
Abstract
Custom CRISPR screens are powerful tools for rapid, hypothesis-driven discovery, but their design is often complex and time-consuming. Green Listed v2.0 simplifies this process with an intuitive workflow for designing custom CRISPR spacer libraries and supports downstream analysis for all users, irrespective of their computational experience. The web application features a user-friendly graphical interface freely accessible at https://greenlisted.cmm.se. Version 2.0 includes significant upgrades to the original 2016 version that were implemented based on user feedback. This includes a new gene synonym tool, expanded library options, optimized output lists, performance improvements, and linked scripts for the rational design of custom CRISPR screen gene sets.
Introduction
Over the past decade, natural CRISPR-Cas systems have been developed into powerful molecular biology tools capable of precise genome editing. 1 Typically, a single-guide RNA (sgRNA) directs the Cas9 endonuclease to a specific genomic region through RNA–DNA interactions, enabling, for example, targeted gene inactivation when designed appropriately. The precise sequence of the part of the sgRNA that binds the genomic DNA sequence, the spacer, is central to any CRISPR experiment. Key publications have identified optimal strategies for designing spacers to ensure high on-target activity while minimizing off-target effects.2–8
In CRISPR screens, a subset or all known genes of an organism are targeted across a cell population. 9 This is commonly achieved through lentiviral delivery of sgRNAs at a low multiplicity of infection (MOI) into cells modified to express Cas9. A low MOI ensures that most cells receive either one or no sgRNA, leaving a population with, on average, one targeted gene per cell after non-transduced cells are removed by selection. The resulting controlled genetic heterogeneity within the cell population is then analyzed to identify genes that influence specific behaviors (e.g., the response to a drug). 10 Next-generation amplicon sequencing of the integrated sgRNA cassette is used to identify such genes by tracking how different genotypes segregate across phenotypic groups, comparing, for example, cells before and after the addition of a drug.
While whole-genome CRISPR screens offer immense potential for unbiased discovery, they are not always practical. For a whole-genome screen in human or mouse, around 80 million successfully transduced cells per condition are suggested (based on ∼20,000 genes, 4 sgRNAs/gene, and 1,000 cells/sgRNA). 11 Consequently, limitations arise in systems that are not scalable, such as in sensitive primary cells, complex models such as organoids, in vivo animal models, or when achieving sufficient transduction efficiency is challenging. In these cases, a custom CRISPR screen targeting a subset of genes offers a viable alternative. These subsets may include candidates identified from OMICS datasets (e.g., genes differentially expressed in response to a drug), specific pathways relevant to the project, or known drug targets where hits can be rapidly translated into clinically relevant contexts. 12
Various whole-genome CRISPR libraries can be purchased at reasonable costs for academic use from sources like the nonprofit Addgene, 13 which also offers libraries targeting broad gene subsets, such as kinases or nuclear proteins. However, custom libraries must be designed and generated by the researcher (or outsourced, often at a significant cost). To address this, we developed Green Listed (version 1.0) in 2016, a web application designed to facilitate the creation of custom screen libraries. 14 Since its release, Green Listed has been widely adopted and significantly facilitated our efforts to perform smaller hypothesis-based screens.15,16 Based on user feedback, we have developed an updated web application version of Green Listed (v2.0), adding several new features, as well as significantly enhancing user experience and efficiency (Fig. 1).

Screenshot of the Green Listed v2.0 graphical user interface. Two gene symbols (cd124 and p53) are displayed to illustrate the functionality of the synonyms tool, and settings “Limit to top” and “Adapter sequences” applied to align with outputs in Figure 2C–E.
Methods
Generation of the Green Listed v2.0 web application
The new web application was developed in several iterations, with each iteration adding new features, including spacer libraries and synonym lists (see the Result section for the full feature list). A set of nonproprietary languages were chosen for development: JavaScript for the algorithms in combination with HTML and CSS to build the graphical user interface. To improve maintainability, no external program libraries or frameworks were used. The application is hosted on an Apache HTTP server.
Spacer libraries and synonyms were downloaded and curated as follows. The mouse and human VBC spacer libraries (top 6 sgRNAs per gene) were downloaded in July 2024 from VBC, 17 and the mouse and human GeCKO v2, Brunello, Brie, Gattinara, and Gouda spacer libraries were downloaded from Addgene 18 or the Broad Institute GPP Web Portal. 19 Nontargeting negative controls were renamed “NegCtrl” in all libraries to harmonize the application. The “ONE_NONE-GENE_SITE” controls (n = 500) in the Gouda and Gattinara libraries (targeting one intergenic region in the genome, serving as controls for dsDNA breaks) were renamed to “CutCtrl” for naming consistency.
The synonyms for human (GRCh38.p14) and mouse (GRCm39) genes were downloaded in July 2024 from Ensembl BioMart,20,21 utilizing the attributes gene name and gene synonyms to create a synonym list.
The code, including .txt files of all embedded reference spacer libraries and the synonym lists, can be accessed via the Green Listed Github page. 22 Videos describing Green Listed v2.0 can be found via the “How to use” tab at the top of the web application.
Evaluation of the synonyms tool
The DepMap public 24Q4 dataset (CRISPRGeneEffect.csv) was downloaded in January 2025 from the DepMap portal,23,24 and the included human gene symbols were extracted using the data.table and stringr R packages. The DepMap gene symbols (n = 17,917) were subsequently used to query the different human reference spacer libraries (GeCKO v2, Brunello, Gattinara, and VBC) in Green Listed v2.0 with the “Use synonyms” feature activated, and the number of productive synonyms recorded.
A compiled file of the used gene symbols in the human reference spacer libraries included in Green Listed v2.0 (GeCKO v2, Brunello, Gattinara, and VBC) as well as the DepMap 24Q4 Gene Effect dataset was generated and the overlap of gene symbols analyzed using the ComplexUpset, ggplot2, and patchwork R packages (dataset, script, and readme file are found at Github 25 ). For the analysis of overlaps, non-targeting controls were removed from all lists and microRNAs (n = 1,864) were removed from the GeCKO v2 list, as no other lists have miRNA targeting spacers.
Expansion of custom gene sets
An R script was created that identifies gene effect correlations in the DepMap CRISPR dataset, as a rational approach to expand gene sets for custom CRISPR screens. The DepMap public 24Q4 dataset (CRISPRGeneEffect.csv), which contains CRISPR screen gene effect values for 17,917 genes from 1178 human cell lines, was downloaded as indicated above. The script is based on the data.table, igraph, ggplot2, and ggraph R packages. The script, explanatory video, and readme file can be found via the “Scripts for analysis” tab at the top of the web application.
Results
The Green Listed v2.0 workflow
A generic custom CRISPR screen project is described in Figure 2A, where an initial gene set of interest is identified, expanded, transformed into a custom CRISPR screen library, and the screen performed. The linked Green Listed v2.0 workflow (Fig. 2B), is divided into four steps, corresponding to the four sections of the graphical user interface:
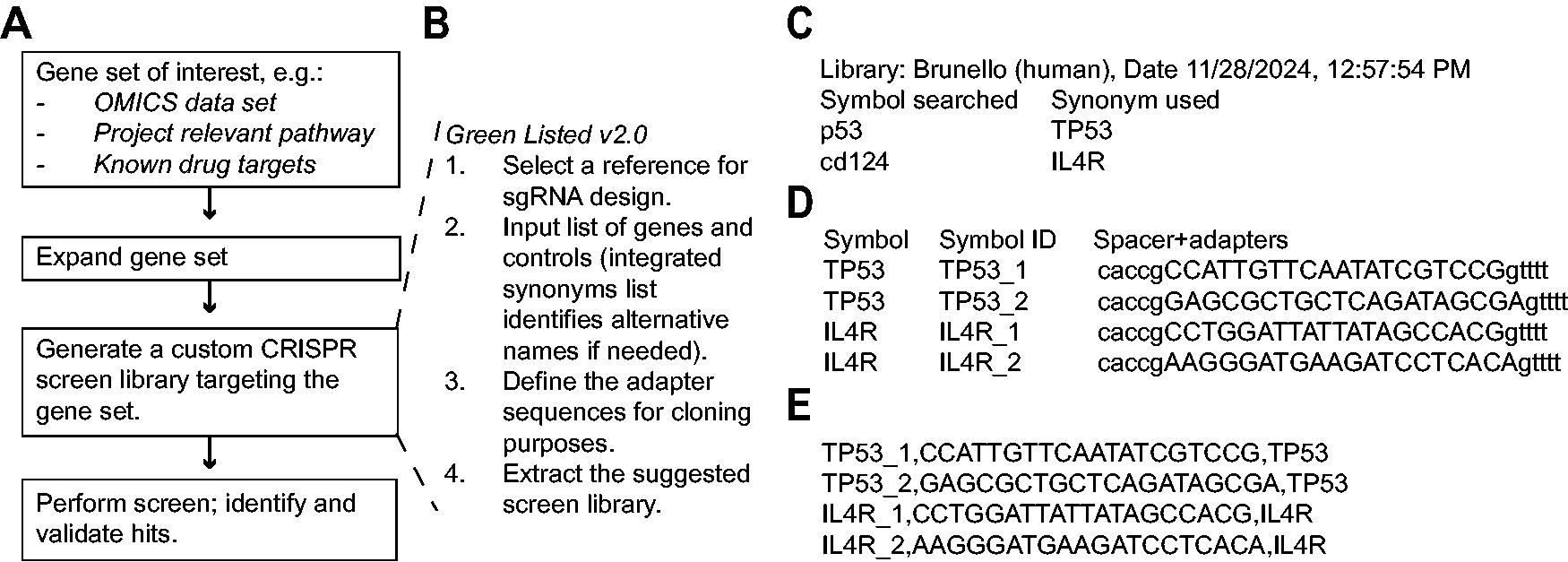
Using Green Listed v2.0 for rapid design of targeted custom CRISPR screen libraries.
First, the user selects a reference library containing suggested spacers from key publications based on various design algorithms.2–5 Alternatively, users can upload their own reference library, enabling the implementation of any custom algorithm or strategy, such as CRISPRa or CRISPRi.
Second, the user inputs the gene symbols to be targeted in the screen. A notable improvement in version 2.0 is the integrated synonym list, which automatically replaces gene names not found in the reference library with corresponding synonyms.
Third, the user defines parameters such as the 5′ and 3′ adapters to be added to the identified spacer sequences for cloning purposes. There is also an option to trim the suggested spacer sequences at either the 5′ or 3′ end, which is useful when user-uploaded libraries include unwanted sequences, such as the PAM. In addition, users can limit the number of sequences selected per gene, which is particularly helpful when incorporating negative controls. Most reference libraries contain 500–1000 negative controls, which may be excessive for smaller screens. Setting these parameters ensures that the output can be directly used for subsequent processing, avoiding extra manual steps.
Finally, after the user presses “Run,” several output lists are generated (exemplified in Fig. 2C–E). Of these, Figure 2C identifies all synonyms used, facilitating traceability in the design process. The “Output with adapters” list (Fig. 2D) contains all the necessary information for ordering the library as an oligo pool, while the “Output for MAGeCK” list (Fig. 2E) is formatted for subsequent analysis of screening results using MAGeCK, a widely used software for CRISPR screen analysis. 26
An integrated synonyms tool
Based on user feedback and our own experience, the most time-consuming and tedious step in designing custom CRISPR screens is the use of alternative gene symbols in gene sets from different sources. It is not uncommon that >10% of gene symbols are not identified when matching two lists of gene symbols (e.g., an RNAseq dataset and a CRISPR screen library). As an example, comparing gene symbols in the four human reference spacer libraries of Green Listed v2.0 (Gattinara, Brunello, VBC, and GeCKO v2), and the (human) DepMap 24Q4 gene list, 16,493 gene symbols were found to overlap between all five lists, 1,510 symbols were overlapping between the four reference spacer libraries, and 910, 780, and 413 unique symbols were found in Gattinara, DepMap 24Q4, and GeCKO v2, respectively (Fig. 3A, also including more overlaps of lower magnitude).
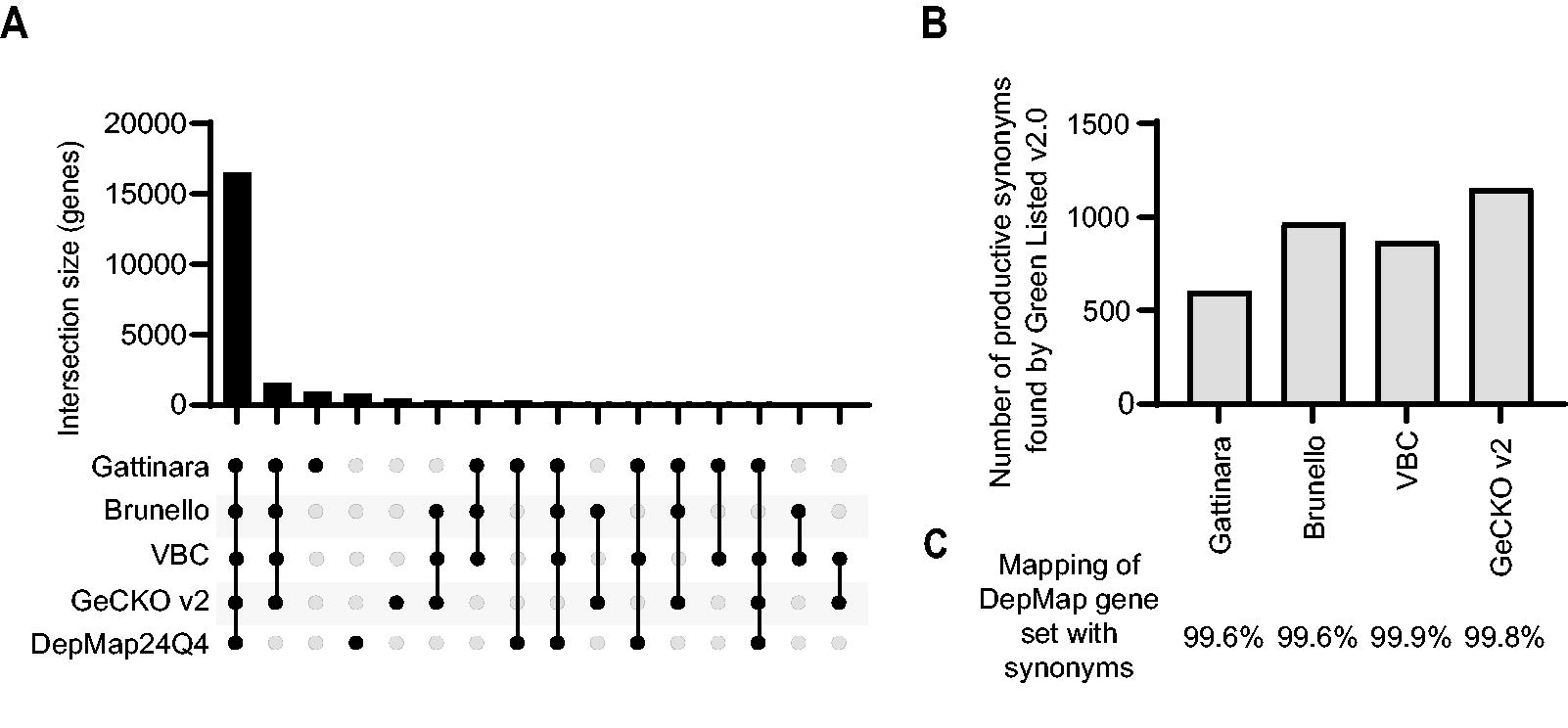
Implementation of the synonyms tool.
For rigorous screen design, identifying synonyms for most of the “symbols not found” is crucial but often challenging, especially when dealing with a large number of them. To address this, a synonyms tool was introduced in the v2.0 update. Mouse and human synonyms were retrieved from Ensembl BioMart, and a compiled file was generated, mapping gene symbols to their corresponding synonyms. When enabled, the feature processes “symbols not found,” cross-references them against the synonyms list, and determines if any match the selected CRISPR library. Running the DepMap 24Q4 gene symbols dataset (n = 17,917) against each of the human CRISPR libraries, the synonyms tool identified 601–1,152 synonyms (Fig. 3B), successfully mapping 99.6–99.9% of the input gene symbols (Fig. 3C).
Running this extensive gene list, identifying spacer sequences and mapping synonyms, takes Green Listed v2.0 less than one second. Notably, only identifying spacers for such lists of genes took several minutes with the previous version.
Expansion of screen gene lists
Another common user request concerns strategies for expanding gene lists to be targeted in a custom CRISPR screen (Fig. 2A). One approach we have explored involves identifying genes that correlate with a user-provided gene list based on DepMap CRISPR screen datasets. These datasets contain gene effect values for 17,917 genes across 1,178 human cell lines (24Q4 version). By generating a correlation matrix through batch analysis, correlations of varying strengths can be identified based on the DepMap dataset. Strong correlations (>0.5 or <−0.5) between two genes indicate that their inactivation has a similar effect across the included 1,178 cell lines, often suggesting some relationship. For example, TP53 and MDM2 exhibit a strong inverse correlation (Pearson correlation coefficient = −0.74), aligning with the well-established role of MDM2 in directly binding to and suppressing TP53 function. 27 To facilitate this type of analysis, we have created a Green Listed v2.0-linked script that generates a graphical representation of global DepMap correlations within a user-provided gene list (Fig. 4A). In addition, text-based output lists define the identified gene list, offering a straightforward expanded gene list for custom CRISPR screen design (Fig. 4B). Notably, this script could also be used in an analysis setting to identify connections in any gene set, for example, a hit list from a CRISPR screen.
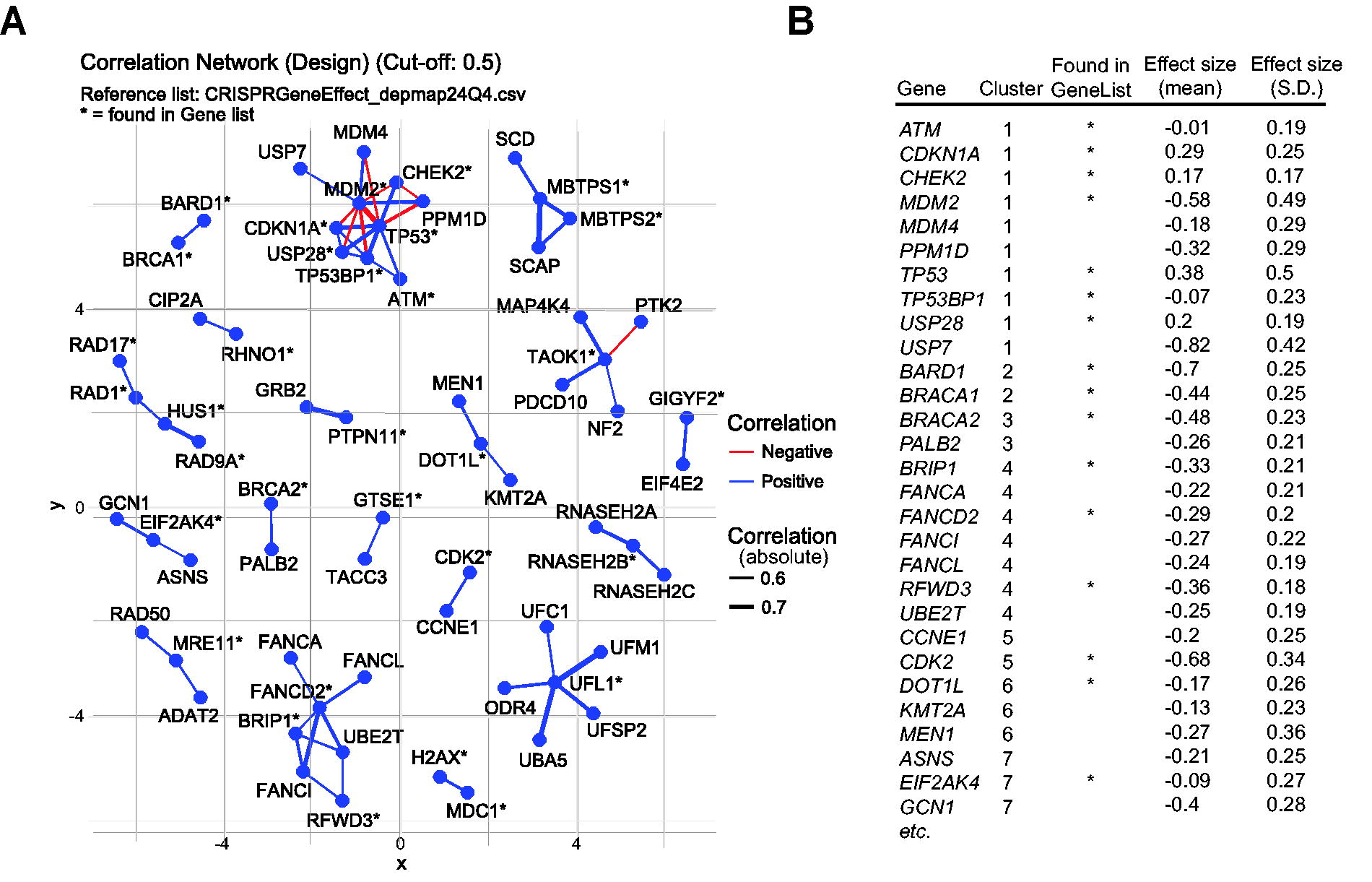
Expansion of gene set based on the DepMap dataset.
Discussion
Custom CRISPR screens serve as a powerful discovery platform when whole-genome CRISPR screens are excessive or not feasible. However, designing spacer libraries targeting a custom gene list can be complicated. A few spacer design tools, such as CRISPick 28 and CRISPR-FOCUS, 29 enable bulk spacer design but limit the number of genes they can target (e.g., 500 for CRISPick and 1000 for CRISPR-FOCUS). In addition, the output from these tools requires further processing of the generated data, such as adding adapter sequences, before the screen library can be ordered as an oligo pool. In contrast, Green Listed v2.0 allows for the generation of spacer libraries without a restriction on the number of targeted genes, supports the design of sgRNAs based on different contemporary genome-wide spacer libraries (that are frequently benchmarked and compared 30 ), and enables the use of user-uploaded spacer libraries for flexibility in design strategy or Cas variant.
The updates of Green Listed v2.0 build on user feedback and our own experience with the original version published in 2016, which has had more than 10,000 users. Significant updates include:
An integrated synonyms tool that identifies and replaces gene symbols not found with matching alternative symbols in real time. This feature significantly reduces the time spent manually identifying alternative gene names. The synonyms applied are indicated in the graphical user interface (Fig. 1) and a dedicated output list (Fig. 2C), allowing users to verify and keep a traceable record of the applied changes. Our benchmarking test indicates that the synonym tool performs well, contributing to accurately identifying 99.6–99.9% of the input gene symbols in our test (Fig. 3C). However, there will always be genes not found that need to be further processed. Alternative sources for identifying synonyms and orthologues include MGI Batch Query,
31
and g:Profiler Orthology.
32
Updated contemporary spacer libraries, including the more compact Gouda and Gattinara libraries described in DeWeirdt et al.,
5
and the VBC libraries targeting sequences corresponding to evolutionarily conserved protein domains described in Michlits et al.
4
In addition, the reference spacer library section allows the user to upload a custom reference spacer list, expanding the applicability to libraries based on, for example, other Cas versions or novel innovations within the spacer design domain. Relevant spacer libraries can be accessed from sources such as Addgene
18
and the Broad Institute GPP Portal.
19
Optimized output lists, allowing for a more streamlined workflow. Five lists are created, and saving these in a dedicated folder is advised to support traceability and reproducibility. The “Output with adapters” list is designed to be sent directly to a company providing the oligo pool synthesis. The “Output for MAGeCK” is designed to be directly used with MAGeCK for downstream analysis of screen results. The “Full output” contains all information about the different spacers provided from the original data source. The “Symbols not found” list identifies gene symbols that were not found in the selected reference library, as well as if synonyms were successfully used. Last, the “Run Settings” list details the settings used in the design process. Performance updates. The updated version was completely rewritten in JavaScript and is significantly faster than the original C# version. In fact, we have yet to see any run taking more than one second to generate an output. This could be compared with the previous version, which often took several minutes to design custom spacer libraries when the input gene list was extensive. Notably, the updated version performs this well, including running the novel synonyms tool in real time, which the previous version did not have.
With the updated release, we also introduce an approach for expanding gene lists to be targeted in a custom CRISPR screen, one of the most frequently discussed topics related to these types of screens. Given the diverse nature of CRISPR screen settings, there is no one-size-fits-all answer to how they should be designed. However, identifying a list of genes/proteins that are differentially regulated when studying the cells of interest in a project-relevant setting has been a productive approach for us 16 and also exploring which components of a relevant pathway that are functionally involved in a phenotype.15,16 Once an initial gene list is established, several methods can be used to expand it in a rational manner. A common approach is to assess whether the list is enriched in specific gene sets, such as Gene Ontology terms, and then supplement the list with additional genes from these enriched gene sets, as discussed in Iyer et al. 12 Alternatively, we here describe a method that leverages DepMap CRISPR screen datasets to identify potential functional connections between genes. The 24Q4 DepMap dataset enables correlation analysis of gene effects across 1,178 included cell lines, allowing for the expansion of a user-provided gene list by including genes that show significant correlations with how they affect the cell lines (Fig. 4). This approach enhances the likelihood of generating more robust hit lists from screens, as detecting multiple hits among linked genes increases confidence in their validity. In addition, it can be used to refine gene lists when size constraints require a more focused selection. In such cases, genes with strong correlations can instead be excluded, yielding a minimal, condensed screen gene list.
This type of correlation analysis can currently be performed in the DepMap portal, but in that case, only for one gene at a time. The script presented here, in contrast, allows for bulk analysis of a list of genes.
Conclusions
Green Listed v2.0 provides a quick and user-friendly solution for designing custom CRISPR screens, facilitating hypothesis-based discovery in scenarios where whole-genome CRISPR screens may be excessive or impractical. The tool is freely available for academic use, requires no registration or login, and is directly accessible using any web browser via https://greenlisted.cmm.se/.
Footnotes
Acknowledgments
The authors are grateful to Feng Zhang, John Doench, and Ulrich Elling for accepting the implementation of their spacer reference libraries into the Green Listed v2.0 web app.
Authors’ Contributions
E.H.: Software—programmed and developed the web app. Z.L.: Investigation and validation—proposed functionalities and beta-tested the web app. B.S.: Investigation and validation—proposed functionalities and beta-tested the web app. D.U.: Software and methodology—assisted with implementation and design. M.H.: Software—assisted with design and software architecture. F.W.: Conceptualization, funding acquisition, supervision, and writing—original draft.
Author Disclosure Statement
F.W. has received consulting fees from SmartCella Solutions and Chiesi outside of the scope of the study.
Funding Information
This project was financially supported by the Swedish Research Council, the Swedish Cancer Society, the Swedish Foundation for Strategic Research, the Wenner-Green Foundations, the King Gustav V’s 80th Anniversary Foundation, and Karolinska Institutet.