
Abstract
Background:
Dyslipidemia due to diabetes is characterized by hypertriglyceridemia and reduced levels of high-density lipoprotein cholesterol (HDL-C) and elevated or normal levels of low-density lipoprotein cholesterol (LDL-C) in type 2 diabetes mellitus (T2DM). The objectives of this Phase III study were to evaluate the safety, tolerability, and efficacy of saroglitazar (ZYH1) 2-mg and 4-mg tablets (Lipaglyn™; Zydus Cadila, Ahmedabad, India) compared with placebo in patients with diabetic dyslipidemia who are not controlled with atorvastatin 10 mg therapy.
Subjects and Methods:
This was a 16-week prospective, multicenter, randomized, double-blind, placebo controlled, three-arm Phase III study in subjects with hypertriglyceridemia (>200 and <500 mg/dL) with T2DM not controlled with atorvastatin 10 mg. The study consisted of a run-in period of 4 weeks of life-style modification followed by 12 weeks of treatment with saroglitazar (2-mg or 4-mg) or placebo tablets. The primary end point was the change in plasma triglyceride level compared with baseline and the placebo arm at the end of Week 12. The secondary exploratory end points were change in lipid profile and fasting plasma glucose at Week 12. In total, 302 subjects were randomized to receive one of the treatments, saroglitazar 2 mg (n=101) or saroglitazar 4 mg (n=99), or matching placebo (n=102). Patients who received study medication and had undergone at least one post baseline efficacy evaluation were included in the efficacy analysis.
Results:
At Week 12, saroglitazar 2-mg and 4-mg tablets significantly reduced mean plasma triglyceride levels by −45.5±3.03% and −46.7±3.02% (mean±SE), respectively, and the difference was significant (P<0.001) compared with placebo. Saroglitazar 2 mg demonstrated significant decrease in levels of non-HDL-C, very LDL-C, total cholesterol, and fasting plasma glucose. Additionally, saroglitazar 4 mg also significantly reduced LDL-C and apolipoprotein B levels. Saroglitazar was found to be safe and well tolerated by patients.
Conclusions:
Saroglitazar appeared to be an effective and safe therapeutic option for improving hypertriglyceridemia in patients with T2DM.
Introduction
D
Cardiovascular risk can be reduced by controlling dyslipidemia, blood pressure, body weight, and hypertriglyceridemia, which is characterized by elevated serum triglyceride (TG) levels and a low concentration of high-density lipoprotein cholesterol (HDL-C). 3,4 Statins are reported to reduce cardiovascular events 5,6 ; however, a considerable residual cardiovascular risk remains among the T2DM patients receiving statin therapy. 7,8
The peroxisome proliferator-activated receptor (PPAR)-α agonists have been reported to improve lipid profile in patients with T2DM, 3 and PPAR-γ agonists are approved for glycemic control in T2DM. 9 It has been proposed that a compound with the effect of both PPAR-α and PPAR-γ agonists might prove beneficial for patients with a range of cardiometabolic imbalances. Several dual PPAR-α and PPAR-γ agonists have been clinically developed; however, none has progressed beyond Phase III development owing to different reasons like lack of efficacy or safety and compound specificities. 10 Therefore, there remains a need for an optimum PPAR agonist that modulates lipid and glycemic control favorably with improved safety profile.
Saroglitazar (ZYH1) is a novel predominantly PPAR-α agonist with moderate PPAR-γ agonistic activity. It was designed to modulate lipid and glucose profiles and ameliorate insulin resistance. Preclinical and clinical (Phase I/II) studies have shown superior or equivalent safety and efficacy profile of saroglitazar compared with marketed fibrates or thiazolidinediones. 11
Therefore this present Phase III study was designed to evaluate the safety, tolerability, and efficacy of saroglitazar in T2DM patients having hypertriglyceridemia that was not controlled with atorvastatin therapy.
Subjects and Methods
Study design
This study was a multicenter, prospective, randomized, double-blind, placebo-controlled, interventional, Phase III study in the subjects with hypertriglyceridemia with T2DM undertaken at 29 centers in India (PRESS VI). The study consisted of a 4-week run-in period involving discontinuation of any antidyslipidemic drugs other than atorvastatin 10 mg; also, patients were put on a dietary and life-style modification program at this time. Following the completion of the run-in period, a double-blind treatment period of 12 weeks was initiated, following which there was a voluntary follow-up visit for safety assessment at 24 weeks. The study was initiated after obtaining the approvals of the Drug Controller General of India and the Independent/Institutional Ethics Committee for each site.
Study population
Subjects were recruited between August 10, 2009 and October 6, 2010 from hospital clinics, and practicing physicians specialized in the treatment of diabetes or cardiology conducted this clinical trial. After the 4-week run-in period, subjects were required to meet the following criteria for inclusion in this trial: age 18–65 years; diagnosis of T2DM; previous treatment with a maximum of two oral hypoglycemic agents; low-density lipoprotein cholesterol (LDL-C) level ≥100 mg/dL; TG level >200 and <500 mg/dL; body mass index >23 kg/m2; and treatment with atorvastatin 10 mg for at least 4 weeks.
Subjects were excluded if they had a history of greater than 5% weight loss in the preceding 6 months, unstable angina, acute myocardial infarction in the preceding 3 months, heart failure classified as New York Heart Association Class III–IV, uncontrolled hypertension, clinically significant edema, thyroid disorder, gallstones, impaired liver (aspartate aminotransferase and alanine aminotransferase ≥2.5 times the upper normal limit [UNL] or bilirubin ≥2 times the UNL) or renal (serum creatinine >1.2 mg/dL) function, ketonuria, myopathies or active muscle diseases (creatinine phosphokinase [CPK] ≥10 times UNL), severe illness such as tuberculosis, human immunodeficiency infection, malignancy, alcohol and/or drug abuse, allergy, sensitivity or intolerance to the study drugs and their formulation ingredients, and participation in any other clinical trial in the preceding 3 months at the time of enrollment. Subjects were excluded if they had been treated within 4 weeks of the run-in period with oral antidiabetes drugs from the glitazone (e.g., pioglitazone or rosiglitazone) and glitazar (investigational products) classes, insulin, lipid-modifying therapy (e.g., fenofibrate) other than atorvastatin 10 mg, thyroid-modulating drug, or anti-inflammatory drugs. Female subjects were excluded if they were pregnant or breast feeding or had initiated hormonal treatment (e.g., hormonal contraceptive, hormone replacement therapy) in the preceding 3 months. All subjects provided written informed consent before randomization to the study medication.
Procedures
Dose selection for saroglitazar was made on the basis of data from a 12-week Phase II dose finding study in subjects with T2DM, which suggested that 2 mg of saroglitazar might be the minimum effective dose and 4 mg the maximum effective dose with an acceptable tolerability profile. 12
At the study initiation, a diet, exercise, and life-style modification plan was discussed with the subjects and implemented on the basis of investigators' recommendation and reinforced at all the subsequent visits.
After the run-in period of 4 weeks, eligible subjects were randomly assigned to double-blind treatment with one of the two doses of saroglitazar (2-mg or 4-mg tablets) or matching placebo in the ratio 1:1:1. The study medication was given once a day before breakfast for 12 weeks.
Randomization was performed using SAS software (version 9.1; SAS Institute Inc., Cary, NC) with a block randomization procedure to avoid imbalance across the treatment groups, and the randomization sheet was placed in a sealed opaque envelope.
Five subject visits were scheduled during the study period of 16 weeks: screening visit (Week −4), enrollment visit (Week 0), Visit 1 (Week 2), Visit 2 (Week 6), and Visit 3 (Week 12).
At the screening visit, the eligible subjects were identified and provided the life-style modification and dietary advice. At the enrollment visit (Week 0), subjects who met all the inclusion and exclusion criteria were randomized to one of the treatment groups. Clinical examinations included assessment of presence of peripheral edema, cardiovascular events, and body weight were performed at baseline (Week 0) and Weeks 2, 6, and 12. Blood samples were collected at each visit for safety laboratory assessment. In addition, electrocardiograms (ECGs) were monitored at baseline and at Week 12.
Study drugs were dispensed to subjects at Week 0 (enrollment visit), Week 2, and Week 6. Study drug compliance was verified by count of drug tablets that were returned on Weeks 2, 6, and 12.
The primary end point for efficacy was percentage reduction in TG, and secondary end points were change from baseline in lipid profile: apolipoprotein A1, apolipoprotein B (Apo B), HDL-C, non-HDL-C, LDL-C, total cholesterol, and very LDL-C (VLDL-C). Exploratory efficacy end points were fasting plasma glucose (FPG).
Safety analysis end points are described in Table 1. Biomarkers used for the assessment of cardiovascular risk included incidence of peripheral edema, high-sensitivity C-reactive protein, CPK, ECG, and two-dimensional echocardiogram (2D-ECHO). Subjects were further monitored for cardiovascular events up to 6 months after the last dose of the study drug by 2D-ECHO, ECG, and clinical history. Suspected and diagnosed cardiovascular events were reviewed and adjudicated by the Ethics Committee and also the Data Safety Monitoring Board. All the laboratory analyses were done by a central laboratory (Ashish Pathology Laboratory, Ahmedabad, India), which is accredited by the U.S. College of American Pathologists and the National Accreditation Board for Testing and Calibration Laboratories of India.
ALP, alkaline phosphatase; ALT, alanine transaminase; Apo A1, apolipoprotein A1; Apo B, apolipoprotein B; AST, aspartate aminotransferase; BUN, blood urea nitrogen; CPK, creatinine phosphokinase; ECG, electrocardiogram; eGFR, estimated glomerular filtration rate; FPG, fasting plasma glucose; GGT, γ-glutamyl transferase; HDL, high-density lipoprotein; hs-CRP, high-sensitivity C-reactive protein; LFT, liver function test; LDL, low-density lipoprotein; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; PCV, packed cell volume; RBC, red blood cells; RFT, renal function test; TC, total cholesterol; TG, triglyceride; TSH, thyroid-stimulating hormone; VLDL, very low-density lipoprotein; WBC, white blood cells.
Statistical analysis
Sample size calculation was performed based on the previous studies done by Zydus Cadila. Assuming a difference in reduction of TG of 21.7% between the placebo and saroglitazar groups and estimated within-group SD of 42% at 12 weeks, 78 subjects per group would be required to provide 80% power with 5% level of significance. Considering a 20% dropout rate, approximately 100 subjects per group were required to be randomized. If there was a dropout of >20%, then additional subjects were to be recruited.
The primary and secondary end points were assessed by analysis of covariance to determine the least square means of change from baseline and 95% confidence intervals. The analysis of covariance model included the treatment fixed effect and baseline as covariates. Comparisons of each saroglitazar dose versus placebo were done at a two-sided significance level of 0.05. All subjects who received at least one dose of randomized study drug and who had an evaluable baseline and at least one evaluable post-baseline value were included in intention-to-treat analysis. All subjects who received at least one dose of a randomized study drug were included into the safety analysis.
Results
Demographics and subject disposition
The disposition of subjects in this study is presented in Figure 1. During this trial 302 subjects were enrolled and randomly assigned to a study group. Baseline demographic, clinical, and laboratory characteristics of the study population after the run-in period were comparable across the treatment groups (Table 2).
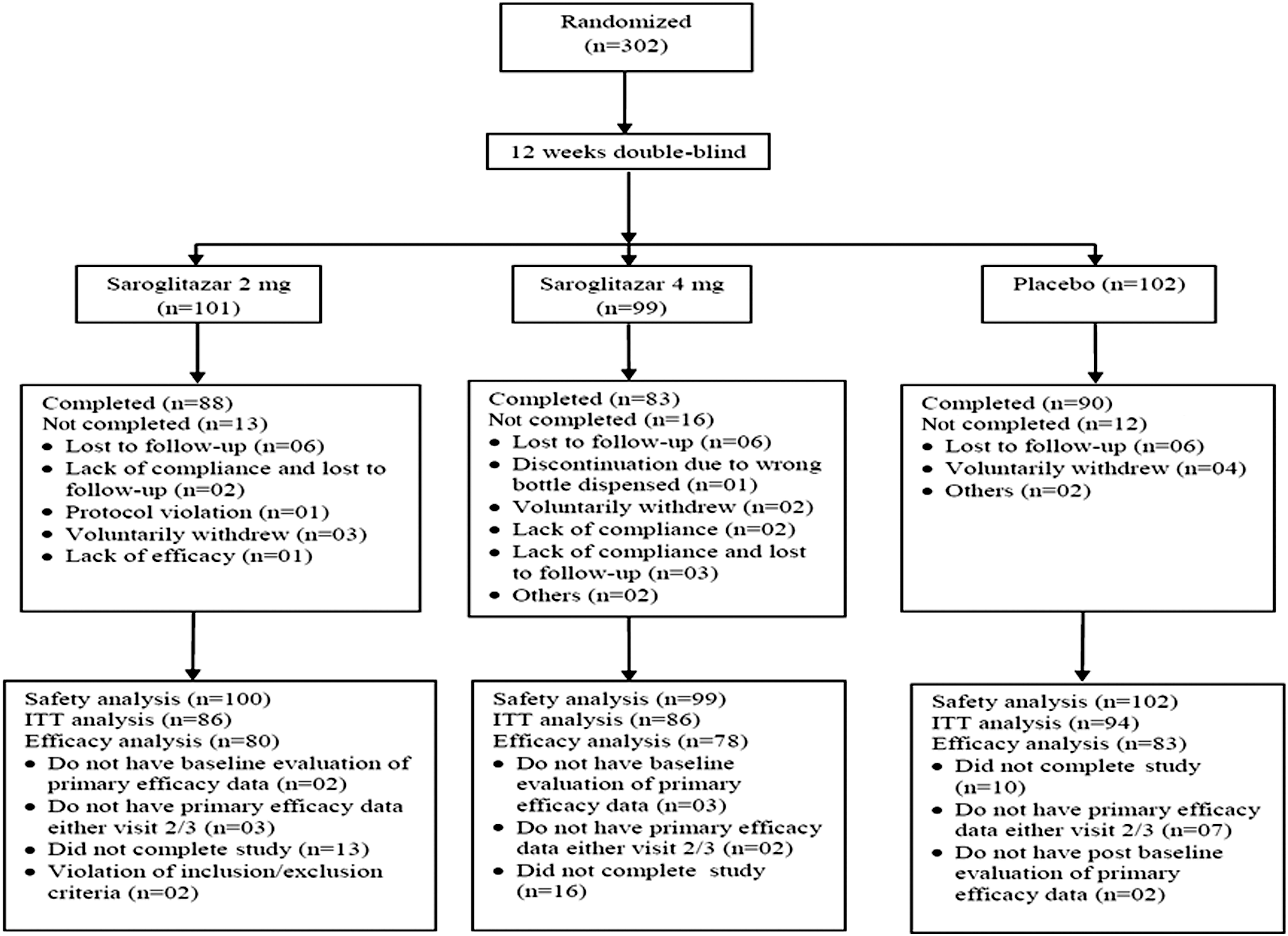
Subject disposition. ITT, intention-to-treat.
Data are mean±SD values or number (%) as indicated.
BMI, body mass index; bpm, beats per minute; HbA1c, glycosylated hemoglobin; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Efficacy analysis
Primary efficacy end point
Saroglitazar 2 and 4 mg decreased TG levels by −45.5±3.03% and −46.7±3.02%, respectively. Mean TG reduction from baseline to the end of the treatment was 132.7±8.30 mg/dL and 139.5±8.29 mg/dL with saroglitazar 2 mg and 4 mg, respectively. This decrease in TG level was statistically significant compared with baseline and placebo (Table 3 and Fig. 2).
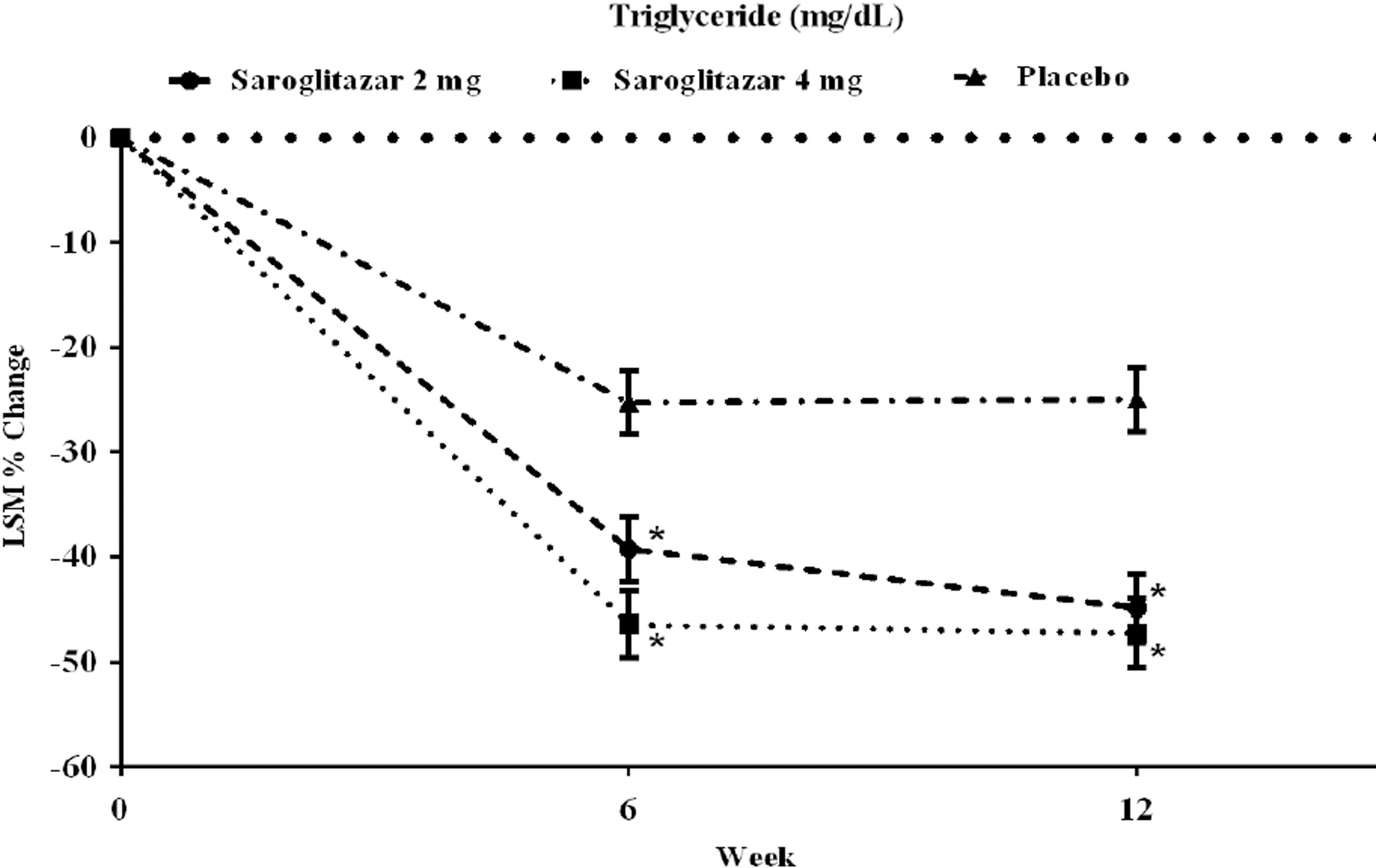
Effects on serum triglyceride levels. Data are the least square mean (LSM) percentage changes from baseline to Weeks 6 and 12 as per protocol analysis. *Significant difference compared with placebo.
Difference significant from placebo value.
Difference significant from baseline value.
LSM, least square mean.
Secondary efficacy end point
As shown in Table 4, non-HDL-C, LDL-C, VLDL-C, total cholesterol, and Apo B levels were significantly reduced compared with the placebo arm at Week 12. Saroglitazar 2 mg also caused a significant decrease compared with placebo in all lipid parameters studied except LDL-C and Apo B. After the 12-week treatment, saroglitazar (2 and 4 mg) caused a significant increase in HDL-C level compared with placebo. There was also a statistically significant decrease in FPG level after 12 weeks of treatment with saroglitazar 2 and 4 mg compared with placebo. The absolute decrease in glycosylated hemoglobin (mean±SE) was −0.3±0.08% with saroglitazar 2 mg and 4 mg and −0.2±0.07% with placebo (decrease not statistically significant).
Data for each parameter are least square mean±SE percentage change from baseline on the first line and mean±SE difference from baseline on the second line.
Difference significant from placebo value.
Difference significant from baseline value.
HDL, high-density lipoprotein; LDL, low-density lipoprotein; VLDL, very low-density lipoprotein.
Safety analysis
Both doses of saroglitazar were well tolerated. There were similar numbers of adverse events (AEs) in the saroglitazar and placebo arms. Most of the AEs were not related to treatment and were mild to moderate in intensity. AEs reported by two or more subjects in the study are presented in Table 5.
Data are number (%) of patients with adverse events. Adverse events presented are those reported by two or more patients in the study.
There were two reports of hospitalization during this study due to chest pain that were classified as serious AEs. One subject was admitted and found to have a normal ECG, whereas another subject underwent coronary artery bypass grafting and informed the investigator subsequently. These two events triggered the adjudication by the Data Safety Monitoring Board for assessment of the cardiovascular event. Both events were resolved and considered unrelated to the study drug.
After 12 weeks of treatment, there was a no significant changes in hemoglobin, liver enzymes (alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, and γ-glutamyl transferase), renal function (creatinine, enhanced glomerular filtration rate, and blood urea nitrogen), CPK, and high-sensitivity C-reactive protein in the saroglitazar and placebo arms. There was no edema or weight gain reported in any of the study arms (Table 6).
Data for each parameter are mean±SD percentage change on the first line and mean±SD change on the second line. The intention-to-treat population (last observation carried forward) was used for analysis.
ALT, alanine transaminase; ALP, alkaline phosphatase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; CPK, creatinine phosphokinase; eGFR, estimated glomerular filtration rate; GGT, γ-glutamyl transferase; hs-CRP, high-sensitivity C-reactive protein.
During this study, subjects were monitored for cardiac events, ECG abnormalities, and cardiac function by 2-D ECHO at the start of the study, at the end of 12 weeks, and at 24 weeks after the last dose of the study drug. There were no AEs reported as far as cardiac safety is concerned.
Discussion
This was a multicenter, interventional, randomized, double-blind, placebo-controlled, parallel-assigned safety and efficacy study to evaluate two doses (2 and 4 mg) of saroglitazar in the treatment of hypertriglyceridemia with T2DM not controlled with atorvastatin 10 mg therapy. Compared with placebo, saroglitazar 4 mg improved the range of lipid profile and glucose parameters.
Saroglitazar 2 mg and 4 mg treatment showed a statistically significant improvement in TG, a primary end point. Saroglitazar 4 mg caused a significant reduction in secondary end points: LDL-C, VLDL-C, total cholesterol, Apo B, non-HDL-C, and FPG. Saroglitazar 2 mg also caused a significant decrease compared with placebo in all lipid parameters studied except LDL and Apo B. Saroglitazar treatment also showed a significant improvement in HDL-C compared with placebo in this study.
Intensive statin therapy does not eliminate the cardiovascular risk associated with low HDL-C or/and high TG levels. 7,8 Saroglitazar has a potential to address the residual cardiovascular risk associated with high triglyceridemia.
Recently evidence has emerged to support the use of Apo B in the management of patients with dyslipidemia. Mechanistically it is important to consider that there is one Apo B molecule per LDL-C, lipoprotein A, VLDL-C, and intermediate-density lipoprotein particle, all of which are atherogenic. Apo B has repeatedly been shown to be a better marker for cardiovascular disease events than LDL-C. Because hypertriglyceridemia is commonly seen in diabetes, knowledge of the Apo B level may guide the aggressiveness with which lipid-lowering therapy is pursued. Based on the available evidence, the optimal level of Apo B can be at least approximately <90 mg/dL or, as supported by the Collaborative Atorvastatin Diabetes Study study, the subject with diabetes ≤80 mg/dL. 13 Therefore measurement of Apo B in response to saroglitazar therapy was studied. Saroglitazar 4 mg treatment showed a significant decrease in Apo B level and has the potential to achieve the target Apo B goal in dyslipidemia due to diabetes. The calculated non-HDL-C has features similar to those of Apo B and LDL-C and is considered an alternative goal of lipid therapy. 14 There was also a significant decrease in non-HDL-C level with saroglitazar treatment.
Saroglitazar has demonstrated a decrease in FPG of up to −27.3 mg/dL compared with baseline, which is significantly better than placebo. This decrease may be due to moderate PPAR-γ agonist activity. The decrease reported in FPG with aleglitazar treatment was −38.91 mg/dL. 10 The glycosylated hemoglobin level decrease with saroglitazar was not statistically significant, probably because of the short duration of the study. The favorable effect of saroglitazar cannot be directly compared with aleglitzar as each clinical study was conducted under widely varying conditions. However, the data still reflect the potential of saroglitazar as an antidyslipidemic and antiglycemic agent.
Although mechanistically PPAR agonism is known to cause hemodilution and a decrease in hemoglobin level following treatment with PPAR agonists, 10 hemoglobin content was not decreased significantly in the saroglitazar arms compared with the placebo arm. Levels of the inflammatory markers high-sensitivity C-reactive protein and CPK were also not increased during this study.
The alanine aminotransferase and aspartate aminotransferase values remain unchanged after the treatment with saroglitazar and placebo. There was a decrease in alkaline phosphatase and γ-glutamyl transferase following saroglitazar treatment compared with placebo. Unlike saroglitazar, treatment with fenofibrate reported abnormal liver function tests in more than 7.5% of subjects, which was significantly higher than in the placebo arm. 15
Unlike other PPAR-α and -γ agonists such as tesaglitazar and aleglitazar, saroglitazar has not been shown to cause a significant increase in creatinine or blood urea nitrogen. PPAR activation by fenofibrate and aleglitazar has shown an increase in creatinine levels 10,16 and decrease in glomerular filtration rate. This has been attributed to hemodynamic effects of PPARs. Saroglitazar showed a minor nonsignificant increase in creatinine content and minor decrease in enhanced glomerular filtration rate in the saroglitazar and placebo arms (Table 6). Saroglitazar has not shown potential for renal toxicity in this study. This might be due to a nonrenal route of excretion of saroglitazar. 17
During this study, there was no significant weight gain in subjects from the saroglitazar arm compared with baseline or the placebo arm. Although edema has been reported with aleglitazar, muraglitazar, and other PPAR-γ agonists in earlier trials, 10 there was no report of edema in the saroglitazar or placebo arm in the present study.
There were no changes in ECG and 2D-ECHO findings during the study period and also up to 24 weeks after the last dose of saroglitazar or placebo. No adverse cardiac event associated with saroglitazar was reported up to 24 weeks after the last dose of saroglitazar or placebo.
Saroglitazar was safe and well tolerated over the course of the 12-week study. Two serious AEs were reported in the saroglitazar 4 mg group; however, they were not related with the study drug. The incidence of AEs was comparable between the saroglitazar and placebo groups. Pyrexia and dyspepsia were the most common AEs reported by subjects in the saroglitazar 2 mg group, and gastritis was the most common AE reported by subjects in the saroglitazar 4 mg group. Most of the AEs were considered to be unrelated to study drugs and were mild to moderate in intensity. It is interesting that the known side effects of the PPAR agonists were not evident in this study.
Saroglitazar yields a favorable improvement in lipid and glycemic parameters and seems to be the first dual PPAR-α/-γ agonist without major safety concern. Saroglitazar is a recent addition to the armamentarium of antidyslipidemic and antidiabetes therapy and has a potential to change the present mode of treatment for dyslipidemia due to diabetes. It is approved by the Drug Controller General of India for marketing.
In this Phase III study the significant changes in lipid and glycemic end point coupled with the favorable safety profile represent promising data for saroglitazar as a therapeutic option in the treatment of hypertriglyceridemia with T2DM. Considering the favorable efficacy and safety profile, a large study to assess the tolerance, safety, and efficacy of saroglitazar in dyslipidemia patients has been initiated recently.
Footnotes
Acknowledgments
This study was sponsored and funded by Cadila Healthcare Ltd., a Zydus Group Company, Ahmedabad, India. The study investigators, in addition to A.B., V.P., and P.J., included the following: Asha Shah, MD, Civil Hospital (now CIMS) (Ahmedabad, India); B. Ganapthi, MD (St. John's Hospital, Bangalore, India); Banshi Saboo, MD (DIA-CARE Clinic, Ahmedabad); Deepak Bhosle, MD (Deogiri Diabetes Centre, Aurangabad, India); Dinesh Kamath, MD (Sudeep Diabetes Care Centre, Bangalore); G.M. Prasad, MD, DNB (Pranav Diabetes Centre, Bangalore); Hemant Phatale, MD, DM (Samrat Endocrine Institute of Diabetes, Obesity & Thyroid, Aurangabad); Jyoti Mannari, MD (Shree Krishna Hospital & Medical Centre, Karamsad, India); Keyur Parikh, MD (The Heart Care Clinic, Ahmedabad); Mala Dharmalingam, MD, DM (Bangalore Endocrinology and Diabetes Centre, Bangalore); Narendra Dedhia, MD (Parakh Hospital, Mumbai, India); Norman Sharma, MD (Choithram Hospital and Research Institute, Indore, India); A. Paneerselvam, MD (Aruna Diabetes Centre, Chennai, India); Rajshree Khot, MD, DNB (Government Medical College, Nagpur, India); Rakesh Sharma, MD (Gurukrupa Hospital, Ahmedabad); K.R. Ravendra, MD (Sreenivasa Diabetic Care, Bangalore); S.R. Arvind, MD (Diancon Hospital [Diabetes Research Centre], Bangalore); Sailesh Lodha, MD, DM (Fortis Escorts Hospital, Jaipur, India); Sanjeev Pathak, MD (DHL Research Centre, Ahmedabad); Satinath Mukhopadhyay, MD, DM (Institute of Postgraduate Medical Education & Research, Kolkata, India); Sunil Ambulkar, MD, Institue of Clinical Endocrinology & Diabetes Care Unit, Nagpur, India; E.M. Surendra, MD (Malliage Health Care, Davangere, India); Tiven Marwah, MD (Le Bonheur Endocrine & Diabetes Clinic, Ahmedabad); V. Shankar, MD (Standard Laboratory and Polyclinic, Bangalore); Y.P. Munjal, MD (Centre of Diabetes & Life Style Disease, Banarai Chandiwala Institute of Medical Sciences, New Delhi, India); and Yogesh Kadam, MD (Poona Diabetes Centre, Pune, India). Dhiraj Gambhire, MD, was a sponsor's medical expert for this study. We thank Bhaskar Vyas, MD, of the Ashish Pathology Laboratory, Ahmedabad, for laboratory support, Rahul Gupta of the Zydus Research Center and Hitesh Chauhan of Cliantha, Ahmedabad, for statistical assistance and data management, and Jayesh Bhatt, PhD, for assistance in preparation of the manuscript.
Author Disclosure Statement
In collaboration with the investigators, Cadila Healthcare Ltd., a Zydus Group Company, the sponsor of the trial, contributed to the design of the study, collection of data, statistical analysis, and interpretation and reporting of the results.
R.H.J. participated in protocol development, planning, supervision of clinical operations, collection of the data, analysis of the data, interpretation of the results, and writing the report. A.B., S.M., and S.J. participated in clinical protocol development, interpretation of results, and preparation of this manuscript. V.P., P.J., and G.J. participated in the conduct of the clinical trial, analysis of data, and manuscript preparation. The authors had full access to all data in the study and had final responsibility for decision to submit this publication.