
Abstract
The discovery of insulin and its clinical application early in the last century dramatically improved the prospects of people with diabetes. However, the limitations of those initial, unmodified insulin preparations were quickly recognized; most notably, their relatively “short action” meant that multiple daily subcutaneous injections were required. This stimulated a concerted effort to modify the properties of insulin in order to extend the duration of its blood glucose-lowering effect, minimize dosing frequency, and decrease the burden of treatment. The first successful attempts to prolong insulin's action were achieved by modifying its formulation with additives such as protamine and zinc, culminating in the production of “intermediate-acting” neutral protamine Hagedorn (NPH) insulin in the 1940s and the lente family of insulins in the 1950s. However, NPH and lente insulins were still associated with several limitations, including considerable variability of effect and a pronounced peak in their time–action profile. In the 1980s, the focus of research moved toward the modification of insulin itself with the aim of producing a “long-acting” insulin that would better satisfy basal insulin requirements over the entire day. Once-daily insulin glargine was the first “long-acting” insulin analog in clinical practice, followed by once- or twice-daily insulin detemir and, more recently, insulin degludec, which is now being evaluated for administration at less frequent intervals. These analogs demonstrate several benefits over “intermediate-acting” insulins, including a lower risk of both overall hypoglycemia and nocturnal hypoglycemia and reduced day-to-day glucose variability, making it more feasible to achieve better fasting and overall glycemic control. Long-acting insulin analogs (insulin glargine and insulin detemir) are now firmly established as key tools in the battle against diabetes, and ongoing clinical research of insulin-based therapy should focus on treatment strategies to maximize their benefits. To date, the clinical experience with insulin degludec is limited but demonstrates it has comparable efficacy to insulin glargine.
Introduction
Prolonging Insulin Action—The Early Years
Initial attempts to prolong the action of insulin involved combining it with gum arabic solutions, 3 oil suspensions, 4 lecithin emulsions, 5 or vasoconstrictor substances, 6 none of which was very successful. 7,8 In 1935, the research groups of Maxwell and Bischoff 9 and Scott and Fisher 10 reported that the addition of certain metal ions, such as zinc, prolonged insulin activity. The following year, the use of strongly basic proteins (protamines) combined with neutral suspensions of insulin was also shown to delay absorption from a subcutaneous depot. 11 During 1936, Hans Christian Hagedorn and colleagues produced the first clinically useful protracted, “intermediate-acting” insulin in the form of a neutral suspension of protamine insulin, 12 although this was found to be chemically unstable. A further preparation using excess protamine and a small amount of zinc, 13 known as protamine zinc insulin (PZI), achieved a prolonged hypoglycemic effect of up to 24 h. 14,15 However, PZI was limited by a greater risk of hypoglycemia, with reports of sudden and severe attacks. 15 In addition, the onset of action of PZI was slow (1–3 h) and required the addition of soluble insulin to achieve an intermediate action, 16 –18 but this admixture was shown to be unstable 19 and had to be administered as separate injections, causing inconvenience to patients. Finally, in 1946, Hagedorn and colleagues introduced crystalline neutral protamine Hagedorn (NPH) insulin, which was a more stable PZI modification combining insulin and protamine in “isophane” proportions (no excess of insulin or protamine) at neutral pH in the presence of a small amount of zinc and phenol and/or phenol derivatives to generate tetragonal oblong-shaped crystals. 20 Unlike PZI, the NPH insulin preparation could be premixed with an intermediate-acting insulin and quickly became, and has since remained, popular as a once- or twice-daily insulin, used alone or in combination with soluble insulin as required. 21
Research and development continued to advance, and other insulin preparations were introduced in an attempt to address the continued need for long-acting agents with a better time–action profile. In the 1930s, globin insulin was developed, 22,23 as well as surfen insulin, which was formulated with a synthetically produced urea as an alternative to protamine. 24 Then in the 1950s, the lente trilogy of insulins, comprising semilente, lente, and ultralente, were developed by Hallas-Møller 25 and colleagues. These were protracted insulin preparations, without any added foreign proteins or synthetic compounds, created by complexing neutral suspensions of insulin with small amounts of zinc ions. The duration of action of members of the lente family of insulins was determined by the physical state, size, and zinc content of the suspended zinc insulin particles.
Traditionally, insulin was extracted from porcine and/or bovine pancreata, which have different solubilities at neutral pH. Insulins of the lente type capitalized on this to achieve different protracted activity. The original lente insulin preparation comprised a 3:7 mixture of amorphous porcine insulin and bovine crystalline particles, with an intermediate timing of action similar to that of NPH insulin. 26,27 Bovine ultralente insulin comprised fairly large crystals (30 μm) resulting in a duration of action similar to PZI. Ultralente is considered to be the first real “long-acting” insulin preparation, requiring concomitant administration with preprandial insulin in subjects with type 1 diabetes and late in the natural history of type 2 diabetes. Additionally, lente-type preparations such as Monotard® (Novo Nordisk, Bagsvaerd, Denmark) and Rapitard® (Novo Nordisk), comprising 100% porcine insulin and 25% porcine/75% bovine crystalline insulin, respectively, were later produced. 28 –30 Throughout this period, considerable efforts were also being made to better purify the various insulin preparations.
During the 1980s, commercial production of human insulin began, initially derived semisynthetically from porcine insulin using an enzymatic process 31 and later biosynthetically using Escherichia coli or yeast (Saccharomyces cerevisiae). 32,33 Subsequently, NPH, lente, ultralente, and premixed preparations were reformulated using human insulin. However, evidence revealed that long-acting human insulin had a shorter duration of action than the animal equivalent. For example, the human insulin version of ultralente (Ultratard® HM) is absorbed much more quickly than its bovine counterpart (Ultratard MC) (both from Novo Nordisk). 34,35 The development of ultralente HM, therefore, deviated from the ideal slowly absorbed preparation that is required to mimic the pattern of basal insulin secretion in people without diabetes. Insulin formulations based on crystalline suspensions, including the lente-type insulins and NPH, are also limited by the need for resuspension. The variability of response seen with these insulins may be a result of a failure to evenly resuspend the insulin within the vial immediately prior to administration. The development and evaluation of biosynthetic human proinsulin, the precursor of endogenous insulin, as a long-acting insulin achieved a significant reduction in variability of effect relative to NPH insulin, while demonstrating a similarly prolonged hypoglycemic action. 36 However, the failure of human proinsulin to demonstrate efficacy benefits over NPH, together with evidence, although not conclusive, of cardiac adverse events led to the termination of its development for clinical use. 36
Exogenous insulin may also be limited by its distribution in the body. Endogenous insulin is secreted from the pancreas into the hepatic portal vein, such that the liver is subjected to relatively high insulin concentrations and is responsible for uptake of as much as 60% of the pancreatic output. When exogenous insulin is delivered subcutaneously, its distribution is more even throughout the body, and the liver may be relatively underinsulinized. Hepatoselective exogenous insulin was generated in the 1990s by covalently linking a thyroxyl group to the insulin molecule. The resulting thyroxyl-insulin analog binds to thyroid hormone binding proteins to form complexes with a molecular size that limits access to the peripheral tissues because of the capillary endothelial barrier, but is more readily accessible to the liver hepatocytes because of the greater permeability of the sinusoid vessels. Small-scale, clinical studies showed that thyroxyl-insulin had an inhibitory effect on hepatic glucose production that was comparable to the value for NPH insulin, but the effect on peripheral glucose uptake was significantly lower than for NPH. 37 Utilization of insulin analog techniques (“designer insulins”) has subsequently been influential in the development of pharmacokinetically optimized insulins for clinical practice.
Retarding Insulin Absorption Through Targeted Structural Modification: The Insulin Analogs
Short- and intermediate-acting insulins have important roles to play in the overall management of diabetes, but the availability of long-acting insulin preparations, effective over a period of 24 h, is better suited to maintain physiological basal insulin levels (Table 1). The advent of recombinant DNA technology enabled researchers to begin to modify the structural properties of insulin with the subsequent introduction of human insulin and later insulin analogs in an attempt to optimize their physicochemical characteristics to better simulate insulin secretory patterns.
NPH, neutral protamine Hagedorn.
Early long-acting human insulin analogs were created through the addition of a positive charges, either by removal of carboxylate anions or by substituting with lysine or arginine using single-chain insulin precursors. 38,39 As a result, the isoelectric point of the insulin was increased from 5.4 toward neutrality. Some of these insulin derivatives crystallize instantly when the pH of their slightly acidic solution is adjusted to neutral pH and thus becoming less soluble at the physiological pH of the subcutaneous tissue. Substitution with basic amino acids at the C-terminus of the B chain promotes crystallization, possibly by promoting faster interaction between hexamers, thereby ultimately slowing absorption because of the delayed disassociation of the insulin molecules from the hexameric state.
Studies continued in the 1980s involving several human insulin analogs, which constituted various amino acid substitutions, found that, of those tested, the GlnB13, ArgB27, ThrB30-NH2 analog had the slowest rate of dissolution at the subcutaneous injection site. 39 It was subsequently found that an additional substitution at residue A21 with glycine conferred improved chemical stability during storage of the slightly acid analog solutions. 38 The resorption of the 125I-labeled GlyA21, ArgB27, ThrB30-NH2 human insulin analog (OPID 174) following subcutaneous bolus injection in individuals with type 1 diabetes demonstrated a slower and more consistent rate of dispersal compared with the human ultralente preparation. 40 However, this insulin analog was not introduced into practice because of the need for escalating doses during clinical trials of NPH. 41 Di-arginine insulin was also evaluated but compared poorly with NPH insulin.
Currently Available Long-Acting Insulin Analogs
Insulin glargine
In 2000, insulin glargine (glargine) became the first long-acting insulin analog to be introduced into clinical practice. Glargine is an analog of human insulin produced by recombinant DNA technology whereby the insulin molecule has undergone two modifications: elongation of the C-terminus of the B chain with the addition of two arginine residues inserted at position B30 and replacement of asparagine with glycine at position A21 (Fig. 1A). 42 The latter modification provides greater stability in the acidic pH of the insulin vial and makes insulin glargine less soluble at neutral pH. As a result, glargine generates an amorphous precipitate in the subcutaneous tissue following injection, retarding its absorption and thereby extending its duration of action. 43
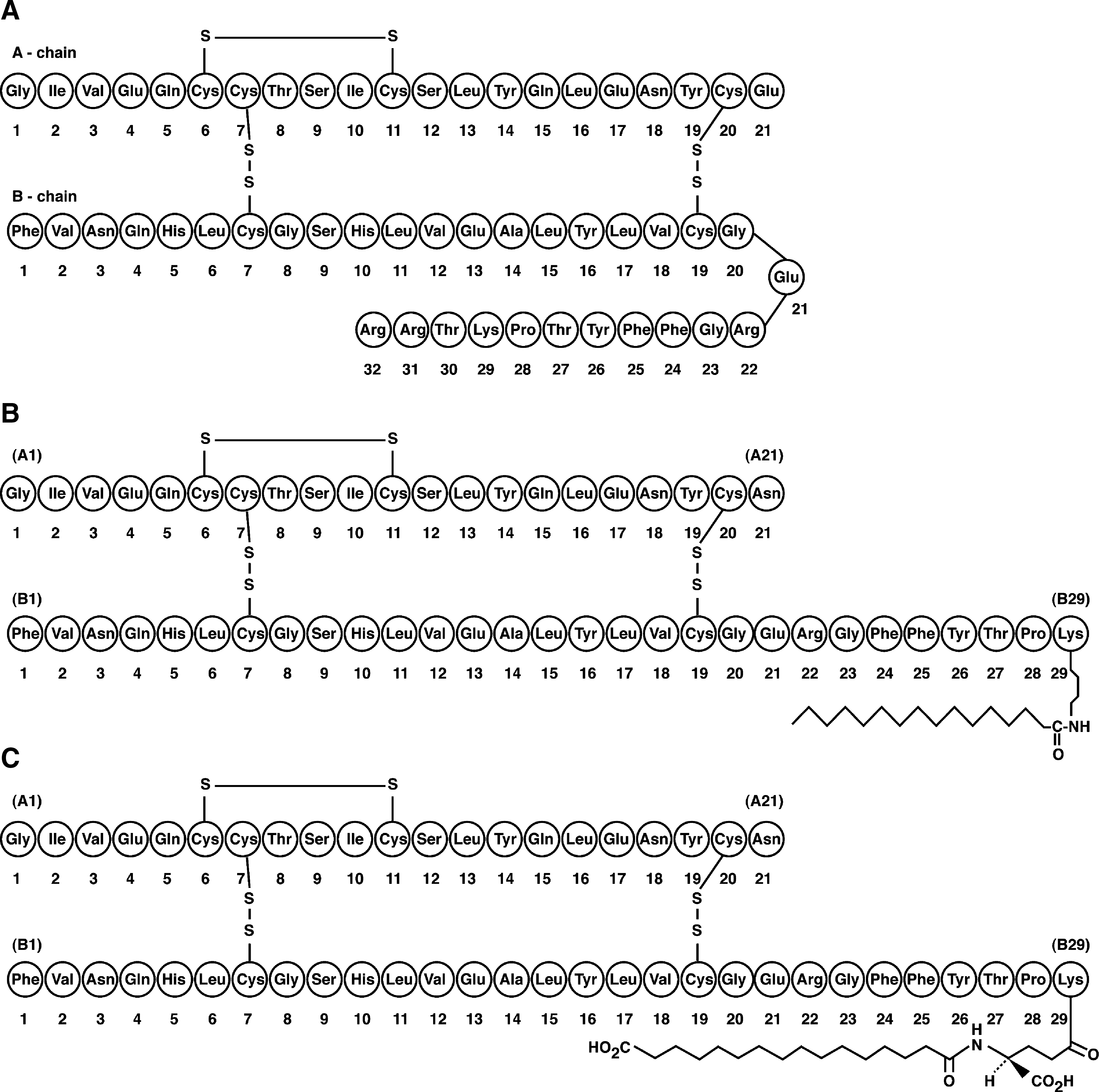
Molecular structure of long-acting insulin analogs: (
Glargine represents a new era in protein engineering, 44 with one amino acid exchange at A21; otherwise it is produced in the same process as human insulin followed by the di-arginine addition step. Insulin glargine represents the first example of a “pro-drug” protein. 45 Insulin glargine itself is active and therefore not truly a prodrug but is needed for precipitation in the subcutaneous tissue. Following injection, glargine undergoes rapid sequential cleavage of the C-terminus of the B chain, forming active metabolites in the subcutaneous tissue. 46 This reflects the process of degradation of proinsulin to insulin in the pancreatic β-cell. A study in healthy volunteers has demonstrated that enzymatic cleavage/maturation of glargine occurs very quickly at the injection site and continues within the circulatory system with the dissociation of the di-arginine molecules to provide fully potent insulin metabolites. 46 When administered on a once-a-day basis, a steady-state condition is achieved within a few days, with no further accumulation in plasma insulin levels. 47,48
The primary rationale behind the development of glargine was to avoid the poor reproducibility and pronounced peak action profile of NPH, while also providing a longer duration of action to better cover the basal insulin requirements over the entire day (Fig. 2). 49 –51 Glargine demonstrated a nearly flat time–action profile and reduced variability compared with NPH, 49,52 with an end of action time of up to 24 h, which is dose dependent and also longer acting than NPH insulin in individuals with type 1 diabetes. 48 –50,53,54 The absence of a clear peak explains the lower risk for nocturnal hypoglycemia and therefore the ability to better dose-titrate in order to achieve lower fasting blood glucose levels in both type 1 and type 2 diabetes. 44 The soluble pharmaceutical formulation of glargine explains the lower variability in both the pharmacokinetics and pharmacodynamics compared with NPH 55 or ultralente insulin, 56 as the latter insulins require homogenization by shaking prior to injection. This finding has been confirmed in a clinical study of people with type 1 diabetes. 57
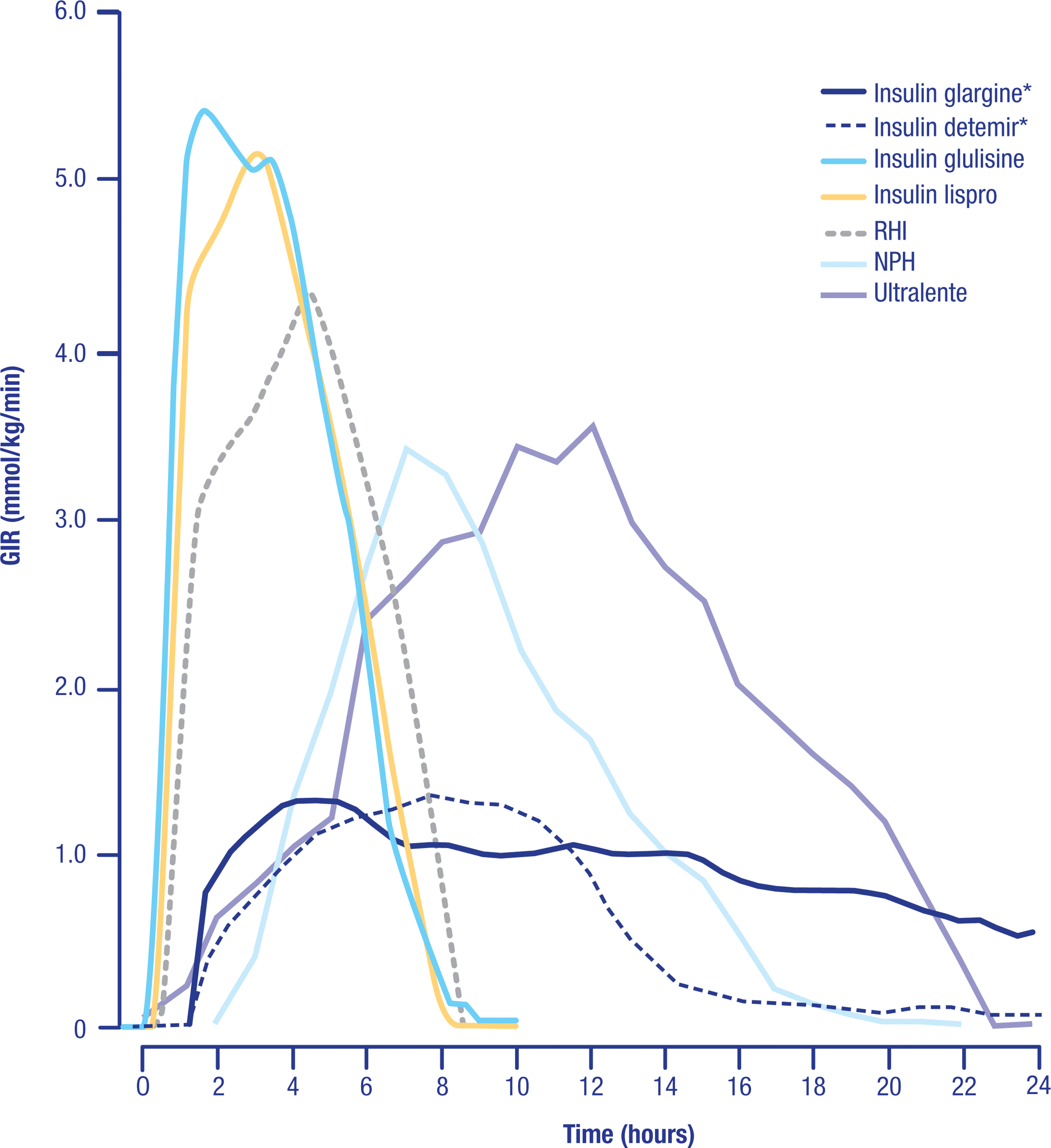
Cross-study comparison of insulin time–action profiles. Data are summarized from Lepore et al., 49 Porcellati et al., 50 and Becker et al. 51 All insulins were dosed at 0.3 U/kg (*a dose of 0.35 U/kg was used). All data are for patients with type 1 diabetes with the exception of glulisine, lispro, and regular human insulin (RHI), which are for obese healthy volunteers. GIR, glucose infusion rate; NPH, neutral protamine Hagedorn.
The most convincing evidence for the suitability of glargine to act as a basal insulin derives from studies that compared once-daily glargine in a multiple daily injection regimen with continuous subcutaneous insulin infusion (CSII). 58 –61 No differences were found between the two regimens regarding the level of glycemic control (A1C) achieved or the frequency of hypoglycemia. In the study by Bolli et al., 62 there was also no difference in plasma glucose variability between the two regimens. The key differences between the two methods included the greater cost (∼3.9 times) of CSII compared with a glargine-based regimen of multiple daily injections, despite the lower (∼20%) insulin dose used in the pump. 61
The physicochemical properties of glargine relate to its safety profile. In vivo, glargine is rapidly cleaved into its active metabolites, which behave similarly to human insulin and thus have a low risk of adverse events related to binding at the insulin growth factor (IGF)-1 receptor, in contrast to what has been observed in vitro when using the parent compound, glargine. 63,64 Clinically, no adverse outcomes have been observed with glargine with respect to microangiopathy, with no higher risk of early worsening of diabetic retinopathy with glargine compared with NPH over and above what may be related to improved glycemic control. 65
More recently, a 5-year, prospective, open-label, multicenter study was conducted in a large number of people with type 2 diabetes comparing the progression of diabetic retinopathy in the NPH- and glargine-treated groups. 66 NPH was administered once or twice a day, whereas glargine was administered once daily with regular insulin introduced as needed. The level of glycemic control was similar at baseline in the two groups, with A1C and fasting plasma glucose (FPG) values for NPH and glargine of 8.3% versus 8.4% and 10.0 mmol/L versus 10.5 mmol/L, respectively. Seven-field fundus photographs taken at screening and at 3, 6, 12, 24, 36, 48, and 60 months of treatment were graded according to the Early Treatment Diabetic Retinopathy Study scale, with a three-step or more change regarded as progression. The prevalence of diabetic retinopathy at baseline was 12.1% versus 15.6% for NPH and glargine insulin, respectively. At study end, both groups achieved a mean FPG level of 7.7 mmol/L with the A1C 0.21% higher in the NPH-treated group. A three-step or more progression in the Early Treatment Diabetic Retinopathy Study score was not significantly different (i.e., 15.7% versus 14.2% in the NPH- and glargine-treated groups, respectively). Thus, in this long-term study, glargine posed no additional independent risk of progression of diabetic retinopathy compared with NPH.
Overall, in individuals with type 1 diabetes, glargine offers lifestyle benefits, improved convenience with only once-daily administration, and flexibility as to timing of injection (morning, pre-dinner, or pre-bedtime). 67 There is no requirement to eat at defined times, and a fast of up to 18 h can be tolerated. 68 Furthermore, it has been demonstrated that subcutaneous absorption of glargine is not affected by exercise. 69 In individuals with type 2 diabetes, glargine also offers improved safety (reduced risk of nocturnal hypoglycemia) and convenience (once-daily administration) when attempting to reach a target A1C level of 7% (achieved in more than 50% of subjects). 70 Although the level of glycemic control achieved does not differ from that achieved with NPH, 70 the lower risk of hypoglycemia allows for a more aggressive dose titration and consequently a greater likelihood of reaching and maintaining a lower glycemic target value over time. Indeed, a meta-analysis of patient data from 11 randomized controlled trials comparing glargine with NPH in type 1 or type 2 diabetes populations showed that the reduction in hypoglycemia with glargine versus NPH was typically underestimated. 71 Analysis of hypoglycemia in these trials did not take into account the differences in A1C levels between the glargine- and NPH-treated groups, and adjustment for endpoint A1C resulted in markedly greater relative reductions in the risk of hypoglycemia with glargine versus NPH. It is important that this study also revealed that the relationship between A1C and hypoglycemia for glargine and NPH diverged at lower A1C levels such that excess risk of hypoglycemia with NPH versus glargine was greater at an A1C of 7% than at 9%.
Insulin detemir
Insulin detemir (detemir) was the second basal insulin analog available for clinical use. Detemir is also produced by recombinant DNA technology, with the deletion of the amino acid threonine at position B30 of the human insulin molecule and the addition of a myristic fatty acid residue to the ɛ-amino group of the lysine residue at position B29 (Fig. 1B). 72 The acylation of the insulin molecule with a fatty acid residue gives the resulting molecule a higher binding affinity to albumin in the subcutaneous, intravascular, and extracellular compartments, which delays its absorption and reduces the variability of its biological activity. 73
Albumin is a multifunctional transport protein that binds reversibly to several endogenous and exogenous substances. 74,75 The first hypothesis for the biochemical concept of albumin binding to protract the effect of insulin was proposed by Kurtzhals et al., 76 capitalizing on the knowledge that many drugs bind to albumin and that their activity profile can therefore be altered by modifying their affinity for albumin. 77 A series of insulin derivatives were produced by acylation of the insulin with fatty acids of varying carbon chain lengths and affinities for albumin to find the optimum profile. 76 The binding affinity of the acylated insulin for albumin relates to the number of carbon atoms on the fatty acid chain, which, in turn, relates to the duration of action of the acylated insulins, such as detemir. 73
In later studies, Kurtzhals 78 proposed that albumin binding, in addition to protracting absorption, improves reproducibility of the time–action profile of acylated insulins. Furthermore, it has been proposed that delayed transcapillary transport of insulin detemir in skeletal muscle occurs primarily via a nonsaturable process, such as passive diffusion via a paracellular or transcellular route, thus delaying the time for the insulin to reach its receptor. 79 Acylated insulin analogs have a much reduced affinity for the insulin receptor, necessitating increased levels of active principle to achieve equivalent biopotency with human insulin preparations. 63 Detemir must therefore be administered subcutaneously at five times the concentration of other basal insulins 80 and, intravenously, at approximately eight times the concentration of human recombinant insulin 81 in order to provide equivalent metabolic effects. In clinical studies, higher doses of the acylated insulins are also required in order to achieve the same metabolic outcome as nonacylated insulins, such as NPH and glargine. 82 –84 Neverthelesss, detemir has consistently demonstrated near-equivalent glycemic control and a lower increase in body weight compared with NPH in type 1 and 2 diabetes at appropriate doses. 85
Comparing glargine and detemir
The efficacy and safety of glargine and detemir have been compared in people with type 2 diabetes, as add-on therapy to oral antidiabetes agents. 84 Rosenstock et al. 84 compared glargine and detemir in a 52-week, open-label, parallel, multinational trial of 582 insulin-naïve individuals. Basal insulin was initiated once daily in the evening, with a starting dose of 12 units; detemir was used additionally in the morning if pre-dinner plasma glucose exceeded 7.0 mmol/L (126 mg/dL). Both detemir and glargine improved A1C (from 8.6% at baseline to 7.2% and 7.1%, respectively; difference, 0.05% [95% confidence interval, −0.11 to 0.21%]), with similar FPG in both groups. Furthermore, the percentage of subjects who achieved A1C <7.0% without experiencing hypoglycemia was also similar (33% vs. 35%, respectively). However, weight gain was less with detemir than with glargine in completers (3.0 vs. 3.9 kg; difference, −0.9 kg [95% confidence interval, −1.6 to −0.2 kg]; P=0.01). In the detemir-treated group, 55% required a twice-daily dose of 1.0 U/kg, whereas the remaining 45% only required a once-daily dose of 0.52 U/kg (overall mean daily dose of detemir was 0.78 U/kg). In contrast, with glargine (once daily in all subjects) the mean daily dose was 0.44 U/kg. This study indicates that glargine and detemir may be equivalent as basal insulins in type 2 diabetes in terms of glycemic goals (lowering of A1C and risk of hypoglycemia) but that detemir required a higher dose with twice-a-day regimen in more than 50% of the cases with slightly lower weight gain.
These early findings were confirmed in a more recent, 24-week, randomized, “treat-to-target” trial comparing glargine (once daily) with detemir (twice daily) in 973 insulin-naïve individuals with type 2 diabetes inadequately controlled on oral antidiabetes drugs. Glargine achieved similar glycemic control to detemir, at significantly lower doses, but with more weight gain. 86 Further studies have also shown detemir (once or twice daily) and glargine (once daily) to be similar in terms of glycemic efficacy and safety when used as part of basal–bolus regimens with short-acting insulin analogs. 87,88 Although absolute weight gain is less in detemir-treated individuals compared with glargine-treated patients, 88 a meta-analysis of 21 studies involving either glargine (17 trials) or detemir (three trials) and one trial comparing glargine and detemir showed that weight gain adjusted for change in A1C was not significantly different between the two analogs. 89
Other Long-Acting Insulin Preparations in Development
The newest arrival to the long-acting insulin analog arena is insulin degludec, currently in Phase III development. Degludec is a soluble, basal insulin analog with a reported duration of action of over 24 h and the potential for a thrice-weekly dosing schedule. 90,91 The molecular structure of degludec retains the human insulin amino acid sequence, apart from the deletion of ThrB30 and the addition of a 16-carbon fatty diacid attached to LysB29 via a glutamic acid spacer (Fig. 1C). 92,93 Under in vitro conditions that mimic the subcutaneous injection site, degludec has been shown to self-associate to form large multihexamer assemblies at physiological pH. 92,93 As absorption rate is influenced by molecular size, this results in the slow release of degludec monomers and an “ultra-long” action profile in type 1 diabetes.
Results from “proof-of-concept,” Phase II, 16-week, open-label, randomized trials in both type 1 and type 2 diabetes have recently been published comparing the efficacy and safety of degludec in various formulations with that of glargine. 94,95 In individuals with type 1 diabetes (n=178), two strengths of degludec (600 and 900 μmol/L) were compared with glargine (600 μmol/L), all in a once-daily regimen, 94 and in combination with insulin aspart (aspart) at meal times. Over the study period, essentially similar results were reported for degludec (600 and 900 μmol/L) and glargine in terms of change from baseline in A1C (–0.6%, −0.5%, and −0.6%, respectively), change from baseline in FPG (–1.60, −2.06, and −0.54 mmol/L, respectively), and the overall shape of the nine-point self-monitored plasma glucose profiles at end point. Rates of confirmed hypoglycemia and nocturnal hypoglycemia with degludec at 600 and 900 μmol/L were numerically lower compared with glargine.
In insulin-naïve subjects with type 2 diabetes (n=178), once-daily degludec, combined with insulin aspart in two different formulations (70/30 [IDegAsp] and 55/45 [alternative formulation]), was compared with once-daily glargine, administered pre-dinner. 95 Glycemic control was similar across all treatment groups (i.e., changes from baseline in A1C were −1.3%, −1.5%, and −1.3% for IDegAsp, alternative formulation, and glargine, respectively), and approximately 50% of subjects in all groups achieved A1C <7.0%. Mean changes from baseline in FPG levels were −4.3, −4.1, and −5.1 mmol/L, respectively, although the 2-h post-dinner plasma glucose increase was lower for both degludec groups than for glargine, which was as expected given the aspart “bolus” component in the degludec treatment arms. The incidence of hypoglycemia and nocturnal hypoglycemia was low and similar across all treatment groups, and increases in body weight were also low and comparable.
The results of a third Phase II randomized trial have also been published comparing once-daily insulin glargine with once-daily (600 or 900 nmol/mL) or thrice-weekly (900 nmol/mL) degludec in insulin-naïve individuals with type 2 diabetes. 96 The data showed comparable improvements in glycemic control and similar, low rates of hypoglycemia across all four treatment groups over 16 weeks of treatment. Changes in body weight were minimal, although a significantly greater change in body weight was observed with once-daily degludec at 900 nmol/mL compared with insulin glargine. Phase III studies with degludec are ongoing.
Two other basal insulin analogs—LY2605541 and LY2963016—are currently in clinical development. Two Phase II studies that compared LY2605541 with glargine in people with type 1 and type 2 diabetes have been completed (registration numbers NCT01049412 and NCT01027871 at ClinicalTrials.gov), and publication of data is awaited. Phase III clinical testing for both insulins is expected to commence in 2011.
Alternative Approaches Providing Basal Insulin Requirements
Insulin pumps have been used for several decades in conjunction with a short- or rapid-acting insulin for CSII, particularly in patients with type 1 diabetes, although the practicalities and expense of traditional pump technology may have limited its use. Recent efforts to simplify these delivery systems has resulted in the development of “patch pumps” that integrate an insulin reservoir, delivery system, and cannula into a small, wearable, disposable, or semidisposable device that adheres directly to the skin. 97,98 In one small-scale, 30-day study of patients with type 1 diabetes, A1C was found to be significantly lower after patients switched from a traditional CSII pump to a patch pump. 99
A novel approach to injectable therapy currently undergoing preclinical investigation is that of a “glucose-responsive insulin formulation.” SmartInsulin™ (SmartCells Inc., Beverly, MA, USA) consists of a modified insulin that is reversibly bound to an engineered multivalent glucose-binding molecule. 100 Glucose in the blood competes with the modified insulin to bind to the glucose-binding molecule, thereby displacing the insulin and releasing it into the bloodstream. 100 Thus, insulin is only released in response to a specific blood glucose concentration, and so, in theory, glucose-responsive insulin formulations may provide basal and prandial insulin requirements with a low risk of hypoglycemia. 96
Conclusions
The two currently marketed long-acting insulin analogs, glargine and detemir, represent the most significant advances in “basal insulin” supplementation since the 1940s and 1950s and introduction of the intermediate-acting NPH and lente insulin family, respectively. Both analogs lower the risk for hypoglycemia and nocturnal hypoglycemia, lower FPG, and reduce day-to-day glucose variability, primarily as a result of their pharmacodynamic characteristics. Glargine provides the added benefit of once-daily administration, whereas detemir can be used once or twice daily depending on individual needs. Degludec, which is currently in Phase III clinical development, has the potential to further broaden the options for diabetes treatment with a possible thrice-weekly dosing regimen. It is now essential to construct further large-scale, longer-term clinical studies to compare degludec with glargine to establish whether the new chemical entity offers additional benefits in both efficacy and/or safety in individuals with either type 1 or insulin-requiring type 2 diabetes.
Footnotes
Acknowledgments
Editorial support for this article was provided by Huw Jones, Ph.D., of Medicus International and was funded by sanofi-aventis.
Author Disclosure Statement
D.R.O. has acted as a consultant for sanofi-aventis and Roche Diagnostics.