/mol]) (Fig. 2C). The estimated treatment difference was −0.9% [95% CI −1.36%, −0.45%] (−9.8 [−14.9, −4.9] mmol/mol) (P=0.0007). Similarly, FPG, measured by glucometer, decreased in liraglutide-treated participants and increased slightly in placebo-treated participants. The estimated treatment difference was −1.44 [−3.61, 0.73] mmol/L (−25.9 [95% CI −65.03, 13.16] mg/dL) but was not statistically significant (Fig. 2D). Throughout the trial, mean body weight remained stable (change from baseline in both groups, −0.5 kg).
Discussion
This study describes the first evaluation of safety, tolerability, PK, and PD of liraglutide, administered as monotherapy or in combination with metformin, in youth with T2D. Liraglutide, in escalating doses over 5 weeks, was safe and generally well tolerated in this population, in this short-term trial. In accordance with previous observations in adults with T2D, 16 mild GI symptoms were the most commonly reported AEs, with no evidence of additional safety concerns.
An important observation in this trial was that most GI AEs in liraglutide-treated youth occurred earlier and at lower doses (0.3 and 0.6 mg) in the escalation process. Fewer GI AEs occurred later at higher doses of 0.9, 1.2, and 1.8 mg; however, this reduction in frequency of AEs can only be confirmed for the nine participants who were escalated to these doses. Similar transient GI AEs were observed in adult T2D trials upon initiation with glucagon-like peptide-1 receptor agonists. 13,16 Although nausea was the most commonly reported GI AE for liraglutide-treated adults, 9 –16 in this trial, diarrhea was around twice as prevalent as nausea (42.9% vs. 21.4% of participants, respectively). However, this observation may have been influenced by the small sample size.
Hypoglycemia was more common with liraglutide (n=5) than placebo (n=1). Four of a total of nine asymptomatic episodes and one documented symptomatic episode occurred after a prolonged fast (>9 h) in liraglutide-treated participants. No relationship was observed between hypoglycemia occurrence and liraglutide dose level or timing of the preceding dose. At present, the observed numerical imbalance in hypoglycemic events remains inconclusive because of the limited sample size and is undergoing further investigation in a larger safety and efficacy trial. 30
The monitoring of pancreatic enzyme levels has become an established practice in clinical trials involving either glucagon-like peptide-1 receptor agonists or dipeptidyl peptidase-4 inhibitors, following postmarketing reports of pancreatitis during T2D treatment with these agents. 31 Diagnosis of pancreatitis required meeting two of the following three criteria: amylase/lipase levels three or more times the ULN, characteristic abdominal pain, and/or characteristic findings of acute pancreatitis on computed tomography scan or magnetic resonance imaging. None of the four cases of lipase elevations that occurred in this trial was above three times the ULN and were not accompanied by adverse events of “abdominal pain.” Thus, as in adult studies, the clinical significance of these lipase changes remains unclear. 32 As in adult T2D trials with liraglutide, 9,13 mean calcitonin levels remained with the normal range for both genders in this study (data on file with Novo Nordisk).
Similar to previous findings in adults, 20,21 the PK profile of liraglutide in youth also demonstrated a dose–concentration relationship. The t max (8 h), t ½ (12 h), and CL/F (1.7 L/h) at steady-state liraglutide 1.8 mg in pediatric participants were consistent with those previously reported in adults (t max, 8–12 h; t ½, 13 h; CL/F, 1.2 L/h). 19,20,24 Using a population PK modeling approach, the pediatric data obtained in this trial were compared with those from two historic trials in adults with T2D. This analysis indicated similar dose–exposure proportionality for liraglutide 0.3–1.8 mg with body weight and gender being significant covariates for liraglutide exposure in both populations. Furthermore, the analysis estimated that the adult liraglutide dose regimen would achieve a similar range of exposure in youth (age, 10–17 years; body weight, 57–214 kg). 33
Investigation of PD was a secondary objective of this study. The effects of 5 weeks of liraglutide treatment on indicators of glycemic control (HbA1c and FPG) and body weight were analyzed. Compared with placebo, liraglutide resulted in glycemic improvement (significantly greater decrease in mean baseline-adjusted HbA1c and a trend toward decreased FPG); there was no discernible effect on body weight. However, the short treatment duration of this trial and the fact that five of 14 participants were not escalated to the highest dose of liraglutide 1.8 mg both preclude drawing firm conclusions about these pharmacodynamic parameters, which will be investigated as primary end points in a long-term safety efficacy trial of liraglutide in youth (Ellipse trial [NCT01541215]).
The limitations of this study are in line with its exploratory nature and include a small sample size and short duration of treatment. The small sample size limited the diversity of subject demographics, resulting in a higher proportion of white, non-Hispanic subjects compared with a previous report. 26 Although trial participants were mostly limited to Tanner stages IV and V, this is consistent with previous observations 3 that T2D is most commonly diagnosed later in puberty.
In the small sample size, having a mix of participants on diet/exercise or metformin at study initiation and during the trial could also affect the interpretation of trial results. In the ongoing safety and efficacy trial with liraglutide in adolescents mentioned above (Ellipse), all patients will enter the trial on a stable dose of metformin. Lastly, not having pancreatic autoantibody testing performed in the participants may have been a limitation, but considering this was a short-term PK/PD and safety trial, it is highly unlikely that the results would have been impacted in any significant way by this limitation.
Completed clinical studies of incretin agents in youth with T2D are limited to two Phase 1, single-dose studies. The first of these evaluated the safety, tolerability, and PK of sitagliptin in 26 adolescents and showed that increasing sitagliptin dosage (50–200 mg) led to greater exposure. 34 In the second study, single doses of exenatide (2.5 μg and 5 μg) were well tolerated by 13 adolescents and were associated with dose-dependent increases in plasma exenatide concentrations and improved postprandial glucose concentrations. 35 The results of these studies are consistent with the findings of our study that an incretin agent such as once-daily liraglutide is safe and generally well tolerated, produces dose-dependent increases in plasma concentrations, and appears to improve glycemic control in youth with T2D. Further controlled trials evaluating the efficacy and safety of incretin agents in pediatric T2D are currently in progress evaluating exenatide, sitagliptin, saxagliptin, and liraglutide. 30,36 –40
As mentioned previously, the execution of drug trials in pediatric T2D has proved a significant challenge, largely because of the relatively small pool of available potential participants. Although the original glycemic inclusion criteria were broadened by protocol amendment, it remained a significant challenge to find qualified participants. In addition, the strict requirement for the use of non-insulin pharmacotherapy at the time of recruitment precluded inclusion of a major proportion of this pool. The SEARCH study reported approximately 31% of glutamic decarboxylase–negative patients diagnosed with T2D (mean time after diagnosis, 11.7 months) to be on insulin therapy. 26 The executional challenges in our trial are common among trials in this population 41 and contribute to the extended trial durations observed at the clinicaltrials.gov Web site.
In summary, short-term treatment with liraglutide up to 1.8 mg was safe and generally well tolerated in 10–17-year-old youth with T2D, with mild GI AEs being most commonly reported. Liraglutide's PK profile in youth was consistent with that in adults, and a trend toward glycemic improvement was observed after 5 weeks of treatment. Because liraglutide demonstrated acceptable safety and tolerability in this exploratory investigation, a larger safety and efficacy trial (Ellipse) has now been initiated in pediatric participants with T2D.
Footnotes
Acknowledgments
This study was funded by Novo Nordisk. The funder participated in trial design, data collection, and analysis. Liraglutide is a proprietary compound manufactured and marketed by Novo Nordisk A/S. The authors gratefully acknowledge the contribution of all the investigators, their staff, and trial participants. The authors thank Irina Nayvelt, PhD, employed by Novo Nordisk, Inc., for medical writing assistance and Watermeadow Medical (Witney, United Kingdom), supported by Novo Nordisk, for medical writing and editorial assistance. The authors also thank C.T. Chang, PhD, previously employed by Novo Nordisk, Inc. for analyzing the data.
D.J.K. was responsible for trial execution (as Principal Investigator), consultation regarding trial design modification, data interpretation, and critical review of the manuscript. T.B. was responsible for trial execution (as Principal Investigator), data interpretation, and critical review of the manuscript. D.J.C., L.V.J., and P.M.H. were responsible for trial design and execution, data interpretation, and critical review of manuscript. S.A. was responsible for trial execution (as Principal Investigator), data interpretation, and critical review and editing of the manuscript.
Author Disclosure Statement
D.J.K. has been an Investigator on a Novo Nordisk trial. T.B. has attended advisory panels for Bayer, Medtronic, and Eli Lilly & Co., has attended speakers' bureaus for Roche, Eli Lilly & Co., Medtronic, and Bayer, and has received research support from Dyamid, Medtronic, GluSence, Abbott, and Novo Nordisk. D.J.C., L.V.J., and P.M.H. are or were all full-time employees of, and shareholders in, Novo Nordisk. S.A. has attended advisory panels for Novo Nordisk, Sanofi-Aventis, and Bristol Myers Squibb, has received research support from Novo Nordisk, and has been a drug safety monitoring board member for Boehringer Ingelheim.
Appendix
The Principal Investigators of the NN2211-1800 Study Group are as follows: in Belgium, J. De Schepper; in Slovenia, T. Battelino; in the United Kingdom, T. Barrett, M. Bone, and T. Randell; and in the United States, S. Arslanian, J. Blumer, M. Christensen, and R. Ferry (at the same single site), L. Hazan, D.J. Klein, X. Lopez, N. Neufeld, P. Toltzis, E. Tsalikian, R.P. Wadwa, and K. Wintergerst.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.