
Abstract
The incorporation of nuclear localization signal (NLS) sequences at one or both termini of CRISPR enzymes is a widely adopted strategy to facilitate genome editing. Engineered variants of CRISPR enzymes with diverse NLS sequences have demonstrated superior performance, promoting nuclear localization and efficient DNA editing. However, limiting NLS fusion to the CRISPR protein’s termini can negatively impact protein yield via recombinant expression. Here we present a distinct strategy involving the installation of hairpin internal NLS sequences (hiNLS) at rationally selected sites within the backbone of CRISPR-Cas9. We evaluated the performance of these hiNLS Cas9 variants by editing genes in human primary T cells following the delivery of ribonucleoprotein enzymes via either electroporation or co-incubation with amphiphilic peptides. We show that hiNLS Cas9 variants can improve editing efficiency in T cells compared with constructs with terminally fused NLS sequences. Furthermore, many hiNLS Cas9 constructs can be produced with high purity and yield, even when these constructs contain as many as nine NLS. These hiNLS Cas9 constructs represent a key advance in optimizing CRISPR effector design and may contribute to improved editing outcomes in research and therapeutic applications.
Introduction
Genome editing and modification technologies have experienced a transformative evolution with the emergence of CRISPR-mediated engineering, offering unprecedented ease and precision in modifying genetic material. The capacity to manipulate the genome using these programmable enzymes has led to groundbreaking advances in various fields including biology, medicine, and agriculture. 1 However, challenges persist, particularly in the context of optimizing the editing efficiency of enzymes such as the widely used Streptococcus pyogenes nuclease Cas9 (hereafter Cas9). This is especially true in therapeutic use cases, when it would be ideal to attain high rates of editing via a relatively low, transient dose of the enzyme delivered via the ribonucleoprotein (RNP) format, which has been used for multiple ex vivo clinical trials 2 and is used in the first FDA-approved genome editing drug. 3 One of the first ways Cas9 was engineered for efficient eukaryotic genome editing was to fuse nuclear localization signal (NLS) sequences to one or both Cas9 termini.4–6 NLS motifs are short (7–16 amino acid) sequences found in many eukaryotic nuclear proteins as well as viral proteins, with the latter class (e.g., SV40) typically exhibiting exceptional potency for nuclear import. Prior work has also engineered the naturally occurring c-Myc NLS (PAAKRVKLD)—itself a potent NLS motif 7 —to further enhance its activity through sequence optimization 8 (the resulting sequence, PAAKKKKLD, is referred to as “c-Myc” throughout). Proteins containing NLS motifs are actively imported into the nucleus by endogenous proteins such as importin-α, and increased NLS density is typically associated with increased nuclear localization, both in terms of the rate of import and the relative partitioning between nucleus and cytosol.9–11 Because CRISPR enzymes are of prokaryotic origin and inherently lack signals for nuclear import, incorporation of NLS motifs via genetic fusion can greatly enhance editing efficiency in the context of eukaryotic cells. 12 The impact of NLS density on editing efficiency is variable and can depend on the delivery strategy, 13 whether the RNP is being continually expressed (vs. rapidly metabolized), and whether or not the target cells are dividing, given the nuclear envelope breaks down during cell division. 14
Beyond the initial goal of shuttling CRISPR cargo to the cell’s nucleus, NLS optimization has persisted as a valuable avenue for improving genome editing efficiency. Several studies have demonstrated that engineered Cas9 variants with assorted, terminally fused NLS motif combinations exhibit superior performance over constructs with lower NLS density.7,13,15–18 NLS-rich Cas12a proteins achieved efficient targeted mutagenesis in human-transformed cell lines (HEK293T, Jurkat, and K562 cells) and primary cells (natural killer [NK] cells and CD34+ HSPCs]. 7 In the case of Mad7, an engineered class II type V-A Cas12a nuclease, a fusion of additional NLS motifs improved editing efficiencies in Chinese hamster ovary cells. 18
NLS-based enzyme engineering has emerged as an important aspect of CRISPR construct design for in vivo delivery contexts. An NLS-rich Cas9 variant bearing six SV40 motifs at its termini—surgically administered in RNP format—exhibited the capacity for self-delivery into neurons of the mouse brain 19 and has since demonstrated activity comparable to an adeno-associated (AAV)-Cas9 delivery system in the same murine context. 20 The same 6xSV40 Cas9 construct (henceforth “6xNLS” here) was a top performer in our development of peptide-enabled RNP delivery for CRISPR engineering (PERC) in human primary lymphocytes, 21 as was a Cas9 construct bearing a trio of different NLS motifs, 22 herein referred to as “t-Cas9.” A complementary study of human and murine lymphocytes found that peptide-mediated delivery of Cas9 or Cas12a worked well with protein constructs featuring six to eight terminal NLS motifs. 23
Electroporation of Cas9 RNP has been the basis for several clinical trials, as well as the first-ever FDA-approved CRISPR therapy, and even via this potent route of delivery there is copious evidence that NLS-based enzyme engineering can be used to attain optimal electroporation-mediated editing rates.7,22,24 Between electroporation, 25 peptide-mediated RNP delivery,21,23 and delivery of RNP in the brain, 19 there is a groundswell of pre-clinical and clinical development employing CRISPR enzymes in RNP format. Furthermore, for all of these RNP use cases, there is direct evidence that NLS-based enzyme engineering can drive optimal editing efficiency. RNP is appealing for therapeutic use because it is inherently transient, which minimizes off-target editing26,27 and decreases risks of cell-mediated immune response to the microbial protein.28,29 The transient nature of RNP may explain why NLS density has such a pronounced impact on efficiency in this context: due to its 1–2 day half-life, 22 the enzyme must quickly reach the nucleus so that it can induce editing before it is metabolized by the cell.
The RNP format is also appealing for therapeutic development because it can be readily manufactured: solid-state synthesis of guide RNA (gRNA) is straightforward, as is recombinant production of CRISPR proteins. However, our lab and others (Jennifer Doudna and Junwei Shi, personal communication) have observed that CRISPR protein yields decrease—sometimes dramatically—if the construct bears many terminally fused NLS motifs. As an example, the high-performing 6xNLS Cas9 construct yields only 0.6–1.5 mg/L in our experience, with similar yields of ∼1 mg/L reported previously. 20 Poor yield of NLS-rich Cas9 constructs may limit their utility in research applications and therapeutic efforts. With this in mind, we were motivated to explore other approaches to increasing the NLS density of Cas9 at sites other than the N- and C-termini. Our generation and evaluation of Cas9 variants with novel NLS configurations aim to induce optimal nuclear localization while permitting efficient protein production.
While terminal multi-NLS sequences have been widely adopted in CRISPR-Cas genome engineering, the impact of introducing NLS motifs within the backbone of a CRISPR-Cas enzyme has not been systematically investigated. Here, we generated and evaluated several novel S. pyogenes (henceforth “SpCas9”) protein variants containing one or more hairpin internal NLS (hiNLS) modules, with each module comprising a tandem pair of NLS motifs separated by short glycine/serine linkers. These modules were introduced at sites in the SpCas9 sequence previously demonstrated to tolerate domain insertion, and the genome editing activity of the resulting hiNLS Cas9 constructs was evaluated in primary human T cells via two delivery methods: electroporation and PERC. Notably, we found that some hiNLS variants significantly improved editing efficiencies compared with a previously reported NLS-rich construct. Furthermore, the recombinant yield of hiNLS Cas9 variants ranged from good to optimal, suggesting these constructs are better suited for large-scale production, in contrast to a previously reported NLS-rich construct. Our analysis of these hiNLS SpCas9 variants establishes potent and manufacture-friendly constructs that could be broadly employed in research and therapeutics.
Materials and Methods
Cell culture
Frozen, isolated bulk T (CD3+) cells were thawed (on day −3 relative to delivery) and cultured overnight in X-VIVO 15 medium (Lonza) supplemented with 5% fetal bovine serum (FBS), 50 μM 2-mercaptoethanol and 10 mM N-acetyl-
Peptides
INF7TAT-A5K peptide was purchased from CPC Scientific (IMMO-008). Peptide was stored lyophilized or as 10 mM stocks in dimethyl sulfoxide (DMSO) at −20°C in a desiccator.
Cloning
All cloning was conducted by Gibson Assembly Master Mix enzyme cloning methods (New England Biolabs). DNA templates were derived by polymerase chain reaction (PCR) amplification and carried out using Q5 High Fidelity DNA Polymerase (New England Biolabs). All primers and gBlocks Gene Fragments used in this work were obtained from Integrated DNA Technologies. Vectors were transformed into XL-Blue competent cells (Agilent Technologies) prepared by UC Berkeley MacroLab. All plasmids used in this work were freshly prepared from 5 mL of XL-Blue cell culture using QIAprep Spin Plasmid Miniprep (QIAGEN). Molecular biology grade, diethyl pyrocarbonate (DEPC)-treated water was used in all assays, transfections, and PCR reactions to ensure the exclusion of RNase activity. All hiNLS plasmids are available upon reasonable request.
Expression and protein purification
In brief, a single colony from a freshly transformed plate of Escherichia coli strain BL21 Star (DE3) cells was used to inoculate 100 mL Terrific Broth (TB) medium supplemented with the appropriate selection antibiotic and 34 μg/mL chloramphenicol. Cells were grown overnight at 37°C in a shaking incubator and 20 mL was used as a starter culture to inoculate 2.5 L baffled flasks containing 1 L TB with antibiotics as before. Cells were incubated at 37°C at a shaking speed of 200 rpm. When cells reached an OD600 of approximately 1.2, flasks were placed on ice for 30 mins prior to adding isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 200 μM. Cells were incubated for an additional 18–20 h at 16°C with shaking. Cells were harvested by centrifugation for 15 min at 4000 rpm (3470 × g). The supernatant was decanted and the cell pellet was resuspended in 15 mL (per 1 L cell culture) ice-chilled lysis buffer composed of 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.5), 1 M NaCl, 10% (v/v) glycerol, 10 mM imidazole, and supplemented with 1 mM PMSF and a protease inhibitor tablet (Pierce Complete Mini, EDTA-free; A32955). Cells were frozen at −80°C until ready to use. Cells were lysed by sonication, and the resulting lysate was clarified by centrifugation at 20,000 g for 30 min at 4°C to remove cellular debris and recover the supernatant. The clarified lysate was loaded onto a nickel resin column, which had been equilibrated with lysis buffer. Subsequently, the column was washed with at least five column volumes of lysis buffer to remove non-specifically bound proteins. The His-tagged proteins were then eluted from the nickel column using an elution buffer containing 20 mM HEPES (pH 7.5), 100 mM NaCl, 10% (v/v) glycerol, and 300 mM imidazole. Fractions were collected and analyzed by SDS-PAGE to monitor protein purity and yield. The His-tag was removed from the purified Cas9 by an overnight incubation at 4°C with Tobacco Etch Virus (TEV) protease. TEV protease cleaves the His-tag, allowing for the isolation of the target protein in its native form. After TEV protease treatment, Cas9 proteins were subjected to an additional purification step using heparin affinity chromatography. A binding buffer for the heparin column, composed of 20 mM HEPES (pH 7.5), 300 mM NaCl, and 10% (v/v) glycerol, was used to load the fractions onto the column. Non-binding proteins were washed away, and the Cas9 was eluted from the heparin column using elution buffer, which contained 20 mM HEPES (pH 7.5), 1 M NaCl, and 10% (v/v) glycerol. The eluted fractions were collected for subsequent purification steps. The final purification step involved size exclusion chromatography (SEC). SEC was performed on an Akta Purifier using a HiLoad 16/60 S200 superdex column for Cas9 with gel filtration buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 10% [v/v] glycerol) at a flow rate of 1 mL/min. Protein was loaded in volumes no greater than 2 mL. Purified proteins were concentrated to ∼50 μM in a buffer of 20 mM HEPES (pH 7.5), 150 mM NaCl, and 10% (v/v) glycerol, and stored at −80°C. Expression and purification of the cysteine-free 6xNLS Cas9 protein was performed by UC Berkeley MacroLab using the same protocol.
Cas9 sgRNA
SpCas9 single guide RNAs (sgRNAs) (Supplementary Table S1) were purchased with manufacturer-recommended standard chemical modifications from Integrated DNA Technologies and resuspended in DEPC-treated water to make a stock of 100 μM. Before use, sgRNAs were diluted to 30 μM in 20 mM HEPES pH 7.5, and 150 mM NaCl. Diluted sgRNA was refolded by warming to 95°C for 5 min and slow cooling to room temperature for 25 min. Glycerol and MgCl2 were added to the diluted guide only after refolding and cooling to a final concentration of 10% (v/v) and 2 mM, respectively. In addition, for sgRNA used in electroporation-mediated delivery experiments only, poly-
RNP formation
For electroporation experiments, Cas9 protein was diluted to 20 μM in “RNP buffer” (20 mM HEPES pH 7.5, 150 mM NaCl, 10% glycerol, and 2 mM MgCl2). For peptide-mediated delivery experiments, Cas9 protein was diluted to 20 μM in “RNP buffer” supplemented with arginine and trehalose to a final concentration of 300 and 100 mM, respectively. Cas9 was mixed with diluted sgRNA in equal volumes yielding 10 μM RNP complex, with the RNP concentration defined by the amount of Cas9 protein and a final molar ratio Cas9:sgRNA of 1:1.5.
Peptide and RNP delivery formulations in T cells
Peptide (10 mM in 100% DMSO) was diluted in DEPC-treated water to 1 mM (resulting in a solution of 90% water and 10% DMSO) and added to RNP, resulting in a volume no greater than 20% of the eventual final volume (e.g., ≤20 μL formulation for a well with 100 μL final treatment volume when mixed with cells in Opti-MEM). The RNP/peptide mixture was added to a 96-well round-bottom plate, and 200 × 103 cells in Opti-MEM (Gibco) per well were added to the RNP/peptide mixture. The final dose of RNP during cell treatment was 50 pmol per well with a final peptide concentration of 10 μM, unless stated otherwise. In all cases of peptide-enabled delivery, the final concentration of DMSO was proportional to the peptide concentration: 0.1% DMSO per 10 μM peptide; this concentration of DMSO was used for non-treated (NT) negative control conditions. After a 1 h incubation at 37°C, 100 μL of treated volume was split in half into two plates, then 150 μL supplemented culture medium was added per well, thus diluting but not removing the treatment. The concentration of additives and cytokines in the recovery medium was such that the final concentrations matched the description above for each cell type.
RNP electroporation in T cells
In a 4D nucleofector (Lonza), 20 pmol of Cas9 RNP was electroporated into 200 × 103 T cells resuspended in 20 μL of P3 buffer and supplement (Lonza V4XP-3032) using the EH-115 pulse code. Cells were incubated for 10 min at 37°C, then rescued with 80 μL of pre-warmed culture medium before diluting for further cell culture as above.
Flow cytometry
Flow cytometry was performed on an Attune NxT flow cytometer with a 96-well autosampler (Thermo Fisher Scientific). Cells were resuspended in FACS buffer (phosphate-buffered saline, 2% FBS, and 1 mM EDTA) and stained with live–dead stain Ghost dye Red 780 (Tonbo Biosciences) and surface marker-targeting antibodies (BioLegend) according to manufacturer-provided instructions. Sampling was at defined volumes (60 μL per well) to quantify cell counts. Cytometry data were processed and analyzed using FlowJo software (BD Biosciences), and graphs were prepared using GraphPad Prism V10.2.
Data
Data values (underlying the figures) are available in Supplementary Table S2.
Results
hiNLS Cas9 design
We aimed to engineer CRISPR-Cas9 for optimal nuclear import—and compatibility with recombinant expression and purification—by introducing NLS sequences in the protein’s backbone, in hopes that we might discover new variants with robust activity and other desirable properties that some NLS-rich constructs lack. This approach represents a departure from previous efforts, which have almost exclusively introduced NLS sequences at one or both termini of a CRISPR protein. Internal NLS sequences have been introduced within the backbone of a CRISPR enzyme in the context of base editor engineering (with NLS sequence fused adjacent to an inserted deaminase domain), 30 but that report did not systematically evaluate the impact of the internal NLS sequences. We were curious if NLS sequences could be inserted at one or more sites within the backbone of SpCas9 to confer more efficient nuclear localization and thereby improve genome editing efficiency. Because the N- and C-termini of Cas9 are proximal to one another in the 3D structure of Cas9, all terminally fused NLS will be found at just one area on the surface of the RNP. We sought to select sites for NLS insertion that were more evenly distributed across the surface of the enzyme since this could increase the RNP surface area capable of productively interacting with proteins that mediate nuclear import. A prior study used randomized insertional mutagenesis to identify regions of Cas9 that tolerated insertions of a PDZ domain without disruption of the enzyme’s DNA binding and cleavage activities. 31 That work identified local clusters of amino acids that demonstrated a high tolerance to insertions, often around flexible loops, the ends of helices, and at solvent-exposed residues. Specifically, regions of high plasticity were profiled at six clusters within the helical recognition (REC) lobe, the linker between the REC and nuclease lobes, the HNH domain, three extended sites in the RuvCIII region, and throughout the PAM interacting (C-terminal) domain. Another study demonstrated that catalytically dead Cas9 (dCas9) can tolerate large single deletions to the REC2, REC3, HNH, and RuvC domains while maintaining near-native levels of DNA-binding activity. 32 Given that orthogonal domains (or combinations thereof) were inserted into the identified sites with minimal functional consequence, we hypothesized that installing hiNLS modules at the insertion-tolerant sites (Fig. 1A) would be unlikely to perturb the enzyme’s native function. We selected four regions within the Cas9 backbone (referred to as sites 1–4) to introduce hiNLS modules either alone or in combination. Examining the three-dimensional structure of Cas9 33 allowed us to identify available surface residues that overlapped with insertion-tolerant sites. The hiNLS modules were installed at four distinct sites, immediately following the respective Cas9 amino acids Gly-205 (helical domain), Lys-468 (helical domain), Ile-1022 (RuvC-III), and Ser-1248 (CTD) (Fig. 1A).
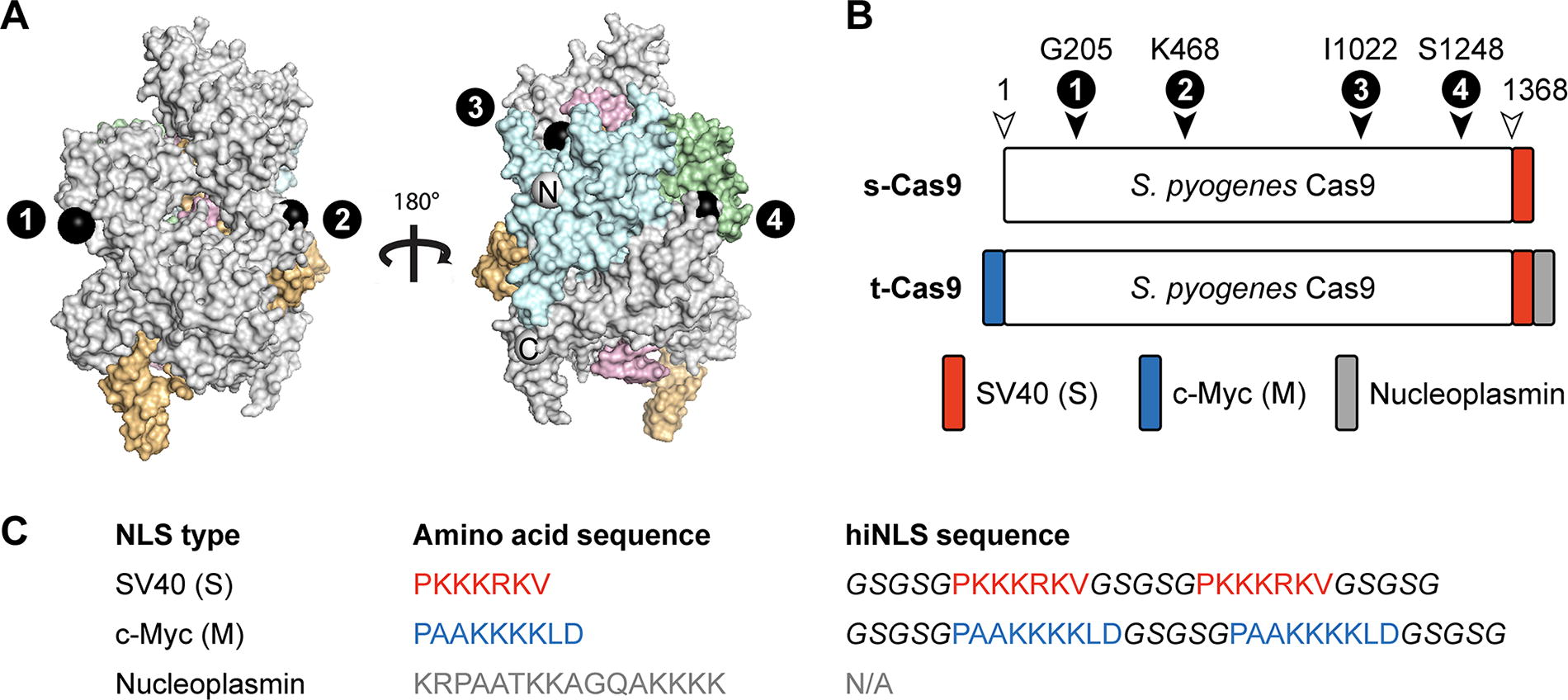
Sequence and position of hiNLS motifs incorporated into CRISPR-Cas9.
Working exclusively with cysteine-free Cas9 constructs to eliminate risks of disulfide-based dimerization, we engineered 15 hiNLS Cas9 variants based on two previously reported Cas9 “backbone” constructs distinguished by their N- and/or C-terminal NLS fusions: s-Cas9 and t-Cas9 (Fig. 1B). The s-Cas9 backbone possesses a single (hence “s”) monopartite SV40 motif at its C-terminus (akin to the 1× NLS Cas9 reported previously 34 ), and the t-Cas9 backbone (Addgene ID# 196245; referred to as 3× NLS-Cas9 when originally reported 22 and later as Cas9-triNLS 21 ) bears a trio (hence “t”) of different terminal NLS motifs: N-terminal c-Myc (M) as well as C-terminal SV40 (S) and nucleoplasmin sequences, respectively. Each hiNLS module features a “linker-NLS-linker-NLS-linker” layout (Fig. 1C) selected to increase local NLS valency and allow each NLS to adopt the extended, linear conformation that is optimal for binding to importin proteins. 35 We hoped that the bivalent architecture would increase the effective affinity between importins and the hiNLS module since dissociation of one NLS motif could immediately be followed by association with the neighboring NLS motif. Inclusion of a single NLS motif at these insertion sites—which are typically loops in the wild-type Cas9 structure—would introduce the risk that the NLS chain would be constrained to a curved loop-like structure, which is incompatible with the ideal conformation for binding an importin protein. We anticipated that the hiNLS layout (29–33 aa; two NLS motifs within three linker regions) would offer more flexibility than the alternative one-NLS layout (17–19 aa; including two linker regions). Finally, to minimize repetitive DNA sequences within the hiNLS modules and the associated risk of plasmid instability, we deliberately shuffled codons to promote nucleotide sequence diversity without impacting the desired amino acid composition. Each hiNLS construct (Fig. 2) is named based on the backbone type (either “s” or “t”) as well as the hiNLS type (an “S” or “M” pair) and the location at position “1” (Gly-205), position “2” (Lys-468), position “3” (Ile-1022), and/or position “4” (Ser-1248).
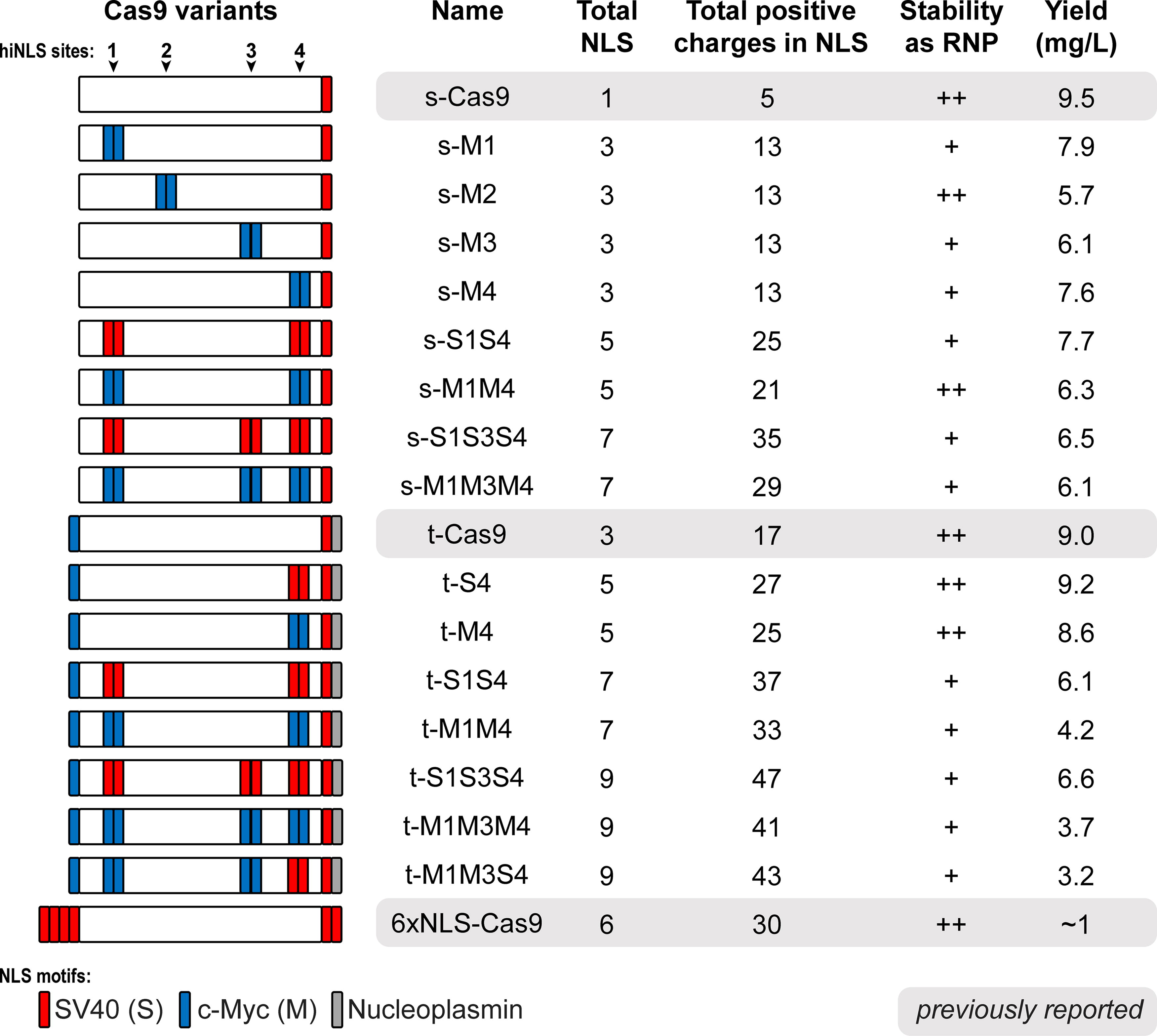
Properties of hiNLS Cas9 constructs. Summary of NLS composition and properties of all the hiNLS constructs designed and tested, alongside s-Cas9, t-Cas9, and 6xNLS-Cas9. The last column shows an expected average yield of protein expressed and purified for each construct. Each scaffold-bearing terminal NLS is denoted in lower case letters (s, SV40; t, triNLS). Type of hiNLS module is denoted in upper case letters (S, SV40; M, c-Myc). The number following each capital letter (S or M) corresponds to hiNLS position in the Cas9 backbone. “+” denotes RNPs that were soluble only in the trehalose/arginine buffer, whereas “++” indicates RNPs that remained soluble in the trehalose/arginine buffer and a standard buffer (lacking additional stabilizers).
Single hiNLS installation in Cas9 can enhance genome editing efficiency
We sought to investigate the impact of strategically inserting a single hiNLS module at four distinct internal sites within Cas9, using RNP-mediated genome editing in primary human T cells as a test case. To evaluate the potential improvements in editing, hiNLS modules—each containing a pair of SV40 NLS motifs—were incorporated at each of the specified internal sites 1–4 within the Cas9 backbone, as depicted in Figure 1A. We assessed the efficacy of the hiNLS Cas9 B2M- and TRAC-RNP variants targeting the beta-2-microglobulin (B2M) and T cell receptor alpha chain (TRAC) loci in primary human T cells (Supplementary Table S1). Knockout efficiencies were evaluated 4 days following intracellular delivery via either electroporation or PERC21 (Fig. 3). In contrast to electroporation, PERC is a hardware-free, non-viral approach to CRISPR RNP delivery that could conceivably be used in vivo, and we view it as a proxy for other in vivo delivery technologies—such as virus-like particles or lipid nanoparticles—that will typically introduce a transient and relatively low “dose” of CRISPR RNP enzyme into cells, potentially heightening the need for potent NLS activity. 36 Peptide-mediated RNP delivery to cultured cells works especially well with NLS-rich CRISPR proteins,21,23 driving our curiosity about the impact of hiNLS constructs in this context. Our experimental design deliberately employed sub-optimal doses of RNP (as compared with doses previously used for electroporation 21 or PERC 37 ) to ensure an informative dynamic range, allowing us to distinguish between the performance of various constructs. Except for s-M2, all single hiNLS Cas9 variants increased B2M and TRAC knockout efficiencies via PERC delivery relative to both s-Cas9 and t-Cas9 backbone constructs lacking hiNLS modules, as measured by fold change in editing (Fig. 3A; compare dashed-bracket “one hiNLS” data with white and gray bars). The normalized fold change (nfc) is in comparison to the average editing efficiency in a given experiment for a single donor to account for biological donor-to-donor variability (Supplementary Table S2). Notably, single hiNLS variants s-M1, s-M3, t-S4, and t-M4 achieved between 47.0% and 48.5% (1.32–1.38 nfc) B2M knockout via PERC, surpassing the 37.9% (1.05 nfc) mediated by the high performing t-Cas9 (Supplementary Fig. S1A). The same hiNLS variants achieved between 42.3% and 48.0% (1.22–1.38 nfc) TRAC knockout via PERC, surpassing the 41.4% (1.19 nfc) mediated by t-Cas9 (Supplementary Fig. S1A and Fig. 3C). Electroporated RNPs showed similar editing trends (Fig. 3B and D; compare dashed-bracket “one hiNLS” data with white and gray bars). Comparably, the single hiNLS variants delivered via electroporation that demonstrated higher B2M knockout rates than t-Cas9 (66.4%; 1.08 nfc) were s-M3, s/t-M4, and t-S4 (73.0%–78.9%; 1.19–1.27 nfc) (Supplementary Fig. S2A and Fig. 3B). Similarly, the same hiNLS variants achieved between 59.2% and 71.3% (1.22–1.49 nfc) TRAC knockout via electroporation, surpassing the 49.9% (1.02 nfc) mediated by t-Cas9 (Supplementary Fig. S4a and Fig. 3d). The s-M2 construct performed poorly (Fig. 3 and Supplementary Fig. S1, S2, S3, and S4), perhaps due to the insertion site’s proximity to the bridge helix essential for the allosteric signal transduction that underlies the nuclease activity of Cas9. 38 Because of this poor performance, hiNLS site 2 was not prioritized in subsequent engineering efforts.
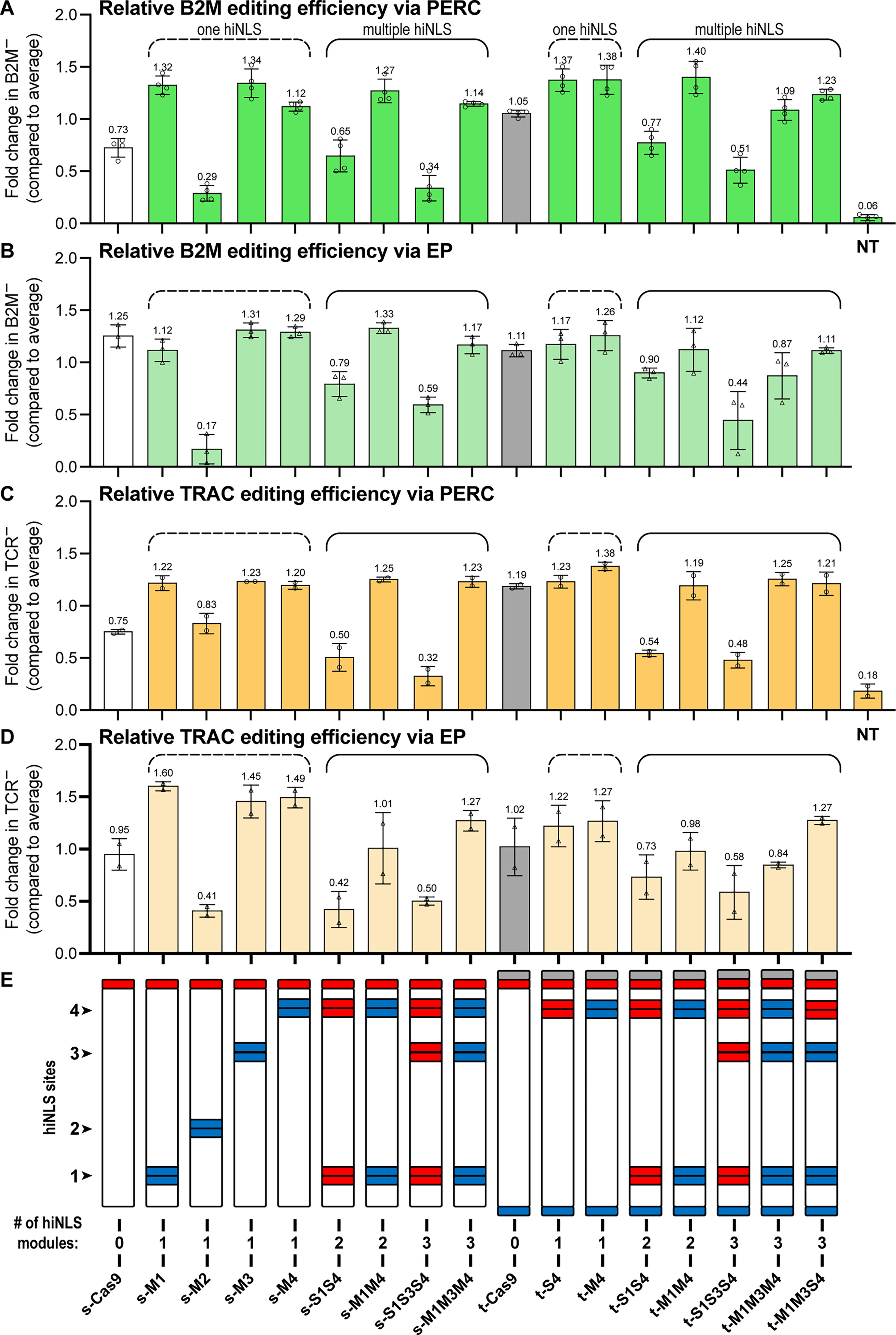
Relative editing efficiencies mediated by hiNLS Cas9 in primary human T cells. Cas9 B2M-RNPs and TRAC-RNPs were delivered via peptides (PERC)
Multiplex hiNLS Cas9 constructs further improve editing efficiency via peptide-mediated delivery
Prior work demonstrated that the t-Cas9 construct (which bears terminal fusions of three types of NLS) promoted improved editing efficiencies as compared with s-Cas9 (which bears a sole NLS) 22 suggesting that increased NLS count and/or increased NLS diversity could boost enzyme activity. Furthermore, under most conditions tested, the incorporation of a single hiNLS (s-M1, s-M3, s-M4) module yielded improved editing efficiencies as compared with the s-Cas9 scaffold. We wanted to explore the potential synergistic effects of incorporating multiple hiNLS modules inside the high-performance t-Cas9 architecture. To determine if the potency of t-Cas9 is influenced by factors beyond the sheer quantity of NLS motifs, we compared the impact of distinct NLS motifs, e.g., SV40 and c-Myc. To test whether combining multiple hiNLS in Cas9 would improve editing, we incorporated multiplex hiNLS modules within the Cas9 backbone and evaluated B2M- and TRAC-RNP knockout efficiencies in primary human T cells. Notably, multiplex hiNLS variants s/t-M1M4, s/t-M1M3M4, and t-M1M3S4 achieved between 39.6% and 49.0% (1.09–1.4 nfc) B2M knockout via PERC, surpassing the 37.9% (1.05 nfc) mediated by the high performing t-Cas9 (Supplementary Fig. S1 and Fig. 3A; compare solid-bracket “multiple hiNLS” data with white and gray bars). Although electroporated multiplexed hiNLS B2M-RNPs showed less pronounced improvement over t-Cas9 (Fig. 3B; compare solid-bracket “multiple hiNLS” data with white and gray bars), s-M1M4 mediated an editing efficiency of 81.5% (1.32 nfc), the highest efficiency resulting from any construct we evaluated (Supplementary Fig. S2A). RNPs delivered via PERC-mediated TRAC knockout efficiencies are higher when using all multiplex hiNLS Cas9 variants except for s-/t-S1S4 and s-/t-S1S3S4 as measured by fold change (Fig. 3C; compare solid-bracket “multiple hiNLS” data with white and gray bars). Multiplex hiNLS variants achieved between 42.0% and 43.8% (0.48–1.5 nfc) TRAC knockout via PERC, slightly surpassing the 41.4% (1.19 nfc) mediated by t-Cas9 (Supplementary Fig. S3A). While the multiplex hiNLS strategy further enhanced editing via PERC, it offered less pronounced improvements in the context of electroporation when compared with the single hiNLS constructs (Fig. 3B, D; compare solid-bracket data with dashed-bracket data). Nevertheless, two hiNLS Cas9 RNP variants (s-M1M3M4 and t-M1M3S4) delivered by electroporation for TRAC KO surpassed t-Cas9 (49.9%; 1.02 nfc), reaching efficiencies as high as 61.0% (1.27 nfc) (Fig. 3D, Supplementary Fig. 4A). Further, we sought to assess the genome-level specificity of the top-performing hiNLS constructs. Because our B2M and TRAC gRNAs do not detectably edit any off-target sites in the human genome, we employed an EMX1-targeting gRNA with a well-established off-target site. 39 We electroporated EMX1-RNP and assessed on- and off-target editing using amplicon-based NGS (Supplementary Fig. S5). This experiment compared t-M4 and t-M1M4 to triNLS as well as 6xNLS, an NLS-rich Cas9 construct that has poor yield via recombinant expression but mediates high-efficiency editing via PERC and electroporation 21 (potentially due to its NLS density). The hiNLS constructs produced the highest editing efficiency at the EMX1 on-target site, with slightly lower efficiency induced by the previously reported triNLS and 6xNLS, which performed similarly to each other. Interestingly, genome editing at the EMX1 off-target site was about twice as likely for hiNLS constructs t-M4 or t-M1M4 (1.09% or 1.40%, respectively) as compared with triNLS and 6xNLS (0.58% or 0.39%, respectively). This observation suggests that placing NLS at non-terminal sites may change the way Cas9 interacts with the R-loop, in turn impacting the enzyme’s interaction with partially mismatched DNA targets. Cas9 variants with improved specificity have been generated via point mutations that selectively remove positive charge from the enzyme surface.40–43 It is conceivable that hiNLS constructs are having the opposite effect: introduction of positive charge may be promoting non-specific interaction with DNA and in turn promoting editing at off-target loci.
c-Myc-rich hiNLS Cas9 constructs are especially efficient
NLS content and diversity may play a role in mediating delivery as well as activity of Cas9 within the cell, thereby affecting the overall efficiency of genome editing. Previous studies have demonstrated that when compared with SV40 NLS, c-Myc NLS has a higher nuclear localization efficiency.7,44 Interestingly, our results do not suggest a direct association between the number of NLS in a given Cas9 construct and the resulting genome editing efficiency (Fig. 4B, D). Nor do the positive charges introduced by the NLS sequences (four per c-Myc motif; five per SV40 motif) clearly predict the performance of a given Cas9 construct (Fig. 4C, E). Our results reveal an apparent trend: constructs rich in c-Myc NLS (Fig. 4A) tend to support higher efficiency genome editing than those rich in SV40 NLS (Figs. 4B–E and 5; note blue symbols). That said, the similar performance of t-S4 and t-M4 suggests a complex situation that does not lend itself to a single set of rules. Although it may seem intuitive that it is generally beneficial to introduce more NLS motifs, our results suggest that the composition and arrangement of NLS sequences, in conjunction with other factors such as the presence of c-Myc-derived motifs, 8 may be a key contributor to the editing efficiency of a given Cas9 construct.
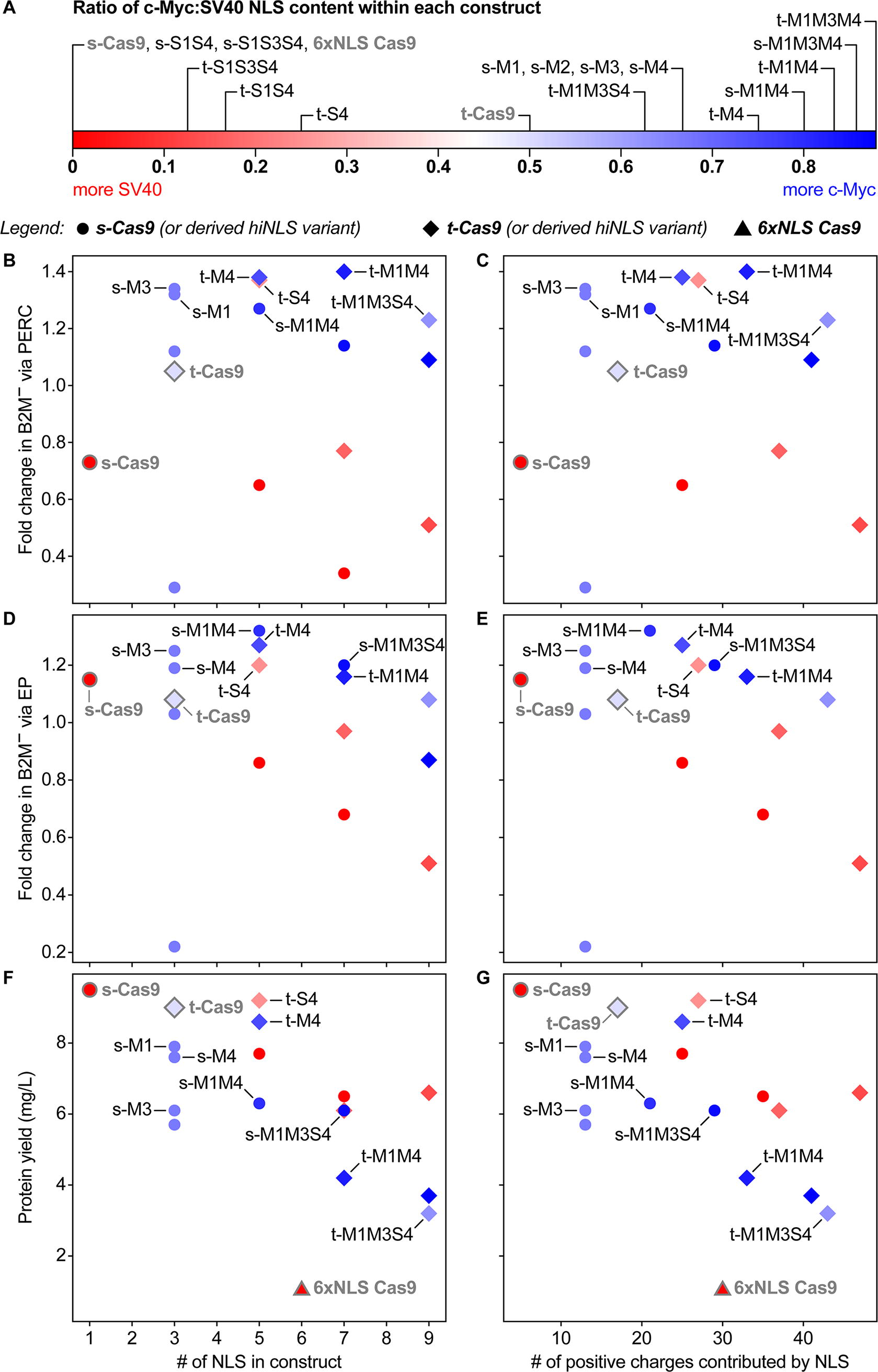
The impact of NLS amount and diversity on editing outcomes and recombinant protein yield.
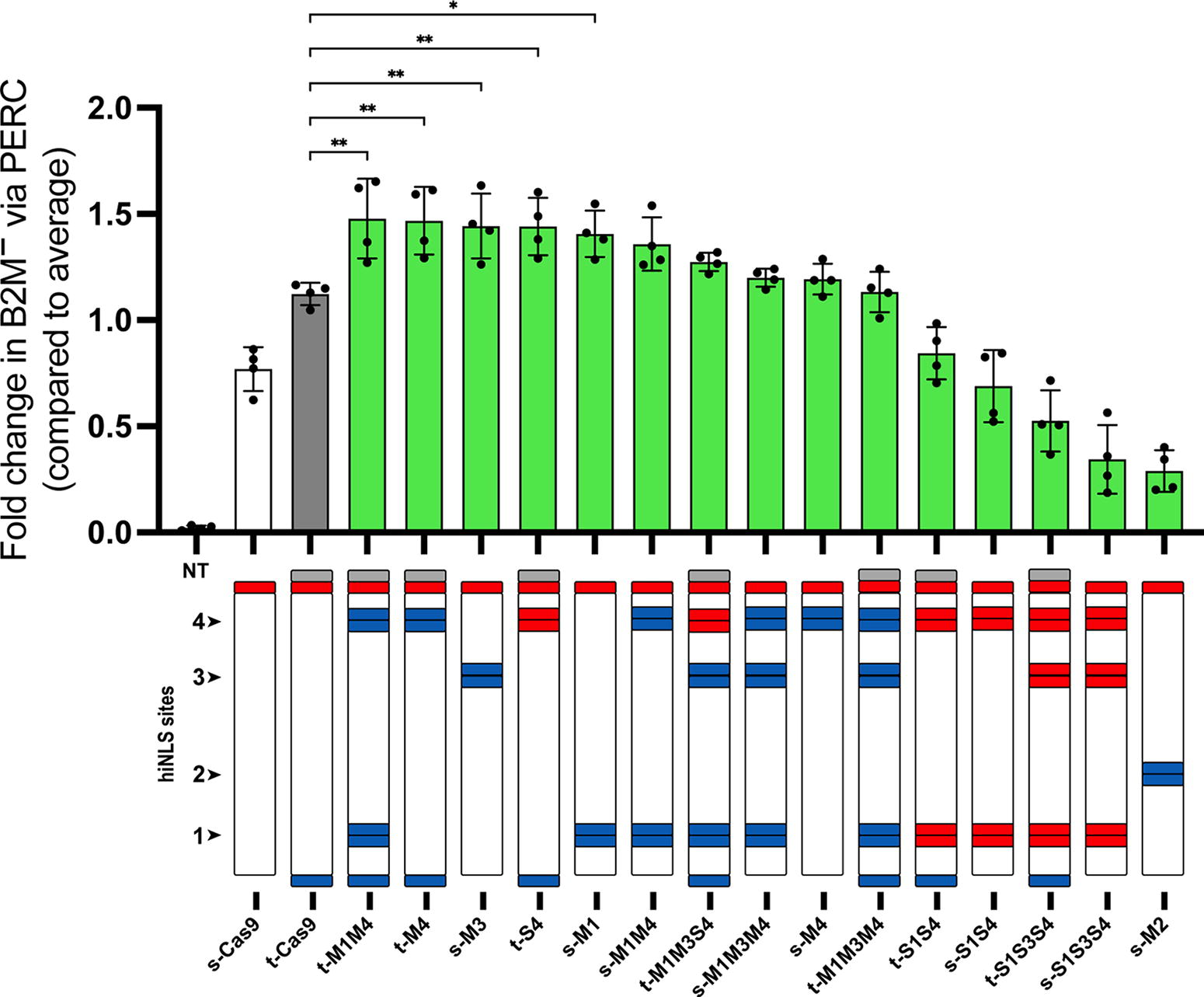
Ranked editing performance of hiNLS Cas9 constructs via peptide-mediated delivery. The B2M knockout efficiency, with hiNLS constructs presented according to their performance. The Cas9 B2M-RNPs were delivered via peptides (PERC), with knockout efficiencies assayed 4 days after delivery by flow cytometry to evaluate surface expression. Here, knockout efficiencies are represented as fold change compared with the average editing efficiency in the experiment. In total, 50 pmol of RNP were delivered. *p < 0.05; **p < 0.01. n = 4 distinct T cell donors. These values are derived from the same experiment underlying Figure 3 and Supplementary Figures S1 and Figure S2.
The hiNLS framework supports increased NLS density without sacrificing Cas9 yield
To assess protein production efficiency, we recombinantly expressed hiNLS Cas9 and terminally fused NLS Cas9 constructs in E. coli and purified each via three-step chromatography. Notably, all hiNLS Cas9 constructs substantially outperformed the highly potent 6xNLS Cas9 in terms of protein yield (Figs. 2 and 4F, G). For instance, hiNLS Cas9s t-M4, t-M1M4, and t-M1M3M4 possess five, seven, and nine NLS sequences, respectively, yet still achieved protein expression levels exceeding between 3.7–8.6 mg/L, while the terminally fused 6xNLS Cas9 yields only 0.6–1.5 mg/L. One hiNLS Cas9, t-S4, bearing five NLS motifs, yielded 9.2 mg/L, and demonstrated superior editing performance in comparison to the highly potent t-Cas9 in both PERC and EP delivery contexts, at two genetic loci. We also performed a qualitative assessment of hiNLS RNP stability. Each hiNLS construct was assembled into an RNP complex, and its solubility was tested in two different buffers: a standard buffer (lacking additional stabilizers) and a specialized trehalose/arginine buffer designed to promote protein stability. As summarized in Figure 2, we used a simple notation wherein “+” denotes RNPs that were soluble only in the trehalose/arginine buffer, whereas “++” indicates RNPs that remained soluble in both buffers. This observation implies that certain hiNLS variants may be more appealing than others, as their ability to remain soluble in the RNP state across different formulations could be advantageous for downstream applications. These findings highlight the capacity for the hiNLS framework to simultaneously improve genome editing activity and capacity for efficient recombinant production.
Discussion
The use of Cas9 constructs with terminally fused NLS sequences has been ubiquitous in genome editing applications. Here we reported the engineering of Cas9 variants featuring hiNLS modules that introduce pairs of NLS motifs into the backbone of the CRISPR enzyme, resulting in improved rates of genome editing in human lymphocytes via two RNP delivery approaches: electroporation and PERC. Strategic positioning of hiNLS within Cas9 constructs promoted enhanced knockout efficiencies at genetic targets such as B2M and TRAC, with preservation of cell viability. The versatility demonstrated across different target genes highlights the robustness and potential translational impact of hiNLS Cas9 variants.
Increasing NLS content and/or diversity in Cas9 has been shown to improve genome editing efficiencies,7,20,22 but in some cases NLS-rich Cas9 has been difficult to generate with high yield via recombinant expression and purification. The hiNLS constructs reported here not only improve editing efficiencies but also support high-yield recombinant production. Our findings suggest that the potency of hiNLS in boosting editing efficiency may not be solely dictated by the sheer number of charges or NLS motifs but rather NLS type and placement. It appears that there exists a point of diminishing returns, indicating a non-linear relationship between the number of NLS motifs in a Cas9 construct and its resulting editing efficiency. While increasing the NLS content or the charge of Cas9 constructs can enhance editing performance to a certain extent, further engineering does not necessarily translate into continuous improvement of outcomes. For example, hiNLS constructs s-M1, s-M3, t-S4, s/t-M4, s/t-M1M4, and s/t-M1M3M4 consistently demonstrated higher editing rates than the potent t-Cas9, while s-S1S3S4 and t-S1S3S4 performed relatively poorly (Fig. 5).
Three hypothetical mechanisms may contribute to these NLS-linked increases in genome editing efficiency. (1) Positively charged NLS helps promote localization of RNP to the negatively charged cell surface, promoting efficiency of intracellular delivery (via electroporation or PERC). (2) Once the RNP cargo has reached the cell interior (cytosol and/or nucleus) NLS content promotes not only rapid initial nuclear import but also ensures that a given RNP is re-imported to the nucleus if it diffuses out. This would amount to an NLS-dependent change in “nuclear partitioning,” shifting the balance between nuclear RNP and cytosolic RNP. This would likely minimize exposure to the cytosolic proteasome and thereby extend the half-life of RNP in the cell, giving each RNP more opportunities to generate productive edits. (3) Positively charged NLS promotes non-specific interaction with genomic DNA, in turn accelerating the search for intended target sites in the genome. This is the most speculative potential contributor due to our limited knowledge of the genomic search mechanisms employed by RNA-guided nucleases, but we note the prior finding that Cas9 variants featuring one or more mutations removing a positively charged amino acid substantially decreased the efficiency of Cas9. 40 This finding is consistent with the idea that positively charged amino acids—8 or 10 of which are found in each of our hiNLS modules—may be generally beneficial for Cas9 activity. Our results suggest that Cas9 constructs rich in c-Myc motifs specifically tend to exhibit enhanced editing capabilities (Fig. 5), suggesting that the composition and arrangement of NLS sequences may play a more nuanced role in determining Cas9 performance than their absolute quantity. These results underscore the importance of considering factors beyond mere NLS count when engineering genome editing enzymes for optimal performance. Using different types of internal NLS sequences in Cas9 may offer advantages by fine-tuning nuclear import efficiency and otherwise enhancing the enzyme’s performance. Different NLS types, such as monopartite, bipartite, or even NLS motifs from diverse origins (e.g., viruses or various eukaryotes) can exhibit distinct affinities for nuclear import machinery, 45 resulting in varying rates of nuclear localization. Overall, the strategic use of different internal NLS types can improve the specificity, efficiency, and versatility of Cas9-based gene-editing technologies. These findings contribute to our understanding of the factors influencing CRISPR-mediated genome editing and may pave the way for the rational design of optimized enzyme variants with enhanced genome editing capabilities.
Novel hiNLS constructs can produce higher editing efficiencies compared with Cas9 constructs bearing terminally fused NLS motifs, revealing a fruitful strategy for loading a CRISPR enzyme with NLS motifs without compromising protein yield via recombinant production. A previously reported NLS-rich construct, 6xNLS Cas919, suggests a potential limitation of incorporating multiple NLS motifs via fusion to the protein termini: the yield of this construct is extremely low following recombinant expression: merely ∼1 mg/L of E. coli culture. 20 Although some of our novel hiNLS constructs generally result in lower yields with increasing NLS density, we nevertheless attained reasonable yields for constructs featuring 5, 7, or 9 NLS sequences: 8.0, 5.7, or 4.5 mg/L on average, respectively (Fig. 2). Two hiNLS constructs that demonstrated robust editing efficiencies across genetic targets (B2M, TRAC, EMX1) and bear five NLS motifs each, t-S4 and t-M4, yielded 9.2 and 8.6 mg/L of E. coli culture, respectively, which is comparable to the high-yielding s-Cas9 (9.5 mg/L) and t-Cas9 (9.0 mg/L) constructs. t-S4 and t-M4 emerged as the most promising hiNLS constructs based on their strong editing performance across delivery methods, favorable recombinant yields, low off-target editing, and superior solubility profiles. Differences in solubility suggest that certain hiNLS constructs may be more suitable for practical applications, where stability in diverse conditions is advantageous. Moreover, the capacity to produce large quantities of these proteins will likely contribute to their utility in research or therapeutics applications. These results underscore the substantial advantage of hiNLS Cas9 in terms of recombinant protein production potential, offering a significant improvement over traditional Cas9 engineering strategies. The hiNLS facilitates the highest NLS counts for Cas9 that we are aware of, and in many cases the impact to recombinant yield is moderate or negligible.
While the increased editing rates of hiNLS Cas9 variants represent a significant advantage, it is important to acknowledge the potential of off-target editing, particularly in therapeutic contexts. In our study, off-target activity was assessed using a gRNA known to have a prominent off-target site. Although some hiNLS constructs exhibited higher off-target editing at this locus, this observation may be less concerning for gRNAs without significant off-target sites in the genome. This highlights the need for careful gRNA design when deploying hiNLS Cas9 variants in therapeutic applications.
Incorporating hiNLS within the Cas9 primary sequence surpasses the limitations of terminally fused NLS tags. The addition of extra amino acids to one or both termini can disrupt protein function and/or its stability. 46 In fact, small changes to the protein sequence, especially at the ends, can alter its overall structure and interfere with its ability to interact with other importin-like molecules, potentially rendering it nonfunctional. Leveraging hiNLS sequences inside of Cas9, rather than placing them on the termini, offers advantages for protein engineering and functional versatility. By positioning the NLS internally, the N- and C-terminal ends of Cas9 remain more available for the fusion of other functional domains or fluorescent tags. Moreover, internal NLS fusion reduces the likelihood of the signal being proteolytically cleaved, because two cleavage events are required to remove the NLS motif from the RNP, instead of one. The use of the hiNLS framework could bolster the integrity of fused NLS motifs as well as terminally fused functional domains (which sometimes rely on NLS motifs as linkers tethering them to the CRISPR enzyme). The hiNLS strategy allows greater flexibility in designing increasingly complex and modular Cas9 systems for diverse genome editing tasks.
The most promising—albeit most challenging—frontier in CRISPR-mediated editing is extrahepatic delivery of in vivo therapeutics, where balancing potency and the trafficking of CRISPR enzymes into the relevant cell types remains an extraordinary obstacle. This need is further heightened by the practical appeal of transient (non-viral) delivery, which imposes a brief window during which a CRISPR enzyme must perform its intended edit. NLS engineering is one way to attain the high specific activity mandated in transient in vivo delivery contexts, but there have been limitations on how many NLS can be introduced before protein production begins to suffer. With hiNLS engineering, we propose a way around these limitations, reaching new heights in NLS density and enzyme potency without substantially sacrificing yield. The hiNLS framework supports the efficient production of potent genome editing tools and candidate therapeutics. Our generation and evaluation of novel hiNLS Cas9 constructs highlights the importance of considering the role of NLS motifs in enhancing activity as well as their impact on recombinant protein yield, informing the design landscape for optimizing Cas9 and other CRISPR enzymes for ease of manufacture and optimal genome editing performance.
Footnotes
Acknowledgments
The authors thank Megan Hochstrasser for critical commentary on this article.
Authors’ Contributions
E.A.N. and R.C.W. conceived the study. E.A.N. and S.U.S. performed experiments, analyzed data, and performed data visualization. E.A.N. and C.J. contributed reagents. S.K.W. performed data visualization. N.K. performed experiments. E.A.N., S.U.S., and R.C.W. wrote and reviewed the article.
Author Disclosure Statement
R.C.W. is a co-founder, shareholder, and compensated consultant of Editpep, Inc.
Funding Information
E.A.N. received support from the Innovative Genomics Institute. S.U.S. and R.C.W. received support from the CRISPR Cures for Cancer.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.