
Abstract
Abstract
Background:
Lentiviral vectors are a common way to introduce RNA interference that cause loss-of-function effects in cells. While commonly used and well respected for their ability to create stably transduced cells, lentivirus can also have unanticipated off-target effects. In this study, we assessed the off-target effects caused by a lentiviral vector on the immortalized choriocarcinoma cell line, BeWo.
Methodology:
We transduced cells using a third-generation lentiviral vector containing a scrambled sequence of short hairpin RNA (shRNA). Cells were treated with lentiviral particles at a multiplicity of infection (MOI) of 25 and selected with puromycin. After antibiotic treatment, cells were allowed to recover from antibiotic treatment, and then, RNA was collected and assessed for their ability to differentiate.
Results:
Compared to wild-type BeWo cells, scramble shRNA cells were less fusible and secreted significantly lower levels of human chorionic gonadotropin (hCG), both hallmarks of differentiation in placental cells. We hypothesized that these effects may have been caused by altered gene expression of human endogenous retroviruses (ERV). Messenger RNA levels of ERVFRD-1 were significantly lower in transduced cells compared to wild-type cells. Additional mRNA levels of ERV were also lower, albeit not significantly.
Conclusion:
These data suggest that lentiviral vectors can cause func-tional disruptions in placental cells, leading to altered gene expression and cellular differentiation deficits. This study provides additional evidence to the growing body of work, demonstrating the negative effects of lentiviral vectors on cellular phenotype and off-target gene expression.
Introduction
Loss-of-function experiments provide important insight into how genes function on a cellular level. RNA interference (RNAi) is a straightforward method to induce gene silencing in a rapid period of time.1 RNAi mimics the post-transcriptional gene silencing utilized by microRNAs (miRNA) to target messenger RNA (mRNA) molecules and induce degradation, preventing translation tothe functional protein and creating a gene “knockdown.”2 Transient transfection of chemically synthesized short interfering RNAs (siRNAs) is a common way to mimic endogenous miRNAs and induce post-transcriptional gene silencing in cell culture.3 However, depending on the cell line, transfection can be inefficient, and, in all cell lines, transfection of siRNA is transient, which makes loss-of-function studies limited to a brief window of time post-transfection.4 Stable transduction of short hairpin RNA (shRNA) using viral vectors can circumvent the short-lived nature of transient transfection.4 Transduction of shRNA integrates foreign DNA into the host cell’s genome, allowing for long-term expression of shRNA.5 Lentiviral vectors are commonly used for stable transduction, due to their ability to transfect both dividing and nondividing cells6 as well as their relatively low ability to trigger an inflammatory response compared to other viruses including adenovirus and herpes virus.4
While popular and effective, lentiviral vectors do not come without their drawbacks. Previous studies have reported unintended effects on cellular differentiation and viability in embryonic stem cells and immortalized cancer cells.7,8 Additionally, most lentiviral vectors currently used for gene therapy are derived from human immunodeficiency virus (HIV-1) with the caveat they made replication incompetent.9 The removal of critical viral accessory genes renders lentiviral vectors incapable of generating replication-competent viruses, leaving both the handler and the host safe from potential infection.10 However, the inability to replicate does not prevent the lentivirus or shRNA from influencing the gene expression of off-target genes.11,12 This dysregulated gene expression can be detrimental to cells and cause cytotoxicity and differentiation defects.7,13 In this study, we aimed to determine if lentiviral vectors also potentially interfere with endogenous gene expression of genes from viral origins, more specifically endogenous retrovirus (ERV) genes. ERV genes are DNA sequences arising from ancient retroviral infections acquired over the last 100 million years.14 Most ERV genes are either nonfunctional or dormant; however, some can still code for protein or be reactivated by viral infections.15 Several papers have reported the ability of HIV-1 to interact with and activate ERV in several cell types.16–18 However, there is little information on how transduction with lentiviral vectors might affect ERV expression and cellular function. Therefore, we hypothesized that lentiviral transduction can influence ERV gene expression and cellular function.
We used the BeWo placental choriocarcinoma cell line. We chose the BeWo cell line as the placenta is abundant in protein-coding ERV. These ERV genes regulate cell–cell fusion, assist in the suppression of maternal immunity, and provide antiviral resistance from exogenous viruses.19 Furthermore, the BeWo cell line is the most prominent cell line model of placental fusion and syncytialization20 and expresses several ERV genes, including ERVW-1 and ERVFRD-1 (encode for proteins Syncytin-1 and Syncytin-2, respectively), two of the most well-studied placental ERV.21,22 For the lentiviral transduction, we used the pLV plasmid vector containing a scrambled shRNA control. Our study revealed that lentiviral transduction of a nontargeting shRNA had significant effects on placental cell differentiation as well as mRNA levels of ERVFRD-1.
Methods
Cell culture
The choriocarcinoma cell line, BeWo, was the cell line used in this study (purchased from ATCC). BeWo cells were cultured using an F12-K medium (ATCC) with 10% fetal bovine serum (FBS) (Gibco) and 0.5% penicillin/streptomycin/amphotericin B (Corning).
Lentiviral transduction
Lentiviral particles containing a scrambled shRNA sequence. Lentivirus was a vesicular stomatitis virus G (VSV-G) pseudo-typed third-generation lentivirus containing enhanced green fluorescent protein (eGFP) and a puromycin resistance gene (VectorBuilder). BeWo cells were transduced with viral particles at an MOI of 25 viral particles per cell and 5 mg/mL of the transduction agent Polybrene (Sigma Aldrich) for 24 h. Two days after transduction, cells were treated with 2 mg/mL puromycin for 72 h. Wild-type, nontransduced cells were used as a control to ensure puromycin sufficiently selected all successfully transduced cells. After 72 h, all wild-type cells were killed. Selected cells were cultured in antibiotic-free media for 1 week before any further experiments were performed.
Reverse transcription quantitative PCR (RT-qPCR)
Three biological replicates of each cell type were grown to approximately 60% confluency at which time RNA was collected from cells using an RNeasy Mini kit (Qiagen). Five-hundred nanograms of complementary DNA (cDNA) was synthesized from RNA using a qScript cDNA Synthesis Kit (Quanta Bio). After cDNA was synthesized, 1 mL of cDNA template was diluted in 19 mL of a prepared master mix containing RNase-free water, PerfeCTa SYBR Green SuperMix (Quanta Bio), and primer sequences specific for each gene of interest. Each reaction was conducted in technical duplicates for each gene. Gene expression was normalized to the expres-sion of ACTB and then relative expression determined as fold change against the wild-type control. RT-qPCR was run for ERVW-1 (Fwd: 50-
Immunofluorescence
Cells were plated at a density of 50,000 cells per chamber in a 4-chamber slide. After 24 h, differentiation was induced using 50 µM Forskolin for 48 h. The cells were then fixed using a 4% PBS paraformaldehyde solution for 15 min and then rinsed three times with PBS. The cells were blocked using a 3% bovine serum albumin (BSA), 10% FBS in PBS solution for 30 min, and then incubated in 1:250 ZO-1 (Abcam, ab216880) antibody for 1 h. The cells were then rinsed three times with PBS before applying 1:1,000 goat-anti-rabbit 488 secondary antibody (Ther-moFisher Scientific, #35552) to all chambers overnight at 4 C. After incubation, cells were rinsed with PBS before mounted using Prolong Gold Antifade Mountant with 4′,6-diamidino-2-phenylindole (DAPI) (ThermoFisher Scientific, P36935). Three biological replicates were used for each cell type.
hCG enzyme-linked immunosorbent assay
Cells were plated into a 12-well plate at 100,000 cells per well and treated with 50 µM Forskolin. The spent cell culture medium was collected after 48 h and centrifuged at 1,500 rpm. Concentrations of hCG in the spent cell culture medium were quantified using an enzyme-linked immunosorbent assay (ELISA) kit specific for beta-hCG (ALPCO Diagnostics, #25-HCGHU-E01). Each cell type was evaluated in three biological replicates.
Statistics
All experimental replicates were performed with at least three biological replicates and technical duplicates. The average of the technical duplicate was used for statistical analysis. To determine significance, a Student's t-test was used to compare between cells. All statistics were performed using GraphPad Prism 9 software. P-values less than 0.05 were considered significant. All error bars denote the standard error of measurement (SEM).
Results
Lentivirus disrupts cellular fusion and induces nuclear translocation of ZO-1
Successfully transduced BeWo cells containing a scrambled shRNA sequence (SC) were treated with the cyclic adenosine monophosphate (cAMP) agonist, Forskolin, to induce differentiation toward the syncytiotrophoblast (STB) lineage. Successful STB differentiation creates a large fused syncytium and a break-down of cell membranes. To assess cellular fusion, we performed immunofluorescent microscopy using the tight junction protein ZO-1 as a marker for cell membranes. Both wild type (WT) and SC undifferentiated cells had discrete, well-formed cell membranes (Fig.1A and C), whereas the differentiated WT cells no longer had defined cell borders, indicating successful cellular fusion (Fig.1B). Interestingly, the differentiated SC cells displayed nuclear translocation of ZO-1 (Fig.1D). Previous studies documented that exposure to HIV-1 can alter ZO-1 gene expression and cause translocation of ZO-1 to the nucleus.23 However, further work demonstrated that the HIV-specific protein transactivator of transcription (TAT) protein is the primary cause of ZO-1 nuclear translocation after exposure to HIV-1.24 As third-generation lentiviral vectors no longer contain the TAT protein,25 these findings suggest that even lentivirus no longer containing the TAT protein can still activate nuclear translocation of ZO-1 in transduced cells.
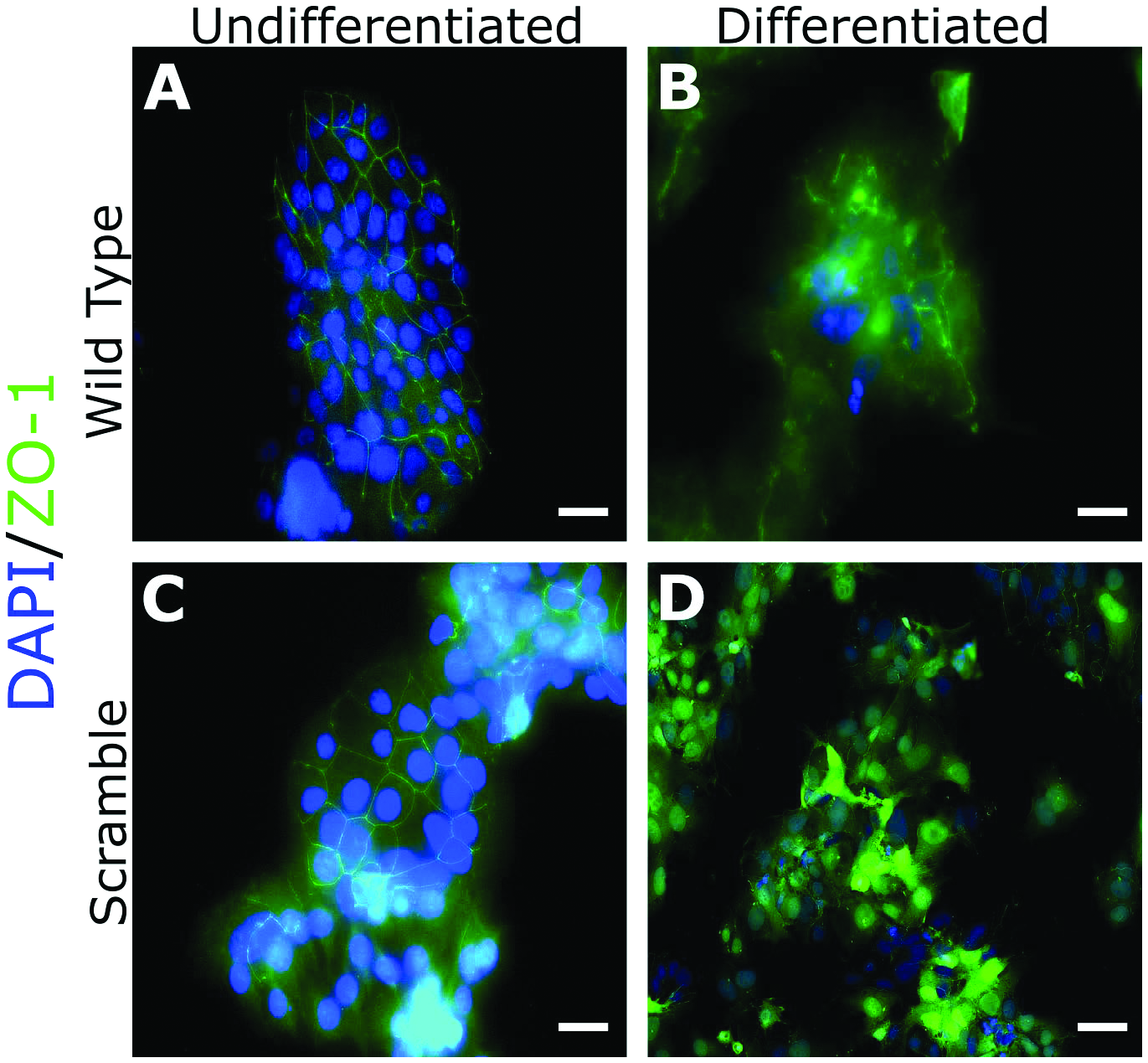
Lentiviral control shRNA vector impairs cellular fusion. WT and SC BeWo cells were cultured in either maintenance (A and C) or differentiation (B and D) medium for 48 h. After 48 h, cells were fixed and processed for immunofluorescent staining using ZO-1 antibody and DAPI mounting medium. These images are representative images taken from three separate experiments. Scale bar = 100 µM.
SC BeWo cells have diminished cellular differentiation and secrete lower levels of hCG
Another marker of successful STB differentiation is the cell’s ability to secrete hCG, which is a hormone critical for maternal recognition of pregnancy in humans. We again treated WT and SC BeWos with Forskolin and collected spent media after 48 h. Using an ELISA, we quantified levels of hCG and found that SC cells secreted significantly lower (p = 0.0001) levels of hCG compared to WT (Fig.2).
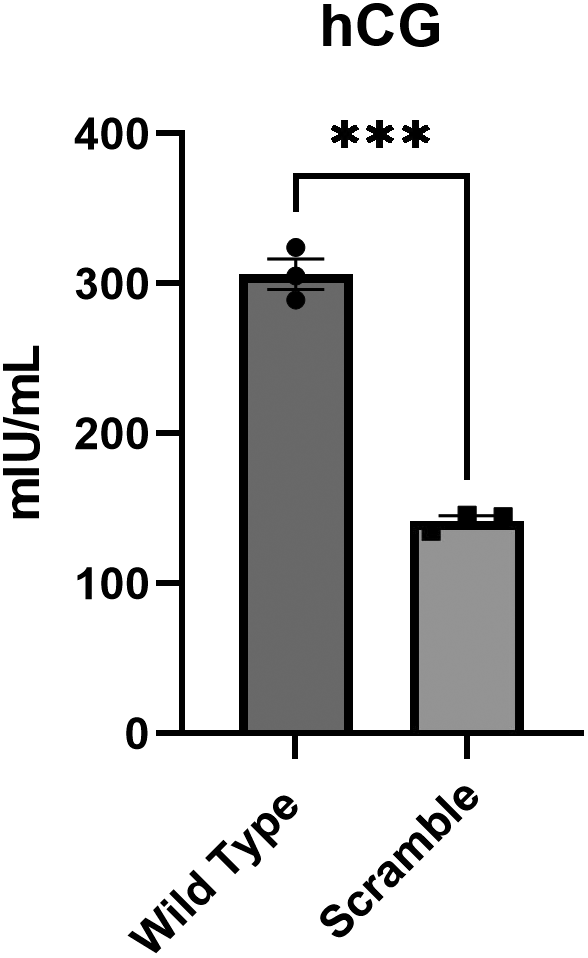
Cells treated with a lentiviral control shRNA vector have impaired cellular differentiation. WT and SC BeWo cells were cultured in differentiation medium for 48 h. After 48 h, spent culture medium was collected, and an ELISA was performed to test for the presence of hCG in culture medium. Three replicates were used, and a Student’s t-test was performed to determine the significance. ***p = 0.0001. Error bars represent SEM.
ERVFRD-1 mRNA levels are significantly downregulated in BeWo NTCs
To determine if the deficits in cellular function were potentially related to ERV, we collected RNA and performed RT-qPCR. There was no difference in ERVW-1 mRNA levels between WT and SC cells. However, ERVFRD-1 was significantly decreased (p < 0.0001) in SC cells (Fig.3). In addition to the well-studied ERVW-1 and ERVFRD-1, the placenta contains other, less characterized HERV. To determine if transduction had an effect on these ERV, we performed RT-qPCR. There were no significant differences in other HERV mRNA levels compared to WT, although ERVV-2 and ERVK3-1 also appeared to be slightly decreased (Fig.4).

ERVFRD-1 is decreased in cells treated with a lentiviral control shRNA vector. RNA was isolated from WT and SC BeWo cells and RT-qPCR performed. ERVFRD-1 is significantly decreased in SC cells compared to WT. All RT-qPCRs were performed with three biological replicates and repeated three times. ****p < 0.0001. Error bars represent SEM.

Lentiviral control shRNA vector potentially affects additional HERV. We assessed levels of other HERV mRNA in WT and SC cells. No other HERV genes were significantly decreased; however, ERVV-2 and ERVK3-1 were lower. All RT-qPCRs were performed with three biological replicates and repeated three times. Error bars represent SEM.
Discussion
Lentiviral vectors are commonly used for the stable introduction of shRNAs that can target and silence mRNAs. While generally considered safe and unlikely to stimulate an immune response in host cells, some studies have reported that lentiviral vectors can elicit an innate immune response.26,27 This immune response is seemingly enhanced when the lentiviral vector encodes an shRNA sequence.28 Beyond immune stimulation, other papers report the ability of lentiviral shRNA control vectors to induce spontaneous cellular differentiation7 and off-target gene silencing, leading to disruption of the cell cycle and apoptosis.8 These effects can be potentially mitigated by treating cells with low concentrations of vector; however, there is limited research on the myriad of ways lentiviral vectors might affect cellular function. This study provides additional evidence that control shRNA containing lentiviral vectors can affect cell differentiation and potentially interact with HERV genes in cells.
As this study used a lentiviral vector containing a puromycin resistance gene to assist with cell selection, it is possible that our findings were caused by the puromycin resistance gene within the vector. Previous studies reported puromycin lentiviral vectors can induce protein misfolding driven by reactive oxygen species.29 An additional study comparing a puromycin vector to a neomycin vector found that only the puromycin vector induced spontaneous differentiation in an embryonic stem cell line.7 Additionally, our lentiviral shRNA control vector contained a U6 promoter, which is a strong RNA polymerase III promoter. There is some evidence to suggest that strong promoters can induce higher levels of shRNA expression, leading to increased levels of cytotoxicity and cellular dysfunction.30 Potentially, using a weaker promoter could ameliorate the differentiation defects we observed; however, very little research has been performed that can validate this hypothesis.
Finally, it has been demonstrated that siRNA can manipulate mRNA and corresponding protein levels of genes unrelated to the targeted gene.31–33 Furthermore, titration of siRNA did not mitigate the off-target effects.31 The genes previously described were related to cell proliferation and viability. Our paper documents off-target effects related to cell differentiation and potential interaction with lentiviral vectors and HERV. Previous research used an envelope-defective HIV-1 strain in combination with a plasmid vector containing the ERVW-1 envelope sequence.34 Cells infected with the HIV-1 virions and cotransfected with ERVW-1 plasmid resulted in the production of infectious virus, indicating that HERV can interact with exogenous retroviruses.34 While it is unlikely that a third-generation lentiviral vector system could interact with HERV to infect a host with HIV, it is important to consider this newly described off-target effect of lentiviral vectors. HERV genes have a number of functions in the human body, including cell fusion, immune modulation, and antiviral resistance.35 Furthermore, dysregulation of HERV is linked to carcinogenesis and activation of proto-oncogenes.36–38
Conclusion
While shRNA and viral vectors are commonly used, there are still many concerns related to the use of shRNA in potential gene therapy, and ensuring that the vector delivery is not causing off-target effects is of the utmost importance. There is a general belief that third-generation lentiviral vectors are relatively safe; however, there is a growing body of evidence that these vectors have effects on cellular function and gene expression. It is essential that choice of promoters, MOI, use of antibiotic resistance genes, and the vector itself should be carefully considered before the execution of experiments. Furthermore, these findings also suggest that a nontargeting shRNA control is not a sufficient control when performing invitro cell culture experiments using viral vectors.