
Abstract
Transfer RNA (tRNA) plays a pivotal role in protein synthesis by mediating the accurate translation of mRNA codons into amino acids. While the aminoacylation process—where aminoacyl-tRNA synthetases (aaRSs) attach cognate amino acids to tRNAs—has been extensively studied, key mechanistic and assumptive gaps remain unresolved. Current models predominantly focus on tRNA–aaRS interactions after initial binding, leaving the fundamental question of how this first encounter occurs, which is poorly understood. Furthermore, the positions and roles of tRNA identity elements (e.g., anticodon, discriminator nucleotide base, D-stem) vary across the whole classification of tRNA molecules, suggesting a dynamic and fluid recognition system rather than a rigid set of rules. This review critically examines these unresolved aspects, highlighting: (1) the limitations of conventional tRNA charging models: neglected negligible probability of aaRS interacting with cognate tRNA out of a large pool, (2) the fluid nature of identity elements across tRNA classes, and (3) potential adaptive mechanism by aaRS to interact with seemingly cognate amino acids in the face of hundreds of thousands of tRNA molecules in the cell. By synthesizing structural, biochemical, and evolutionary evidence, it is proposed that tRNA-aaRS recognition is a continuously adaptive process in sense of tRNA charging. Addressing these gaps could redefine our understanding of translational fidelity and open new avenues in the context of cellular function and protein synthesis.
Introduction
Transfer RNA or tRNA is a small 70–100-nucleotide-long RNA molecule that serves as the adaptor between the genetic code carried by mRNA and the amino acids that are building blocks of proteins. 1 Therefore, the understanding of tRNA structural features is fundamental to exploring its paramount role in the process of aminoacylation, which is also known as tRNA charging. Aminoacylation refers to the process of linking the appropriate amino acid to its corresponding (cognate) tRNA molecule, thereby ensuring the correct pairing of amino acids with their corresponding codons during protein synthesis. Hence, each tRNA molecule is charged with a specific amino acid by an enzyme called aminoacyl-tRNA synthetase (aaRS). There are 20 different aaRSs for 20 proteinaceous amino acids. Thereby, they recognize both the specific amino acid and their corresponding tRNA molecule that carries the anticodon. 2 Over the years, extensive research has shed light on the diverse structural characteristics of tRNA molecules across all domains of life. The structural features of tRNA encompass its primary, secondary, and tertiary structures. The primary structure refers to the specific sequence of nucleotides that make up the tRNA molecule, while the secondary structure involves the formation of nucleotide base pairs among them and the characteristic cloverleaf shape. 3 The tertiary structure, on the other hand, is due to the complex folding of tRNA, influenced by interactions between different regions of the molecule. 3 Despite the significant progress made, there are still many unanswered questions about this tRNA charging and amino-acylation. This model applies when tRNA is bound to aaRS, but how does a tRNA initially make statistically efficient contact with an aaRS amid hundreds of thousands of other tRNAs and aaRSs? On a hit-trial basis or is there any other molecular or biochemical marker that induces this interaction? Or is there connectivity among different aaRSs? Based on which aaRSs can exchange their cognate tRNAs after attachment? Provided that aaRS can also recognize other tRNAs apart from their cognate ones. This review aims to elaborate the complex process of tRNA charging and highlight a neglected side of this process.
Secondary structure of tRNA
The primary structure of tRNA is its nucleotide sequence. The secondary structure of tRNA is formed by base-pairing between complementary nucleotides of the sequence. The clover leaf model is the two-dimensional representation of the secondary structure of tRNA, which consists of five distinct regions:
Acceptor stem
This is the region at the 5′ end of the tRNA. It is formed by base-pairing between the first two nucleotides of the tRNA and the last two nucleotides of the anticodon. The acceptor stem contains a CCA sequence at 3′ end, which provides a binding site for the amino acid that is charged to the tRNA. 4
D-loop
This is a loop that contains the modified base dihydrouridine (D). This loop is located between the acceptor stem and the anticodon stem loop. The D-loop is often involved in the interaction of tRNA to the aaRS. 5
Anticodon stem loop
This is a loop that contains the anticodon, which is a three-nucleotide sequence that is complementary to a codon in mRNA. It responsible for recognizing the cognate codon in mRNA during protein synthesis for the incorporation of cognate amino acid in a growing polypeptide chain. 5
Variable loop
This is a loop that is located between the anticodon stem loop and the TΨC-arm. The length and sequence of this loop vary between different tRNAs. The variable loop may contain additional modified bases, which can play a role in the folding and stability of the tRNA molecule. 6
TΨC-arm
This is a short arm that contains the modified base pseudouridine (Ψ). This arm is located at the 3′ end of the tRNA. The TΨC-arm is involved in the interaction of tRNA to the ribosome and the initiation of protein synthesis. 5
The cloverleaf model is a simplified representation of the global structure of tRNA. In the cell, the tRNA molecule is folded into a more complex three-dimensional structure that resembles an inverted L-shape. This tertiary structure is stabilized by a number of noncovalent interactions, including hydrogen bonding, base stacking, and electrostatic interactions. 7 The tertiary structure of tRNA is discussed in the next section. Figure 1 is the schematic representation of the clover structure of tRNA. In addition to the five regions described above, there are a number of other features that contribute to the global structure of tRNA. Modified nucleotides that have been chemically altered after they are incorporated into the tRNA molecule;7–8 bulges and loops that are regions of the tRNA molecule that are not base-paired. Bulges and loops provide additional flexibility to the tRNA molecule, which is important for its function in protein synthesis.7–8

Schematic representation of canonical clover leaf structure of tRNA with its multiple structural features.
Tertiary structure of tRNA
The tertiary structure of tRNA Figure 2 is the L-shaped folding of the molecule. The L-shape is composed of two helices; one formed by the acceptor stem stacked on top of the TψC stem and the other formed by the anticodon stem stacked on top of the D-stem. 4 The variable loop is located between these two major domains. The folding of the D and TψC loops is necessary to create the L-shape of tRNA.2–5 In addition, non-Watson–Crick base pairs and base triplets help to stabilize the three-dimensional structure of tRNA. The significance of the L-shaped folding of tRNA lies in its function in protein biosynthesis. The 3′ terminus of tRNA is separated from the anticodon triplet by more than 70 Å (angstrom), which allows for formation of a general binding domain for aminoacyl tRNA synthetases.3–6 The sites for interaction with mRNA, ribosome, and ribosomal peptidyl transferase, where peptide bond synthesis occurs are located on different regions of the tRNA molecule. 3
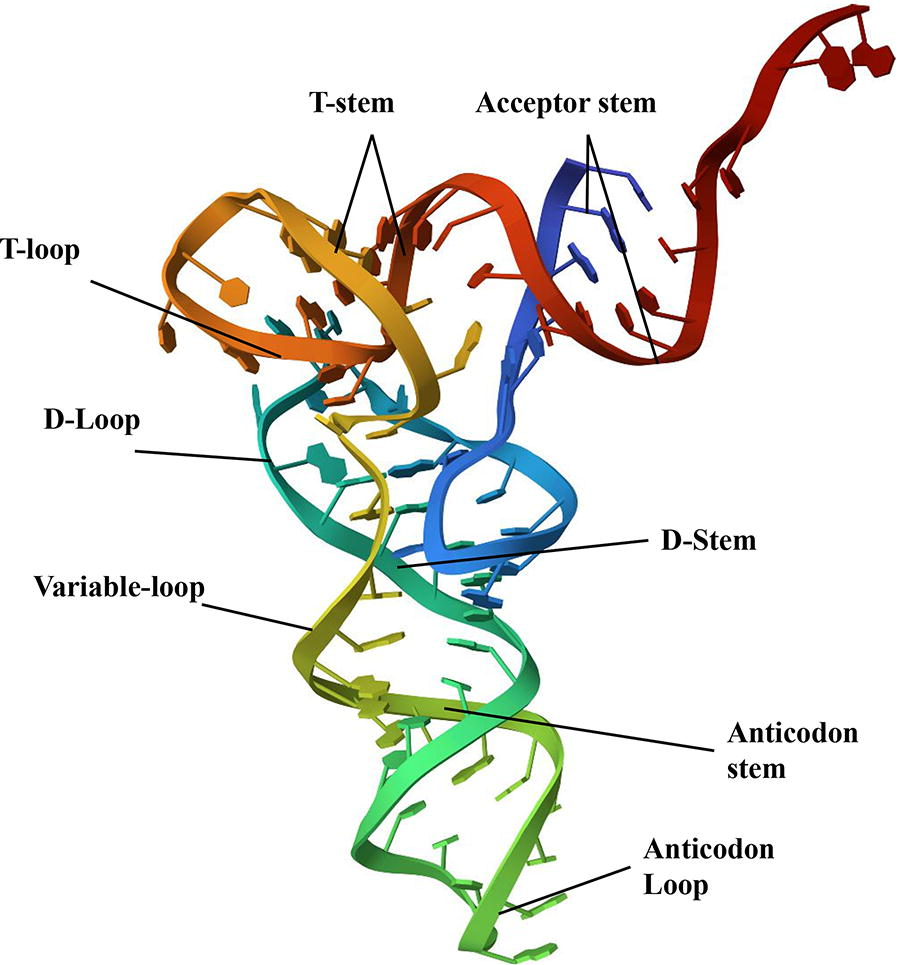
Conventional tertiary structure of a tRNA. Image obtained from Protein Data Bank. 3
Gene diversity and evolution of tRNA
The genes encoding tRNA molecules display significant diversity among organisms and even within a single species. Bacteria typically harbor tRNA genes in clusters on their chromosomes, often possessing multiple copies of these genes. The number of tRNA genes in bacterial genomes spans from 28 to 120, with an average of approximately 59. 9 In archaea, tRNA genes are found in arrays, with the number ranging from 10 to 29, including clusters with 20 or more genes, representing around 17% to 46% of the total tRNA genes in archaeal genomes. 10 Eukaryotic genomes generally contain several hundred tRNA genes, with human genomes having approximately 170 and mouse genomes having around 22,000. In eukaryotes, the number of tRNA genes typically ranges from the high 20s to low 50s. 11 The majority of eukaryotic cytosol contains distinct types of tRNAs, as most of the 61 sense codons possess their own corresponding tRNAs. 12 Insights into the mechanisms and pathways of tRNA evolution can be gleaned from several models. The Minimal Aminoacyl Ribozyme Model proposes the existence of a primitive chemical pathway predating modern aaRSs. 13 Experimental evidence supports the formation of aminoacyl adenylate under prebiotic conditions and the selection of ribozymes capable of self-aminoacylation and aminoacyl transfer. The Minihelix Model suggests that tRNA evolution involved the emergence of minihelices or minigenes, with the acceptor stem and TΨC arm recognized by tRNA synthetases. 13 Amino-acylated minihelices could have played a role in the development of homochirality in proteins. The Genomic-Tag Hypothesis proposes that tRNA-like structures initially evolved as 3′ terminal motifs that facilitated replication in the RNA world. These structures, found in various RNA viruses and retroviruses, serve multiple functions. The Double Helix Model posits that the modern cloverleaf structure of tRNA arose from the duplication of a hairpin structure. 13 The duplicate form retained the top half’s role as a recognition RNA element, while the bottom half underwent subfunctionalization. The split tRNA and intron-containing tRNA model suggests that symmetric tRNA resulted from the fusion of two separate hairpin RNA genes, with introns possibly representing remnants of this fusion. 13 Phylogenetic studies indicate a later addition of the bottom half of tRNA, and split tRNA genes are found in some archaeal genomes, linking them to intron-containing tRNA. 13 These models may represent different evolutionary mechanisms but suggest the involvement of multiple regions of tRNA molecules in aminoacylation during the translation process.
Determination of tRNA identity
The identity of a tRNA molecule is established through several elements, including the anticodon, D-stem, variable arm, acceptor stem, and additional tags. First, the anticodon is a critical component of tRNA identity, with each nucleotide in the anticodon contributing specificity to tRNA-codon interaction during translation. For example, U34 in the first position of the anticodon will pair with A in the codon but not G, but it is not the only factor in the recognition of tRNA.14–15 Second, the D-stem, formed by base pairing between the D-arm and D-loop, provides an extra layer of identity information for tRNAs with the same anticodon.14–16 Many serine tRNAs share a UCU anticodon but differ in D-stem sequences, allowing each to selectively bind only serine and not confuse it with lysine or arginine.14–17 In addition, some tRNAs also possess additional “tags” in loops or bulges, such as the A-box, B-box, or TΨC loop, that provide further nucleotide sequences through which tRNA identity is established.14–17 These identity elements work together to achieve the precise matching of tRNA to amino acid that is essential for faithful translation and meaningful protein function.14–21 This suggests that recognition of tRNA is a multiple-layer mechanism. Following sections will dive deeply into this argument. A major research gap in this section can be observed that the positions of the identity element are not identical in all types of tRNA molecules, instead they are different in different tRNAs; the nature of base is not in question. However, this gap remains unexplored.
Role of anticodon in tRNA recognition
The anticodon serves as a unique recognition site for each tRNA, specific to the corresponding amino acid. Anticodon involvement is evident in both eukaryotic and prokaryotic tRNAs, particularly the first structural class with short V-loops. 22 These “anticodon-dependent” tRNAs require it for acceptor function, regardless of V-loop and D-stem length. However, in the second class (e.g., tRNASer, tRNALeu, tRNATyr), the anticodon’s role may be secondary or absent. Class-I tRNAs are considered to be evolved earlier than class-II. 22
Recognition tRNA without anticodon
Identity elements are distinguishing features of a tRNA molecule that facilitate the charging of cognate amino acid by aaRSs. Anticodon is not the only fundamental identifier for attachment of cognate amino acid to its corresponding tRNA by aaRS because aaRSs can catalyze the aminoacylation of minihelices, which are smaller, stem-loop RNA fragments derived from tRNAs that contain the acceptor stem but not an anticodon stem.23–25 Second, synthase urzymes can acylate tRNAs with cognate amino acid without anticodon binding domain which suggests that acceptor stem recognition precedes anticodon recognition. 26 Moreover, the acceptor stem houses a set of unique identifiers referred to as acceptor stem code that causes amino acids to bond to hydroxyl group (OH) at adenine nucleotide (A) of the 3′ CCA terminal sequence. 26 In addition, a discriminator base that is present at position 73 (N73) and 5′ to CCA sequence also acts as an identifier for correct aminoacylation.27–28
Positive identity elements promote tRNA charging
Identity elements present in acceptor stem, D loop, T loop, and variable arm play an important part in tRNA recognition. 29 These elements can include distinct nucleotides, base pairs, and various structural features of tRNA that are recognized by cognate aaRSs prior to aminoacylation (Table 1).
Various positive identity elements and their positions on different tRNA molecules
Unclear role of discriminator base (N73) in tRNA recognition
The base N73 in tRNA just before the conserved CCA nucleotides (N74-76) is called discriminator base and plays a key role in determining the specific amino acid that the tRNA will bind to during aminoacylation. 50 The discriminator base helps shape the three-dimensional structure of the tRNA to allow for correct binding of the cognate aaRS. 51 Moreover, different aaRS enzymes show preference for different bases at position 73 (Table 2), allowing for specificity in charging the correct amino acid. 15 It was observed that altering the U73 (uridine at N73) discriminator base in E. coli tRNAGly resulted in diminished acceptance of glycine, indicating its role as an identity element. However, it has been found that the archaeal A. pernix tRNAGly does not rely on the presence of discriminator base A73 (adenine at N73). 52 In E. coli, the N73 discriminator position is not required for tRNAGln recognition. 41 tRNAs corresponding to the same amino acid can possibly have different nucleotides at N73 position i.e., tRNAGlu1 contains G73 while tRNAGlu2 contains A73 as seen in Helicobacter pylori. 53 tRNAs for leucine, isoleucine, and valine generally have a pyrimidine base (C or U) at this position (however not constantly), which allows binding of the cognate isoleucyl and valyl-tRNA synthetases. 51 The identification of the nucleotide base at position 73 through tRNA sequencing thus provides valuable insight into the aminoacylation specificity of the tRNA. 50 It can be deduced that N73 base is not a primary identity element and presence of a particular base at this position for some charged tRNAs is a coincidence. It is also possible that it is not a primary but a supplemental identity element required for confirming recognition.
N73 Nucleotide bases of different tRNAs in different species
Negative identity elements prevent tRNA charging
To ensure the fidelity of protein synthesis, tRNA molecules have evolved a number of mechanisms to prevent mischarging with the noncognate amino acids or binding to a wrong codon in the mRNA. One such mechanism is the presence of negative identity elements in the tRNA molecule. Negative identity elements in tRNA (Table 3) are structural features that prevent a tRNA from being mischarged by noncognate aaRSs and participate in identity. 67 The negative identity elements in tRNA are different from positive identity elements. Positive identity elements are features of the tRNA that the cognate aaRS recognizes directly. 55 On the other hand, negative identity elements prevent the binding of noncognate aaRSs to the tRNA. 55
Negative identity elements in different tRNA molecules across various species
In the acceptor stem of tRNA, the N73 acts as an identity determinant, however it is also a critical negative identity element that prevents charging of the tRNA with the wrong amino acid. In addition to the N73 base, other negative identity elements in the acceptor stem include the presence of a G-C base pair at N1-72 and a U-A base pair at N2-71. These base pairs contribute to the stability of the acceptor stem and prevent the binding of tRNA noncognate aaRSs.68–70
Chemical modifications influence tRNA charging
Chemical modifications are essential for the stability, proper folding, aaRS recognition, and protection from stress damage. 76 Collectively, they fine-tune tRNA-ribosome interactions impacting translation efficiency and fidelity. 76 Usual chemical modifications are methylations, thiolations, pseudouridylation, and acetylations. Methylations are chemical modifications that involve the addition of methyl groups (CH3) to specific nucleotides in tRNA molecules. 15 Thiolations refer to the addition of a thiol group (SH) to specific positions in tRNA molecules, typically at the 2-thiouridine or 4-thiouridine positions. 76 Pseudouridylation involves the conversion of uridine (U) nucleotides in tRNA to a modified nucleotide called pseudouridine (Ψ). 77 Acetylations involve the addition of acetyl groups (COCH3) to specific positions in tRNA molecules. 78
Structural organization of aaRS
The structural organization of aaRS is a complex network of domains that govern tRNA selection, charging with amino acids, and hence the fidelity of protein synthesis. The N-terminal and C-terminal domains provide stability, while the anticodon-binding domain ensures accurate pairing of amino acids and codons. The editing domain rectifies errors, and the connective peptide regions enable communication among these domains. Finally, the signature sequence guides the enzyme to the correct tRNA.
Active site domain
The active site domain serves as the catalytic center, where the amino acid is bonded to the cognate tRNA molecule. Both class I and class II aaRSs contain an active site domain with a common fold that binds with amino acid, adenosine triphosphate (ATP) and the 3′-terminus of the tRNA. 79
N-terminal domain
The N-terminal domain plays a crucial role in determining the specificity of the aaRSs for cognate tRNAs, acting as a determinant for recognizing and binding the appropriate tRNA molecules while preventing noncognate tRNAs from being charged.80–82
C-terminal domain
The C-terminal domain of aaRSs also assumes a pivotal role in modulating tRNA binding and aminoacylation activity across various enzymes as seen in the case of methionyl-tRNA synthetase (MetRS), 83 lysyl-tRNA synthetase (LysRS), 84 archaeal leucyl-tRNA synthetase (LeuRS), 85 human methionyl-tRNA synthetase (hErpES), 82 and alanyl-tRNA synthetase (AlaRS). 86
Anticodon-binding domain
This domain is responsible for recognizing the anticodon of the tRNA and ensuring that only the cognate amino acid is attached to the tRNA. It typically contains a binding pocket that interacts specifically with the anticodon loop of the tRNA.82,87,88
Editing domain
This domain is responsible for removing incorrectly attached amino acids from the aminoacyl-tRNA. It typically contains an active site that can recognize and hydrolyze noncognate amino acids, preventing them from being incorporated into the growing peptide chain.82,89
Connective peptide 1 and 2
The connective peptide 1 and 2 regions facilitate communication and coordination between different domains, ensuring efficient enzymatic activity. 86
Signature sequence
aaRSs contain signature sequences that contain conserved amino acid residues among different aaRSs. These signature sequences typically match 4–11 amino acids and are found in several synthetases of the same class. High-resolution structural data reveal that the signature sequences in at least two synthetases adopt a common “mononucleotide binding fold-like structure.” 87 This structural motif is composed of two regions: “motif 2” containing 23-31 amino acids and “motif 3” containing 29-34 amino acids. 87 These two motifs are located adjacent to the active site of aaRS, near the binding regions for the acceptor stem of tRNA and amino acid. 88
Each aaRS possesses unique domain architecture, facilitating the binding of cognate tRNA, amino acid, as well as editing and proofreading processes.89–90 However, aaRS possess regions that bear structural similarity to other aaRS enzymes. These structural similarities can lead to aaRS enzymes recognizing and charging noncognate tRNA molecules (Table 4). This suggests that aaRS recognition is fluid, allowing non-cognate tRNAs with physicochemical similarity to be charged, highlighting a major loophole in the conventional model of tRNA charging.
aaRS enzymes that recognize and charge both cognate and noncognate tRNA molecules
aaRS recognition of cognate amino acid
Amino acid fits into a specialized pocket in the aaRS, where its shape, charge distribution, and hydrophobicity play a critical role in determining its compatibility.109–119 Within this pocket, the aaRS employs a fine-tuned system of “positive” and “negative” interactions. Positive interactions involve the recognition and binding of functional groups on the correct amino acid, solidifying its position as the chosen one. On the other hand, negative interactions prevent the binding of incorrect amino acid substrates, ensuring accuracy in the selection process. The precision of amino acid selection relies on various mechanisms (Table 5) such as electrostatic interactions, hydrogen bonding, and hydrophobic interactions. These interactions are orchestrated by specific parts of the aaRS, guiding the delicate dance of recognition and ensuring that each amino acid is chosen with great accuracy.109,112,122–126
Mechanism by which each aaRS recognizes the cognate amino acid for the corresponding tRNA molecule
Mechanism of tRNA charging
Conventionally, all aaRSs are considered to charge tRNAs through a two-step mechanism Figure 3 that begins with the activation of amino acid and is followed by esterification of the activated aminoacyl group to the tRNA. 141 First, the amino acid forms an activated aminoacyl-adenylate intermediate (amino acid + adenosine monophosphate) with the hydrolysis of ATP that involves nucleophilic attack of amino acid carbonyl group on alpha phosphate of ATP, releasing inorganic pyrophosphate. The resulting aminoacyl-adenosine monophosphate (AMP) complex is stabilized kinetically and thermodynamically, and allows for attachment to the cognate tRNA.118,142,143 In the second step, the activated aminoacyl-group is transferred from aminoacyl-adenylate to the 2′ or 3′ hydroxyl group at the 3′ end CCA sequence of the corresponding tRNA. Accurate positioning of the aminoacyl-adenylate and tRNA mediated by binding of the tRNA acceptor stem and additional tRNA identity elements to various domains of aaRSs enables efficient attack of the tRNA 3′ hydroxyl on the an-hydride linkage. The reaction proceeds through a pentavalent transition state before breaking down to form the amino-acylated tRNA product and an AMP. After aminoacylation, the charged tRNA (tRNA + amino acid) is released from the aaRSs to join the translation machinery.118,142,143

Aminoacylation of tRNA by aaRS, showing the binding of the amino acid and tRNA molecule, the activation of the amino acid by ATP, and the transfer of the activated amino acid to the tRNA molecule.
Proofreading can occur either before the amino acid is transferred to the tRNA (pretransfer editing) or after the formation of the mis-acylated tRNA (post-transfer editing). Aminoacylation provides kinetic as well as chemical proofreading to enhance accuracy. Chemical proofreading monitors the transition state of the esterification reaction, increasing the energy barrier for mis-acylation relative to correct charging. This relies on stabilization of the transition state by geometric and electrostatic complementarity between synthetase and cognate tRNA. Aminoacyl-adenylate hydrolysis can also occur between the two steps, rejecting incorrectly activated amino acids before tRNA binding.
Together, kinetic and chemical proofreading help ensure fidelity of tRNA charging despite the relatively high rates of amino acid activation.140–143 Structural dynamics also facilitate the mechanism of tRNA charging. Conformational changes within aaRSs upon aminoacyl-adenylate and tRNA binding geometrically align the reactive groups for efficient esterification. The tRNA acceptor stem itself undergoes conformational changes i.e., straightening upon binding to align 3′ OH of terminal adenosine (A) nucleotide of CCA acceptor of tRNA for precise nucleophilic attack on the amino acid substrate, optimizing geometry for in-line nucleophilic attack. These induced-fit interactions mediated by dynamic domain movements of both aaRS and tRNA enable the required precision of the aminoacylation reaction.93,140–143
Substrate-assisted editing is one of the editing mechanisms ensuring translational fidelity as observed in recent studies. In this mechanism, the substrate of an aaRS which is either the tRNA or the aminoacyl-adenylate can induce the removal or hydrolysis of a noncognate amino acid.144–146 For example, the noncognate amino acid is edited by the phosphate group of its own aminoacyl-AMP complex as reported in MetRS. 144 Furthermore, CAA sequence at 3′ of the tRNA are also involved in post-transfer editing. 145 Similarly, a study found that that the 2′ OH group of the adenine base (A76) in tRNAThr acts as a mechanistic base, facilitating hydrolysis by positioning a water molecule near the ester bond of Ser-tRNAThr. 146 Another example of substrate-assisted editing is observed in LysRS which charge tRNALys with lysine but may mischarge it with toxic ornithine as these amino acids differ in only one methylene group. 146 It is observed that phosphate in the ornithinyl-adenylate causes self-cyclization of ornithine resulting in its removal. 146 Certain amino acid resides in the active site domains of aaRSs also participate in editing mechanisms. Aspartate residue when present in specific positions in the active site of MetRS is reported to be involved in hydrolyzes of noncognate amino acids; a similar mechanism is suggested for editing in homologous enzymes such as LeuRS, IleRS, and ValRS. 147 Similarly, ThrRS has an editing active site that hydrolyzes Ser-tRNAThr, which is formed when tRNAThr is misaminoacylated with serine. 145 A similar mechanism is observed in ThrRS, where specific cysteine and histidine residues in the editing domain catalyze the editing process. 144
Comparative mechanisms of aminoacylation by class I and class II aaRSs
The two classes of aaRSs, while catalyzing the same fundamental reaction, exhibit significant differences in their structural organization, substrate interactions, and kinetic properties. Class I aaRS enzymes (ArgRS, CysRS, GlnRS, GluRS, IleRS, LeuRS, MetRS, TrpRS, TyrRS, and ValRS) possess active sites characterized by the Rossmann fold (α/β structure) for nucleotide binding, whereas Class II aaRS enzymes (AlaRS, AsnRS, AspRS, GlyRS, HisRS, LysRS, PheRS, ProRS, SerRS, and ThrRS) possess an antiparallel β-sheet fold (antiparallel β-sheet) active site. 148 Furthermore, the active site architecture differs, with the nanospaces in Class I and Class II exhibiting opposite curvatures, a feature influenced by the conformation of ATP within the active site.149–150 Despite these distinct overall folds, some evidence suggests a potential evolutionary relationship between the two classes, possibly originating from a single ancestral gene. 150 The identification of a structurally conserved zinc-binding domain shared between some members of both classes further supports this possibility. 151
A fundamental difference lies in the way ATP is bound within the active site. Class I aaRSs bind ATP in an extended conformation through HIGH (His-Ile-Gly-His, involved in ATP binding) and KMSKS motif (Lys-Met-Ser-Lys-Ser, stabilizes the transition state), while Class II aaRSs bind it in a bent conformation by motif 1 (part of the dimer interface), motif 2 (contains a conserved Arg residue critical for ATP binding), and motif 3 (stabilizes the tRNA acceptor stem), stabilized by interactions with magnesium ions and conserved carboxylate residues.151–152 The mechanisms of ATP recognition also differ: Class I aaRSs utilize backbone hydrogen bonds, whereas Class II aaRSs employ arginine residues to form salt bridges, a structural motif known as the Arginine Tweezers.150–152 The distinct ATP binding modes likely influence the subsequent steps of amino acid activation and tRNA charging by affecting the orientation and reactivity of the substrates.
The two classes also exhibit contrasting strategies for interacting with the tRNA molecule. Class I aaRSs bind to the minor groove of the tRNA acceptor stem and attach the amino acid to the 2′-hydroxyl group of the terminal adenosine where some later shift it to the 3′-OH (e.g., TyrRS, TrpRS).153–155 Conversely, Class II aaRSs bind to the major groove of the tRNA acceptor stem and typically aminoacylate the 3′-hydroxyl group of the same adenosine, with phenylalanine-tRNA synthetase (PheRS) being an exception that uses the 2′-hydroxyl group.153–156 Class I aaRSs often identify cognate tRNA through anticodon loop while class II aaRSs recognize through acceptor stem.153–157 These contrasting tRNA binding surfaces and aminoacylation sites highlight a major divergence in the mechanisms employed by the two classes, likely reflecting their co-evolution with specific sets of tRNA molecules.
Kinetic studies reveal further distinctions between the two classes. Class I aaRSs are generally rate-limited by the release of the aminoacyl-tRNA product and often display burst kinetics.157–160 In contrast, Class II aaRSs are typically rate-limited by the amino acid activation step and do not exhibit burst kinetics.157–160 While Class I enzymes generally follow a random substrate binding order, the specific order for Class II aaRSs requires further investigation based on the provided information.157–160 These differences in kinetic profiles suggest that the catalytic cycles of the two classes are regulated at different stages and might reflect variations in their efficiency or interaction with other cellular components.
At the molecular level of the catalytic reaction, a significant difference is observed in the nucleophilic attack during amino acid activation. Class I enzymes (except TrpRS) utilize the syn-oxygen atom (the oxygen atom positioned on the same side as the ribose ring of ATP) of the amino acids’ carboxyl group to attack ATP, whereas Class II enzymes utilize the antioxygen atom (The oxygen atom positioned opposite to the ribose ring of ATP).159–161 This stereochemical difference is correlated with the distinct relative arrangements of ATP and the amino acid within the active sites of the two classes. This fundamental variation in the initial chemical step underscores the distinct active site environments and substrate orientations in Class I and Class II aaRSs.
Class I aaRSs, such as IleRS, ValRS, and LeuRS, often possess a distinct editing domain, the CP1 domain, which is inserted into their catalytic Rossmann fold. This domain is primarily involved in post-transfer editing, where it hydrolyzes mis-acylated tRNA, ensuring that only the correct amino acid remains attached.161–162 The “Double-Sieve Model” provides a framework for understanding this process, suggesting that the catalytic site acts as a coarse sieve, initially accepting amino acids with some degree of similarity to the cognate one, while the editing domain functions as a finer sieve, specifically hydrolyzing the incorrect products.
Class II aaRSs generally do not have a universally conserved editing domain such as the CP1 domain in Class I. 163 However, several Class II aaRSs, including threonyl-tRNA synthetase (ThrRS), alanyl-tRNA synthetase (AlaRS), and phenylalanyl-tRNA synthetase (PheRS), are known to possess proofreading activity through a second active site.164,165 For example, ThrRS has an N-terminal domain that functions as an editing site, capable of hydrolyzing mischarged Ser-tRNAThr. 142 Lysyl-tRNA synthetase (LysRS), which belongs to Class II in most organisms, also exhibits proofreading activity. 163 In some cases, editing and synthesis in Class II LysRS occur within the same active site, involving pretransfer hydrolysis of the noncognate aminoacyl-adenylate.164–165
Research gap in conventional process of tRNA charging
There is a pool of hundreds of thousands of 22 types of aaRS and 61 types of tRNAs in a cell and the same goes for amino acids. Statistically, each aaRS cannot pick its cognate amino acid and tRNA in the first attempt. The conventional model of tRNA charging works when tRNA is already attached to and interacting with aaRS, but how is tRNA initially attached to aaRS? On a hit and trial basis or is there any other molecular or biochemical marker? Or is there connectivity among different aaRS enzymes? Based on which aaRS can exchange their cognate tRNAs after attachment? The same question applies to aaRS picking its cognate amino acid as aaRS act as a base for tRNA-amino acid complex. Another question, the fluid nature of identity elements remains neglected. Another gap that needs to be discussed is the binding of tRNA or amino acid that induces the adaptive change in aaRS to bind to the other component. The question that an aaRS has to bind to a gigantic pool of tRNAs and amino acids before binding to its cognate ones is not discussed and always neglected. Further research is needed to solve all these mysteries.
Conclusion
While the mechanisms of tRNA charging and aminoacylation have been well-documented, confusing research gaps persist, particularly regarding the initial interactions between tRNA and aaRSs. The conventional process model assumes that tRNA is already bound to aaRSs during the charging process, yet it does not address how this initial attachment occurs. The possibility of an adaptive mechanism by aaRSs remains largely unexplored. Understanding whether aaRSs utilize biochemical markers or engage in a trial-and-error process to establish these connections could reveal significant insights into the efficiency and accuracy of protein synthesis. Addressing these gaps will not only enhance our understanding of tRNA biology but also provide deeper insights into the evolutionary processes that have shaped these essential molecules in the context of cellular function and protein synthesis.
Footnotes
CRediT Statement
A.R.R.: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing—original draft, Writing—review and editing.
Author Disclosure Statement
The author has no conflicting interests to declare.
Funding Information
The author received no funding.