
Abstract
Objectives:
This study aimed to examine healthcare provider (HCP) recommendations and patient preferences for the insulin pen versus vial-and-syringe in patients with type 2 diabetes mellitus (T2DM) and to assess clinical end points and safety outcomes.
Subjects and Methods:
Using a randomized, open-label, crossover design, in total, 405 insulin-naive adults with T2DM from 60 centers received basal insulin glargine in one of two device treatment sequences (2 weeks of pen followed by 2 weeks of vial-and-syringe, or vice versa). The primary end point, patient device preference, was evaluated at Week 4 (end of the crossover period) using the Insulin Injection Preference Questionnaire. Patient preference and HCP recommendation were assessed with one global item and three subscale items (blood glucose control, reluctance to use insulin, and long-term insulin use) using a 5-point scale ranging from 1=not preferred or not recommended to 5=preferred or recommended. Patients were then re-randomized to either pen or vial-and-syringe for further observation (6, 10, and 30 weeks) to evaluate clinical end points (glycosylated hemoglobin [A1C] and fasting blood glucose levels) and safety outcomes (hypoglycemia and adverse events).
Results:
Patients reported a significant preference for pens over vial-and-syringe, and HCPs strongly recommended pens over vial-and-syringe (both P<0.001). Consistent response patterns were observed by HCPs and patients for the three subscale items. Fasting blood glucose, A1C levels, and the incidence of hypoglycemia were comparable in the two groups.
Conclusions:
Patients preferred pens over vial-and-syringe, with the pen device also recommended by HCPs, when initiating basal insulin treatment in insulin-naive patients with T2DM.
Introduction
T
Improved glycemic control benefits patients with diabetes and provides an individualized, patient-centered treatment strategy to reduce and maintain target glycosylated hemoglobin (A1C) levels as recommended by European and American diabetes and endocrinology associations. 10,11 Early initiation of insulin treatment in patients with type 2 diabetes mellitus (T2DM) who do not reach target A1C levels with diet and/or oral antidiabetes agents alone can reduce the risk of developing diabetes-related micro- and macrovascular complications. 12,13 However, many patients are reluctant to start insulin therapy owing to their negative attitudes toward insulin use (e.g., permanence, restrictiveness, problematic hypoglycemia, and personal failure). 14,15 Considering the ongoing challenges associated with insulin therapy, 16,17 additional studies are needed to understand how HCP recommendations may influence patient preferences for an insulin delivery system. Shared decision-making between HCPs and patients may help patients improve glycemic control, reduce the risk of hypoglycemia, and prevent the occurrence of long-term complications associated with T2DM. 18,19
The primary objective of this study was to evaluate patient preferences for administration of basal insulin glargine via a pen device versus vial-and-syringe in insulin-naive patients with T2DM. Secondary objectives included HCP recommendations for either device, diabetes-related clinical end points (changes in fasting blood glucose and A1C levels), and safety outcomes (hypoglycemia, adverse events) following administration of insulin glargine using the pen device and the vial-and-syringe system.
Subjects and Methods
Patients
This study enrolled insulin-naive adult patients (18–85 years old, male or female) with a confirmed diagnosis of T2DM who were receiving treatment with a combination of two or three oral antidiabetes drugs but required the addition of basal insulin to maintain glycemic control. Patients were excluded from the study if they had received insulin therapy in the preceding 12 months (apart from during periods of hospitalization), if they were using glucagon-like peptide 1 agonists, or if A1C levels were <7.0% or >10.0% at the screening assessment. Patients with dementia, severe visual or manual dexterity impairment, and/or any concomitant disease or medication thought to interfere with treatment or affect the ability to answer self-assessment questionnaires were also ineligible. The study was conducted according to the principles of Good Clinical Practice Standards and in accordance with the ethical guidelines of the Declaration of Helsinki. All patients provided written informed consent before enrollment.
Study design
The study design for the randomized, multicenter, crossover, open-label study conducted across 60 centers in the United States is shown in Figure 1. Insulin glargine was administered once daily, in the morning or in the evening, whichever was more convenient for the patient, with the time of injection remaining unchanged for the duration of the study. Patients continued their baseline oral antidiabetes treatment throughout the study. After a 1-week screening period, eligible patients entered a 4-week crossover period (Week 0) and were randomized to receive basal insulin glargine via a pen device (SoloSTAR® [sanofi-aventis, Paris, France]) or by vial-and-syringe for 2 weeks, after which time patients were switched to the alternative insulin delivery method for the remaining 2 weeks. Patients had been trained by their HCPs or site staff in the appropriate use of both devices prior to randomization at Week 0 and prior to switching devices at Week 2. In both treatment arms, the starting dose of insulin glargine was 0.2 U/kg of body weight. Patients were asked to self-monitor their fasting blood glucose levels on a daily basis in accordance with the standard of care after initiation of insulin in insulin-naive patients. Blood glucose monitors were provided by the study's sponsor, and training in their use was provided by the site staff at the start of the study. During the 4-week crossover phase, the dose of insulin glargine could be adjusted at the study investigator's discretion in line with standard clinical practice.
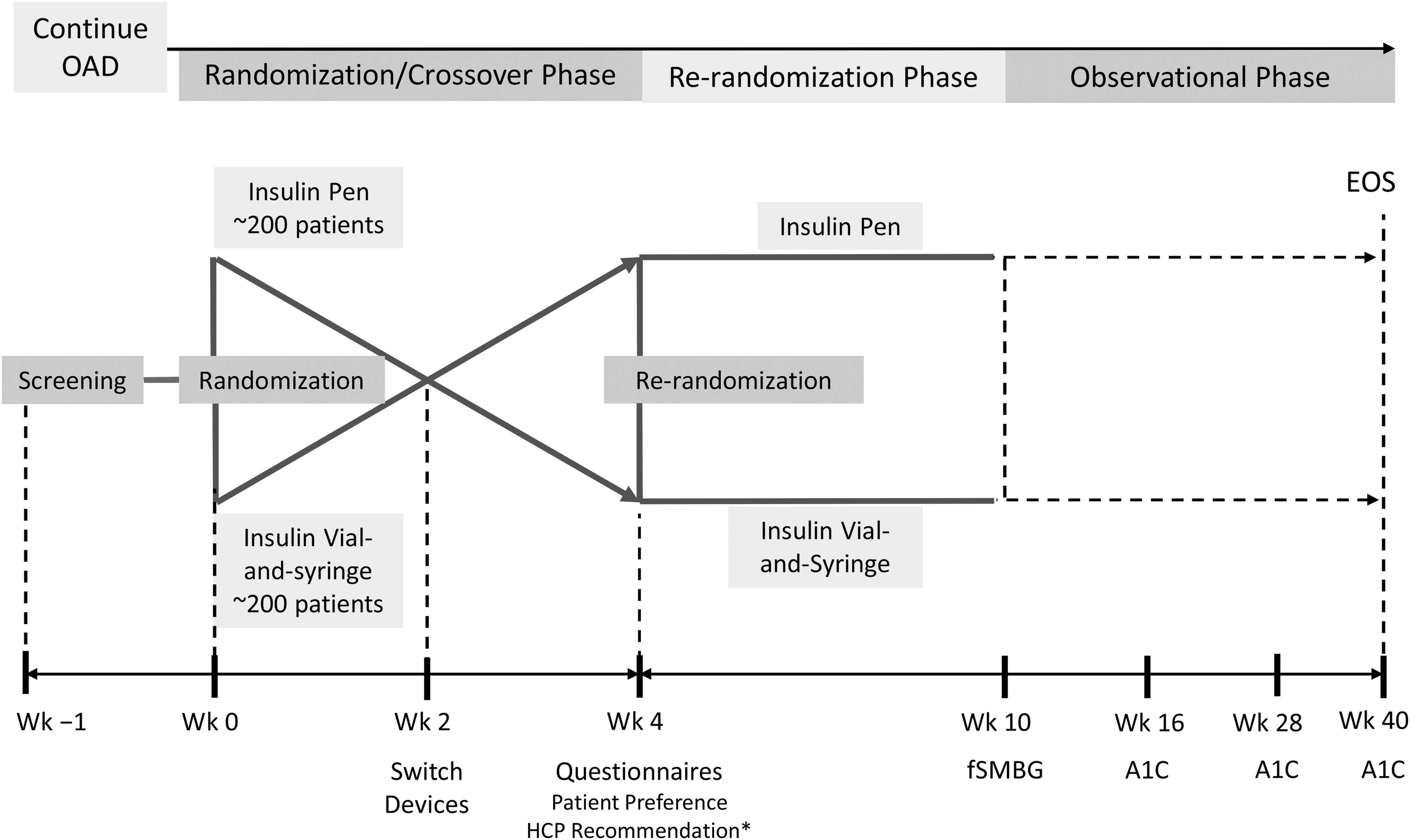
Study design. *Each investigator and one or two additional healthcare providers (HCPs) at each site who were involved in this study will complete an HCP Questionnaire after their last randomized patient has completed the randomization/crossover phase (Week [Wk] 4). A1C, glycosylated hemoglobin; EOS, end of study; fSMBG, fasting self-monitored blood glucose; OAD, oral antidiabetes drug.
Patients who had a mean 3-day fasting blood glucose level of ≥110 mg/dL 3 days before the end of Week 4 were re-randomized to receive insulin glargine via either the pen device or vial-and-syringe for an additional 6 weeks (re-randomization phase) for the evaluation of fasting self-monitored blood glucose (fSMBG) levels. During this phase, patients could adjust their own insulin dose using a titration algorithm provided. At Week 10, patients entered an observational phase of 30 weeks, during which they continued to use the insulin delivery method to which they had been re-randomized. Any insulin dose adjustments that occurred in this phase were recommended by the study investigators. A1C levels were determined at Week 16, 28, and 40 by a central lab.
Delivery system preference outcomes
The primary end point for this study was patient preference for insulin pen versus vial-and-syringe at the end of the crossover phase (Week 4). Preference measures were obtained using a previously developed instrument, known as the Insulin Injection Preference Questionnaire (IIP-q),
20
which assesses preference for the insulin delivery system. Patient preference was assessed using the difference in scores (using a 5-point scoring scale, from 1=not preferred to 5=preferred) for the pen and the vial-and-syringe methods of insulin delivery. A one-item global measure of preference and a three-item subscale measure of preference were evaluated for patient preferences (Fig. 2) and HCP recommendations (Supplementary Fig. S1; online Supplementary Data can be found at
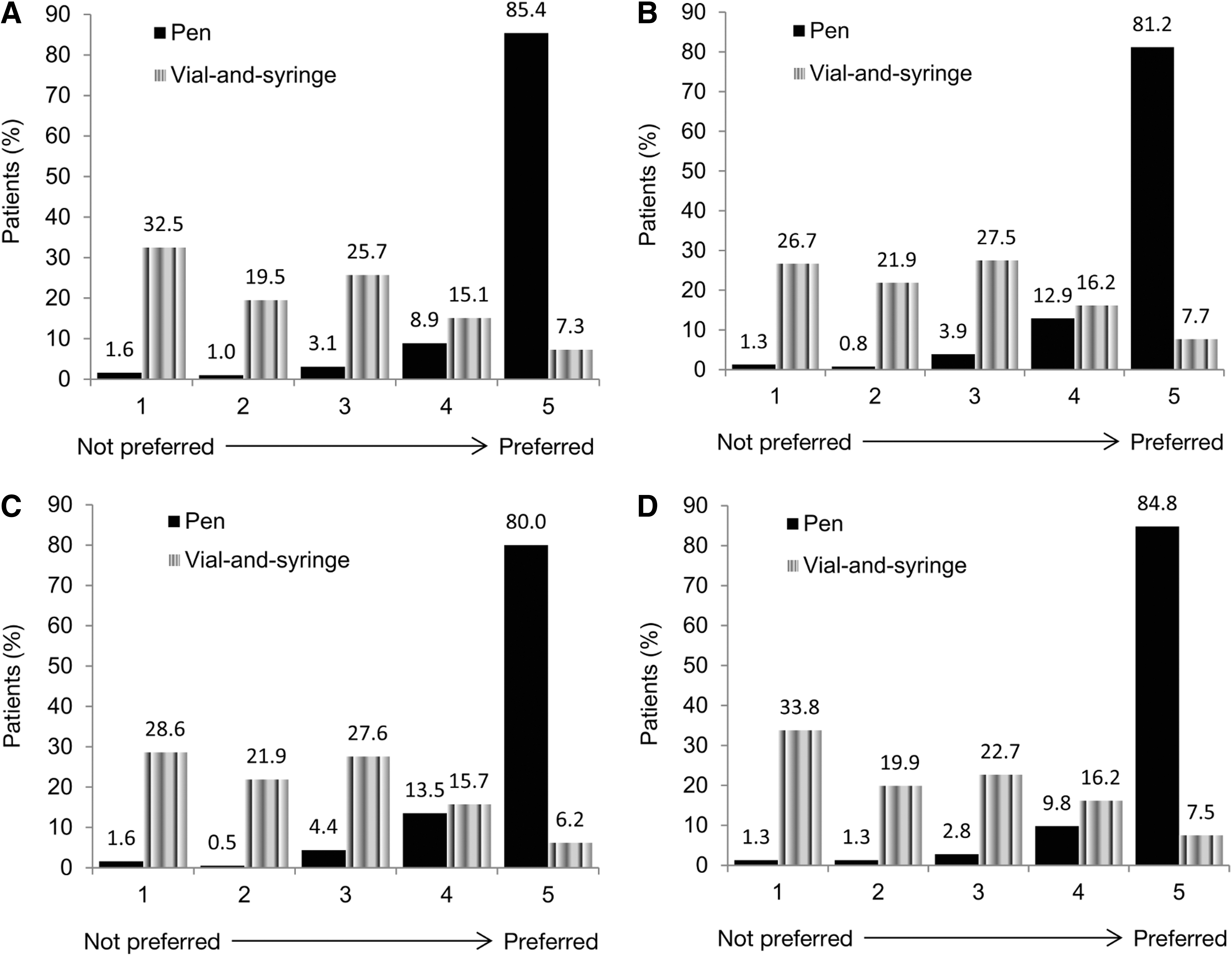
Patient questionnaire scores of pen versus vial-and-syringe preference rating, graded on a 5-point scale from 1 (not preferred) to 5 (preferred), for each insulin delivery device system at Week 4:
Clinical outcomes
The following clinical outcomes were assessed as secondary end points: • Change in fSMBG during the re-randomization phase (Week 4–10) (main clinical outcome) • Proportion of patients who achieved a fSMBG of <110 mg/dL by Week 10 • Change in A1C levels from re-randomization (Week 4) to the end of the observational phase (Week 40) • Proportion of patients achieving A1C levels <7.0% at Week 40 and the time to first observation of an A1C level <7.0% during the observational phase (Weeks 10–40) • Change in insulin glargine dose during the re-randomization phase (Weeks 4–10) • Time to discontinuation: the percentage of patients who discontinued therapy during the re-randomization phase (Weeks 4–10) and during the observational phase (Weeks 10–40). Reasons for discontinuation were also evaluated.
Safety outcomes
The safety outcomes in this study were the incidence of hypoglycemic events, reported adverse events, and vital signs.
The overall incidence of hypoglycemic events (event rate/patient-year) was determined based on fSMBG values below prespecified thresholds and included the following: the total incidence of symptomatic hypoglycemia; nocturnal hypoglycemia (events that occurred between midnight and 6:00 a.m.); severe symptomatic hypoglycemia (symptomatic hypoglycemic events with a blood glucose level of <36 mg/dL that required assistance or with prompt recovery after oral carbohydrate, intravenous glucose, or glucagon administration); and serious hypoglycemia (events that involved diabetic coma/loss of consciousness or seizure/convulsion).
Statistical methods
Sample size assumptions
At minimum, 130 evaluable patients per arm were required for the study to have sufficient statistical power to detect clinically meaningful differences in preference scores, using the paired t test of equal means (two-sided significance level 0.05).
Based on the assumption that 260 subjects would be eligible for the re-randomization phase, there would be approximately 80% power to detect a difference of 22.7 mg/dL in the change in fSMBG from baseline to Week 10 between pen and vial-and-syringe arms, assuming an SD of 65 mg/dL. Assuming a rate of 35% of nonevaluable patients, in total, approximately 400 patients were required.
Analysis populations
The modified intent-to-treat (mITT) population for the Patient IIP-q analysis consisted of all randomized patients who received at least one dose via both insulin delivery devices and completed the questionnaire at Week 4, irrespective of compliance with the study protocol and procedures. The mITT population for re-randomization consisted of all patients who were re-randomized (at Week 4), received at least one dose after re-randomization, and had both a re-randomization baseline (Week 4) assessment and at least one post–re-randomization baseline assessment of fSMBG, irrespective of compliance with the study protocol and procedures. The mITT population was used to ensure the presence of a completed questionnaire for both devices in order to perform the comparative analyses.
The safety population was defined as all patients who were randomized and exposed to at least one dose of insulin glargine after randomization/re-randomization (for the crossover and re-randomization phases) or during the observational phase.
The HCP population for the IIP-q analysis consisted of HCPs who had treated at least one randomized patient during the crossover phase (who had received at least one dose of insulin glargine via both insulin delivery devices during the crossover phase) and who had completed the HCP Questionnaire.
Efficacy analyses
Analysis of variance was carried out on the primary end point of overall patient preference. The observed preference scores for each delivery system were used as the response variable to compare pen and vial-and-syringe groups. The analysis of variance model included device, sequence group, and study period as fixed effects with the patient as a random effect for each sequence. Differences in patient preference scores of >0.5 were considered clinically meaningful. The secondary end points, including the three-item subscales of preference, and the composite scores from these, were analyzed in a similar manner.
Secondary outcomes from the HCP Questionnaire were analyzed using a paired t test with the 95% confidence interval (CI) calculated for the difference in HCP preference scores.
The continuous clinical efficacy end points (e.g., change in fSMBG from Week 4 to 10) were analyzed using analysis of covariance to compare the pen and vial-and-syringe groups. The analysis of covariance model included the delivery device as a fixed effect and the corresponding re-randomization baseline value as a covariate.
Comparisons of categorical end points (e.g., percentage of patients who reached the target fSMBG of <110 mg/dL at Week 10) between pen and vial-and-syringe groups were achieved using the Cochran–Mantel–Haenszel test taking into account the re-randomization baseline fSMBG/A1C categories cutoff by median.
The time to first observation of an A1C level of <7.0% was analyzed using survival analysis methodology, with the visit at Week 4 used as the starting time point. The comparison of the cumulative distribution function of each device group was achieved using a log-rank test.
Safety analyses
The incidence of hypoglycemia was estimated and summarized descriptively. For the re-randomization and observational phases, the incidence of hypoglycemia was analyzed using logistic regression models. The annual hypoglycemia event rate was summarized descriptively, and event rates were compared between devices using Poisson regression, accounting for overdispersion if applied.
Results
Demographics and baseline characteristics
In total, 623 patients were screened, from which 405 eligible patients were randomized and entered the crossover phase (randomized population; Supplementary Fig. S2). Baseline patient characteristics for the analysis population were similar (Supplementary Table S1). In the crossover phase, 202 patients were randomized to receive insulin glargine by pen followed by vial-and-syringe, whereas 203 patients were randomized to receive insulin glargine initially via vial-and-syringe before switching to the pen device after 2 weeks. The mITT population comprised 390 patients. Of the 383 patients who completed the crossover phase, 333 patients entered the re-randomization phase (mITT population, n=314 patients), and 312 patients were exposed to pen or vial-and-syringe during the observational phase (Supplementary Fig. S2).
Insulin exposure
In the safety population, the mean (SD) average daily insulin dose was 22.6 U (7.7 U) among 396 pen users and 23.0 U (8.0 U) among 399 vial-and-syringe users during the crossover phase. The mean (SD) average daily doses for the pen device and vial-and-syringe were 37.4 U (14.7 U) and 34.8 U (16.2 U), respectively, during the re-randomization phase (n=165 for each system) and 46.6 U (21.1 U) and 44.7 U (25.1 U), respectively, during the observational phase (n=159 pen users and n=153 vial-and-syringe users). The average daily dose was calculated as the sum of actual daily doses divided by the number of days of exposure using each device during each study phase.
Patient preferences
The primary end point of patient preference was assessed at the end of the crossover phase (Week 4) in the mITT population. The mean overall patient preference score based on the one-item preference question was significantly higher for the pen than for vial-and-syringe (least-square [LS] mean difference in overall preference score, 2.30; SE, 0.07; P<0.001), suggesting that patients strongly preferred the pen device. At Week 4, 85.3% of patients gave the highest score (5=preferred) to the pen compared with 7.5% of patients using the vial-and-syringe (Fig. 2).
High preference scores were also reported for each of the three subscale questions relating to patient preference, namely, preference for controlling blood glucose (LS mean difference, 2.16; SE, 0.07; P<0.001), preference for overcoming reluctance to use insulin (LS mean difference, 2.21; SE, 0.07; P<0.001), and preference for long-term use of insulin (LS mean difference, 2.31; SE, 0.07; P<0.001). The individual scores for each patient and subcategory are presented in Figure 2B–D.
HCP recommendations
Analysis of responses to the HCP recommendation questionnaire (n=135) found that significantly higher mean recommendation scores were observed for the pen compared with the vial-and-syringe, indicating that HCPs also recommended the pen device over the vial-and-syringe delivery system (mean difference, 2.2; SD, 1.1; P<0.001). At Week 4, 85.2% of HCPs gave the strongest recommendation score to the pen compared with 3.0% for the vial-and-syringe delivery system (Supplementary Fig. S1).
Similar to the findings for patient preference, HCPs also reported high recommendation scores for the pen compared with the vial-and-syringe for the three preference subscale questions: recommendation for controlling blood glucose (mean difference, 2.0; SD, 1.0; P<0.001), recommendation for overcoming reluctance to insulin (mean difference, 2.3; SD, 1.1; P<0.001), and recommendation for long-term use of insulin (mean difference, 2.1; SD, 1.1; P<0.001). The individual scores for each HCP Questionnaire subcategory are presented in Supplementary Figure S1B–D.
Clinical end points
In the mITT population, the changes in fSMBG and A1C values, insulin dose, and percentage of patients reaching A1C goal of <7.0% and fSMBG target of <110 mg/dL were similar in the two groups throughout the study (Table 1).
Change in A1C to Week 40 was not a prespecified end point.
A1C, glycosylated hemoglobin; fSMBG, fasting self-monitored blood glucose; LS, least square; mITT, modified intent-to-treat; NA, not applicable.
Discontinuations
Details of patient discontinuations during the different phases of the study are summarized in Supplementary Figure S3. In the observational phase of the study, there was a trend toward fewer patient discontinuations in the pen group (P=0.0699). The mean (SD) time to discontinuation was 206.5 (25.9) days among the 159 patients in the pen group and 199.5 (39.9) days among the 153 patients in the vial-and-syringe group.
Safety outcomes
Hypoglycemia
The incidence of hypoglycemia during each phase of the study is shown in Table 2. The overall incidence of hypoglycemia (any severity) was consistently, but not significantly, higher in the vial-and-syringe group than in the pen group during the crossover and re-randomization phases of the study and almost identical in both groups during the observational phase. No serious hypoglycemic events were reported during the study.
Hypoglycemia with fasting self-monitored blood glucose (fSMBG) or any self-monitored blood glucose <70 mg/dL and occurring between midnight and 6:00 a.m. was classified as nocturnal.
Severe hypoglycemia is classified as those events requiring assistance and either a blood glucose level of <36 mg/dL or prompt response to countermeasures.
Serious hypoglycemia was defined as events with coma/loss of consciousness or seizure/convulsion.
The most common treatment-emergent adverse events (TEAEs) during the crossover phase among pen and vial-and-syringe users were infections and infestations (7.1% and 3.8%, respectively), gastrointestinal disorders (2.3% and 3.8%, respectively), nervous system disorders (2.8% and 2.8%, respectively), respiratory, musculoskeletal, and connective tissue disorders (2.5% for both), thoracic and mediastinal disorders (2.3% and 1.3%, respectively), and skin and subcutaneous tissue disorders (1.5% and 1.8%, respectively).
The most common TEAEs during the re-randomization phase among pen and vial-and-syringe users were infections and infestations (18.2% and 10.3%, respectively), musculoskeletal and connective tissue disorders (6.7% and 5.5%, respectively), gastrointestinal disorders (5.5% and 4.8%, respectively), and nervous system disorders (4.8% and 3.0%, respectively).
The most common TEAEs during the observational phase among pen and vial-and-syringe users were infections and infestations (34.0% and 32.7%, respectively), gastrointestinal disorders (12.6% and 14.4%, respectively), nervous system disorders (12.6% and 10.5%, respectively), and respiratory, thoracic, and mediastinal disorders (10.7% and 7.2%, respectively).
CI, confidence interval; NA, not applicable; OR, odds ratio.
Treatment-emergent adverse events
The incidence of reported treatment-emergent adverse events increased with longer exposure and was similar in both groups during each phase of the study (Table 2).
Discussion
This open-label, randomized, multicenter study assessed patient and HCP preference of a pen device compared with the traditional vial-and-syringe method of administering insulin glargine to insulin-naive patients with T2DM. Additionally, the study evaluated the clinical benefit of the two methods of delivery. The overall patient preference for the pen device was significantly higher than that for the vial-and-syringe (P<0.001) and surpassed the predefined clinically meaningful difference. Likewise, HCPs strongly supported the use of the pen device by patients with T2DM. Clinical outcome measures of efficacy (fSMBG and A1C values) improved from baseline for both devices and were similar in both patient groups, as was the number of hypoglycemic events. There was a nonsignificant trend toward fewer discontinuations in the pen group.
The findings reported here corroborate previously published studies. 3,21 A randomized crossover study of 121 patients with diabetes reported that 74% of patients had a preference for prefilled disposable insulin pens (to administer premixed insulin analog) over vial-and-syringe; factors reported to influence patient preference for device included ease of overall use, readability of the insulin dose scale, more confidence in dose accuracy, and improved discretion for use in public situations. 3 Similarly, initiating basal or rapid-acting insulin treatment using a pen rather than vial-and-syringe was associated with better treatment persistence and adherence, lower levels of hypoglycemia, and comparable clinical efficacy. 21 –23
An important consideration in the choice of insulin delivery system is the potential impact of the device on diabetes-related clinical outcomes. In our study, diabetes-related clinical efficacy (fSMBG and A1C levels) and safety (hypoglycemia and adverse events) outcomes were similar among patients who used the insulin pen device and those who used the vial-and-syringe system despite the high patient preference for the pen device. It should be noted that in this study, a relatively low percentage of patients in each of the treatment groups reached the traditional glycemic targets of A1C <7.0% and fSMBG <110 mg/dL. Perhaps the use of a treat-to-target approach would result in more aggressive dosing, which could potentially unmask the outcome benefits of the preferred, more convenient insulin pen delivery system. Previous studies have reported comparable A1C control, changes in fSMBG levels, and four-point blood glucose profiles and a reduced risk of hypoglycemia among patients using pen devices compared with vial-and-syringe for the delivery of basal 21 or premixed 3 insulin.
Considerations
Taking account of patient treatment preferences and individualizing patient care are of great importance in the management of a chronic condition such as diabetes. 11,24 An increased understanding of patient preferences may lead to better treatment compliance and improve long-term outcomes. This study did not evaluate treatment compliance and did not have a long-term time frame; however, future studies comparing pen and vial-and-syringe delivery methods could add compliance to the study end points and extend the study duration to allow a fuller evaluation of the impact of patient preference on treatment.
Our finding that HCPs would recommend that their patients initiate basal insulin therapy using a pen device rather than a vial-and-syringe was consistent with a previous study in which physicians who were unfamiliar with insulin delivery methods found the insulin pens easier to handle, preferable to use, and more accurate in delivering insulin doses compared with vial-and-syringe. 9 Physician recommendations were highly influential in patients' decisions to use an insulin pen.
This was the first study to use a specific patient preference questionnaire—the IIP-q—in a prospective, randomized clinical trial with a crossover design, which allowed the unbiased comparison of the two insulin delivery systems. Our study revealed a consistent response pattern between HCP recommendations and patient preferences for insulin delivery devices regarding blood glucose control, the reluctance to use insulin, and long-term insulin use. A mutually shared decision process between HCPs and patients may improve patient adherence behavior and other patient-reported outcomes.
Limitations of our study include the short duration and the one-time evaluation of patient preference. Even though fSMBG and A1C end points were not suited to a 2×2 crossover design, the difference in time points will make it harder to associate patient preference with clinical outcome data.
The use of an mITT study design has certain limitations, one of which is the conceptual link between bias introduced by intent-to-treat analyses and bias due to loss to follow-up. In this study, patients were included in the mITT analysis if questionnaire data from both devices were available (primary end point). Although it is possible that the excluded patients may impact the study outcomes, the rate of excluded patients was very low, with <4% (n=15) of randomized patients not included in the mITT analysis. Among those excluded were patients who did not complete the crossover but for whom a valid questionnaire was obtained.
Conclusions
We conclude that the use of the insulin pen device for the introduction of basal insulin glargine treatment in insulin-naive patients with T2DM is preferred by patients and recommended by HCPs.
Footnotes
Acknowledgments
This study was funded by Sanofi U.S., Inc. The authors wish to thank Hailing Guo, MSc, for her assistance with the data collection and analysis. The authors received editorial/writing support in the preparation of this manuscript provided by Tessa Hartog, PhD, of Excerpta Medica, funded by Sanofi U.S., Inc.
Author Disclosure Statement
A.A. has served as a consultant to Sanofi U.S., Inc. and Novo Nordisk, Inc. S.L.S. received financial support from Sanofi U.S., Inc. for her role in the study design, data interpretation, and manuscript preparation. J.G. and L.T. are employees at Sanofi U.S., Inc. S.K.G. has participated in advisory panels for Sanofi U.S., Inc., Eisai, Halozyme, Valeritas, Medtronic, Novo-Nordisk and Dexcom, Inc. and has received research support from Sanofi U.S., Inc., Novo Nordisk, Inc., Eli Lilly and Company, MannKind, Medtronic, Halozyme, and Cebix.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.