
Abstract
The potato family includes a highly diverse cultivar repertoire and has a high potential for nutritional yield improvement and refinement but must in line with other crops be adapted to biotic and abiotic stresses, for example, accelerated by climate change and environmental demands. The combination of pluripotency, high ploidy, and relative ease of protoplast isolation, transformation, and regeneration together with clonal propagation through tubers makes potato highly suitable for precise genetic engineering. Most potato varieties are tetraploid having a very high prevalence of length polymorphisms and small nucleotide polymorphisms between alleles, often complicating CRISPR-Cas editing designs and strategies. CRISPR-Cas editing in potato can be divided into (i) characterization of target area and in silico-aided editing design, (ii) isolation and editing of protoplast cells, and (iii) the subsequent explant regeneration from single protoplast cells. Implementation of efficient CRISPR-Cas editing relies on efficient editing at the protoplast (cell pool) level and on robust high-throughput editing scoring methods at the cell pool and explant level. Gene and chromatin structure are additional features to optionally consider. Strategies and solutions for addressing key steps in genome editing of potato, including light conditions and schemes for reduced exposure to hormones during explant regeneration, which is often linked to somaclonal variation, are highlighted.
Introduction
Potato: Importance and potential
Potato is in terms of human consumption the third most important food crop in the world after rice and wheat (FAOSTAT, New Food Balances, last updated March 28, 2022; https://www.potatonewstoday.com/2022/03/28/fao-updates-global-potato-statistics/) and is in relation to consumption and nutritional value, based on calories generated per acre, the most efficient food crop on earth. The importance and future potential of potato, as evidenced by food security metrics and including both rural and industrial agri-food systems, have recently been reviewed. 1 Current and future climate changes, environmental challenges, and a growing world population call for developing crops with increased resilience to biotic and abiotic stress while maintaining or even increasing yields. This prompts for the introduction of traits for improved resilience against various pests and increased robustness to drought, flooding, salinity, or temperature, but also for traits conferring consumer-demanded high nutritional value and characteristics such as flavor, aroma, or shape. Traits and a list of known target genes of importance for tailoring crops, including potato, with superior product quality through the use of genome editing has recently been reviewed. 2 Similar reviews focused on potato may be found in previous work3,4 and with more general potential for designer crops and domestication outlined in previous work.5,6 Due to its relative ease of genome modification and propagation, potato may serve as a model for field trials of genome edited crops in the Northern hemisphere 7 and enters as a principal crop in cis-genesis discussions and the European green deal. 8
Key features
The complex potato genome, sexual versus clonal propagation of potato
Some crops, including cassava 9 and potato, can be maintained and multiplied through clonal propagation of tubers or roots, allowing for preservation of both existing and ectopically introduced traits, for example, generated by precision gene editing technologies. 10 Cultivated potato is autotetraploid (2n = 4x = 48) and highly heterozygous with a genome of 12 chromosomes 11 and its maintenance is essential to avoid inbreeding depression, including reduced fertility, and productivity. 12 Sexual reproduction by seed is associated with unpredictable segregation and high recombination rates, potentially resulting in loss of existing traits, for example, laboriously acquired from earlier breeding efforts.6,10
Pluripotency and transformation of potato
Some broad-leaved species are distinguished by their relative ease of manipulation and pluripotency, 13 which in the case of potato14,15 and tomato 16 allows for isolated single-leaf protoplast cells (protoplasts) to be regenerated clonally into explants. In the context of gene editing, potato cells may be transformed by agrobacterium-mediated transformation,17,18 ballistic bombardment of meristems or embryos 19 or by polyethylene glycol (PEG)-mediated transformation of single, usually leaf-derived, protoplasts. 20 Whereas agrobacterium-mediated transformation involves stable integration of the construct into the genome, ballistic bombardment and PEG-mediated transformation allow for the transformation of nonintegrative transiently expressed DNA constructs14,15 or DNA-free ribonucleoprotein (RNPs).
The combination of protoplast isolation and transformation, pluripotency, and relative ease of regeneration together with the ability for clonal propagation through the tubers makes potato highly suitable for precise genetic engineering (see summarization in Fig. 1).
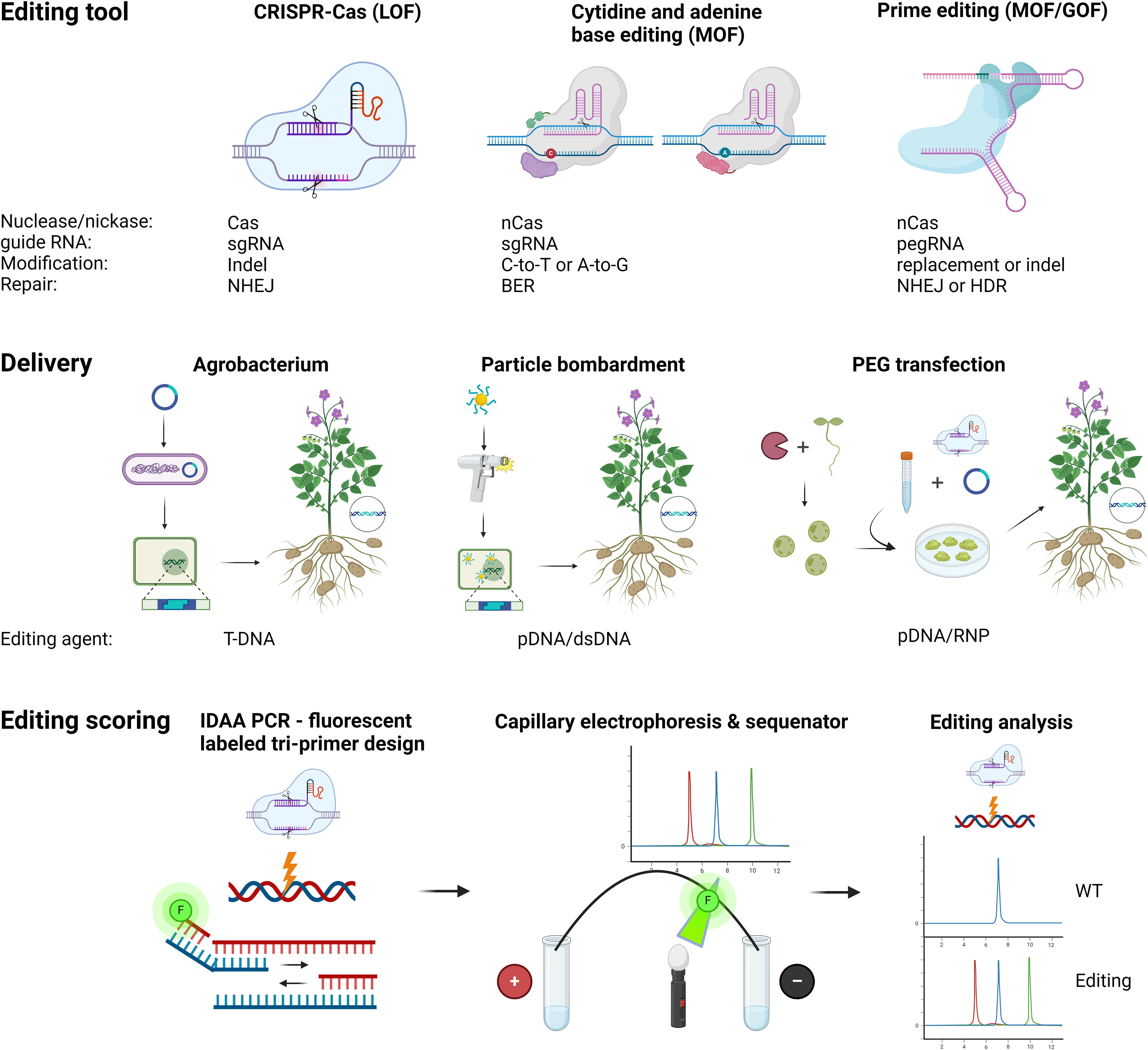
CRISPR-Cas editing and editing scoring. Classical genome editing in plants involves a sgRNA guided Cas dsDNA nuclease with subsequent induction of the non-homologous end-joining (NHEJ) DNA repair pathway, frequently resulting in loss of function (LOF) mutations. Modulation or gain of function (MOF/GOF) may be obtained through the use of sgRNA guided base editors (BEs), for example, cytidine and adenine BEs, which employ a nickase version of the Cas enzyme (nCas) with subsequent induction of the base excision repair (BER) pathway, or by prime editor RNA (pegRNA) guided prime editors (PEs), with subsequent induction of the NHEJ or the homology directed Repair pathway (HDR), depending on cell type. While BEs have been applied successfully in plants (optimizations devised in study by Westberg et al. 21 and precision reviewed in study by Molla et al. 22 ) successful applications of PEs in plants are at present absent or scarce at best. Various mostly PCR-based editing screens exist 23 of which the Indel Detection of Amplicon Analysis (IDAA) appear to be robust and very suitable for organisms with high ploidy and complex genomes, such as tetraploid potato.24,25 IDAA PCR involve a three-primer design including a fluorescently labeled, for example, fluorescein amidite (FAM), universal primer or a two-primer design with one of the gene specific forward or reverse primers being fluorescently labeled. ABE, adenine base editor; BE, base editor; CBE, cytidine base editor; IDAA, Indel Detection Amplicon Analysis; dsDNA, double stranded DNA; GOF, gain of function; Indel, Insertion/Deletion; LOF, loss of function; NHEJ, nonhomologous end-joining repair pathway; MOF, modulation of function; nCas, Nickase Cas; pDNA, plasmid DNA; PEG, polyethylene glycol; pegRNA, prime editing guide RNA; RNP, ribonucleoproten; sgRNA, sequence guide RNA; T-DNA, transfer DNA. Image created with Biorender.com.
Edited plants, for example, the tetraploid potato cultivars Desiree, King Edward, 13 and Wotan 24 have been generated through PEG-mediated transformation of protoplast using transient nonintegrative constructs13,21,24 or RNP15,25 or agrobacterium-mediated transformation of integrative CRISPR-Cas constructs. 18 Schemes for optimizing gene editing efficiency at the protoplast (cell pool) level24–26 and editing scoring have also been devised.23,24,27 Here we summarize these schemes and provide optimizations of current potato transformation and regeneration protocols, which include reducing the number and length of regeneration steps and thus exposure to hormones often linked to somaclonal variation, and discuss identification and mapping of target genes in potato.
Results and Discussion
Gene targeting
Identification and selection of target gene(s) with predicted loss-of-function consequences included, often require considerations relating to gene families, phylogeny, and prediction of orthologues of genes often originally identified outside of the Solanaceae family. For example, one of the most known susceptibility (S) genes, DMR6, has several homologs in potato, and while one homolog, the StDMR6-2 gene, was found to be involved in anthocyanin synthesis, the functional S gene, StDMR6-1, was identified by agrobacterium-mediated CRISPR-Cas-based editing with subsequent scoring of pest resistance.18,28
Genome editing is often used for functional validation experiments of gene candidates obtained from omics (e.g., transcriptomics, proteomics, and metabolomics) studies, hitherto with omics studies in potato carried out under controlled growth conditions.29–34 Now, omics data from potato field studies are emerging which may prove useful and potentially provide even more robust agronomic value.32,33
The seemingly nonstraight forward task of correct assignment of orthologues and the limited number of available well-annotated potato genomes favor targeting genes in smaller gene families, and prompt for allele-specific sequencing of all alleles of the target gene in the relevant genotype background before embarking on gene editing (Fig. 2). 35 Allele-specific sequencing may facilitate fast validation of the CRISPR generated mutations and provide means of generating CRISPR designs that reduce off-target mutations.
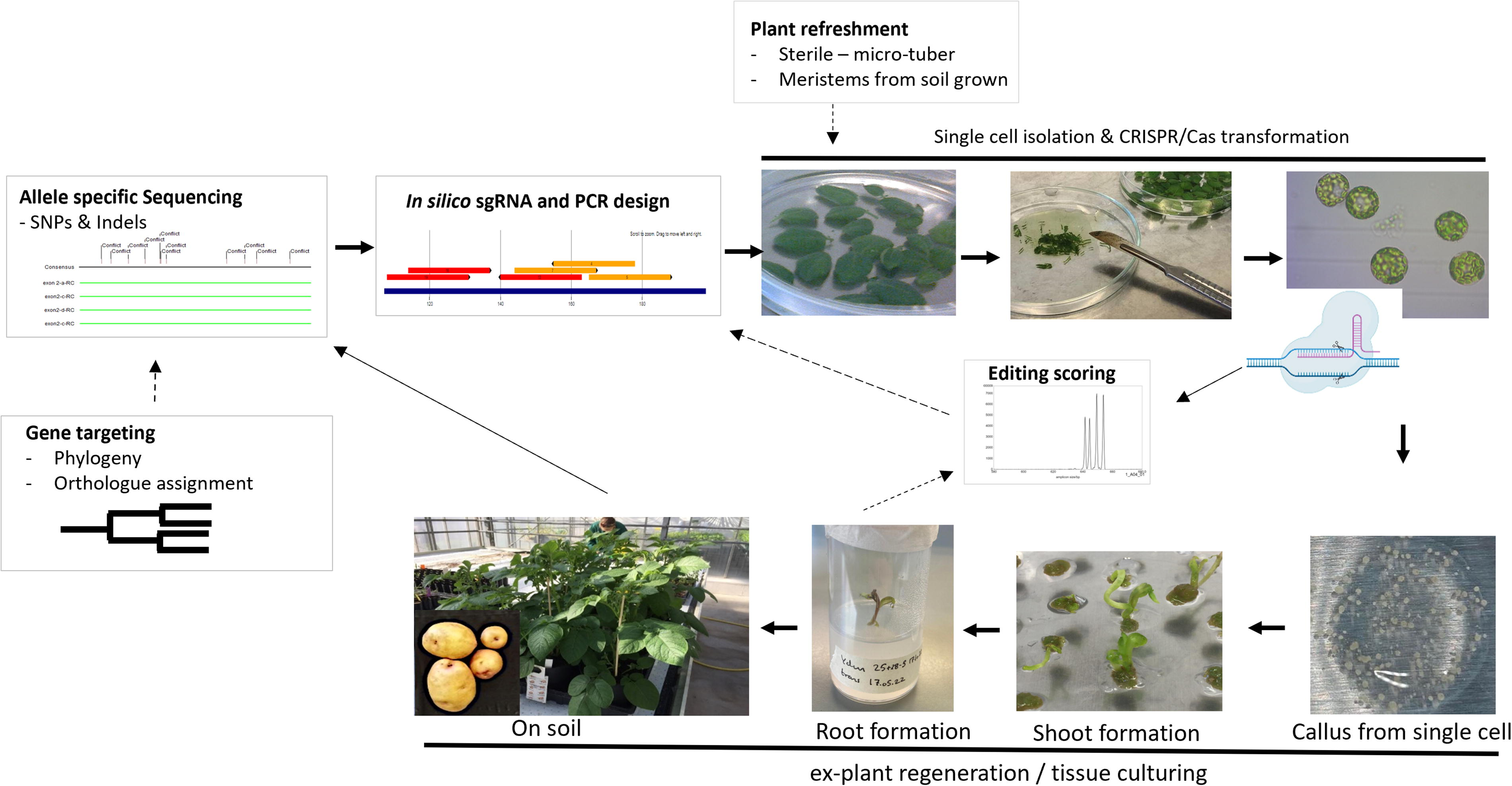
Single cell CRISPR-Cas gene editing and plant regeneration in tetraploid potato-workflow. The workflow involves (i) full allelic characterization of the target region; (ii) sgRNA and diagnostic PCR editing scoring designs; (iii) isolation of protoplasts from leaves and polyethylene glycol (PEG) mediated transformation, (iv) optimization of editing efficiencies via robust high-throughput editing screen for scoring high efficiency sgRNA(s), (v) isolation of and explant regeneration from single edited protoplast cells. Each step (in silico and in vitro) contributes to successful and efficient gene editing. Once step (i) and to (ii) have been carried out, optimization of editing in the target gene may start. Here obtaining high editing efficacies through scoring a range of sgRNAs, at the protoplast pool level will, in particular in multiplex settings, hugely impact the chance of obtaining the desired edited explant and limit the downstream cell culturing workload. Optional inputs to this workflow include the often non-trivial task of correct assignment of orthologous target gene(s), described in the ‘gene targeting’ section and refreshment of start out material, both of which are of importance for successful and efficient gene editing. Optional inputs and potential iterations are indicated by dotted arrows. Thick arrows indicate the main workflow, whereas thinner arrows indicate additional workflows. The figure is partly modified from Johansen et al. 24 and image adapted from Biorender.com.
Genome editing in potato—considerations and optimizations
Implementation of gene editing in tetraploid potato may be subdivided into three main steps: (i) full allelic characterization of the target gene and in silico assisted sgRNA selection, (ii) editing of the protoplast pool, and (iii) explant regeneration from single edited protoplasts, via callus formation and the use of shoot and root inducing sets of hormones (summarized in Fig. 2).
We have optimized and included add-ons to established protoplast transformation (ii) and explant regeneration (iii) protocols, which are detailed at the end of this article. The following focuses on the identification and full allelic characterization of target genes in the complex potato genome, in silico assisted sgRNA selection, establishment and use of efficient editing screens, and considerations regarding editing optimizations and chimerism in explants.
Full allelic characterization of target gene and in silico assisted sgRNA selection
Tetraploid elite germplasms are extremely genetically diverse with a reported small nucleotide polymorphism (SNP) prevalence between two cultivars of 1 per 29 bp, 36 as corroborated in recent in-depth characterizations of the Granular Bound Starch Synthase (StGBSS) 1, Glucan Water Dikinase (StGWD) 1, and downy mildew resistant 6 (StDMR6-1) genes in three elite cultivars Ydun, Desiree, and Wotan, which revealed a 2- to 3-fold higher SNP prevalence when compared to the heterozygous diploid RH reference genome sequence24,25 (see also Fig. 3A). This complicates sequence guide RNA (sgRNA) and editing diagnostic PCR designs, where full allelic nucleotide identity normally is required for both facilitating editing of all alleles and for assessing editing in all alleles via PCR amplification of the target region(s).
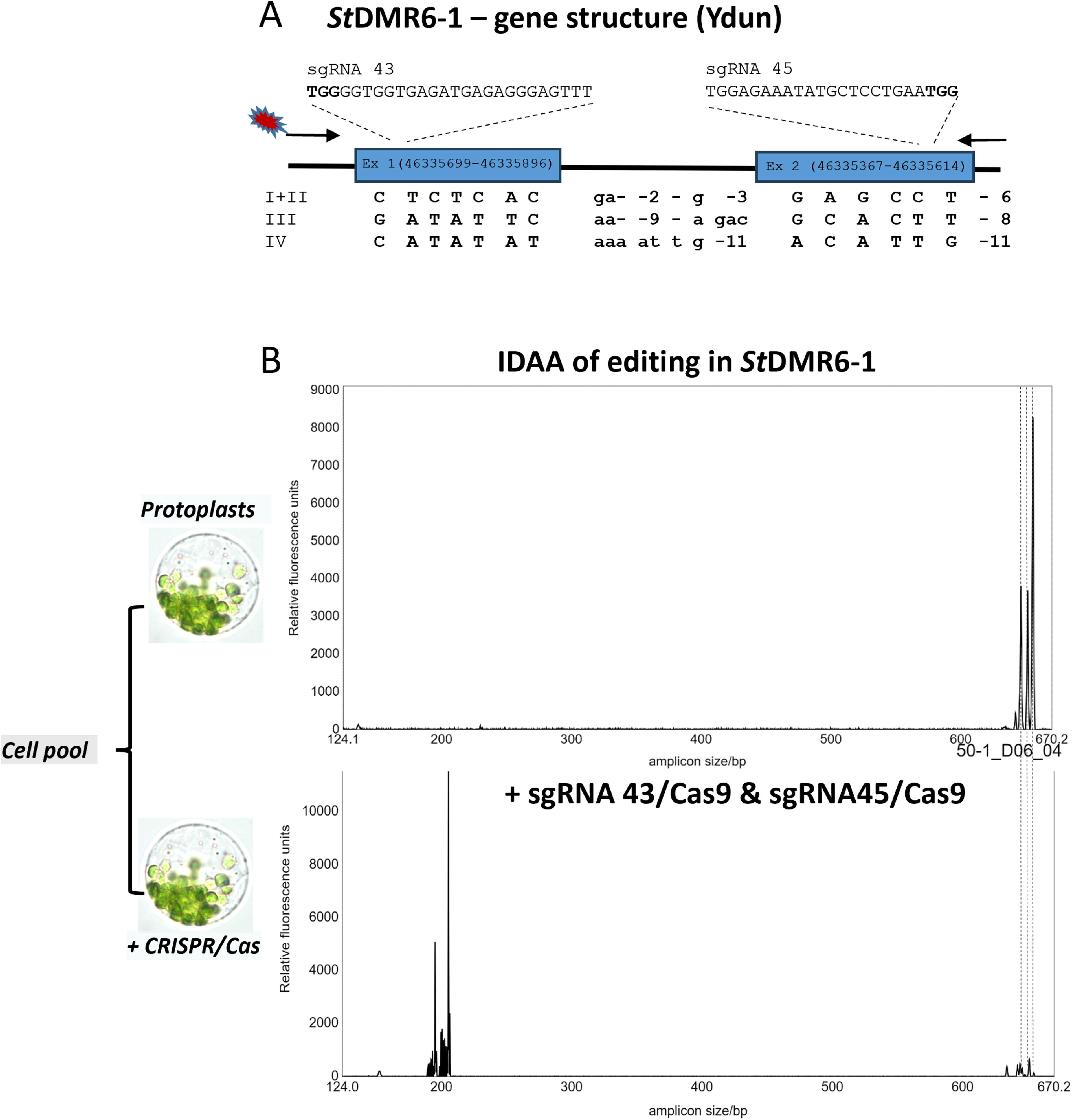
Editing and editing scoring in complex genomes. Indel Detection Amplicon Analysis (IDAA) is a fast, reliable and high throughput method for scoring editing in complex genomes. Naturally occurring length (Indel) polymorphisms in the target region may be incorporated in IDAA PCR amplification designs to ensure amplification of all alleles and thus enable scoring of allele specific editing at both the cell pool and the callus/explant level. IDAA PCR amplification of target regions which include naturally occurring Indels frequently provide a reliable fingerprint of amplification of all of the four alleles as indicated by number of peaks and height of individual peaks, that is, 1:1:1:1 (4 Indel sizes, similar peak height), 1:1:2 (3 Indel sizes, with two alleles having the same Indel size yielding double intensity/peak height), 1:3 (2 Indel sizes, one of triple intensity/peak height), or 1:1 (2 Indel sizes of double intensity/peak height, each from two alleles). The ability to assess all alleles allows for an early on focus on efficient sgRNAs with capacities for editing all alleles and on identifying calli/explants with editing in all alleles.
Obtaining reliable allele-specific sequence information of all four alleles in target genes is therefore normally a prerequisite for obtaining efficient full allelic or, if desired allele-specific, editing. Regular PCR amplification of target regions often results in generation of chimeric fragments between the four alleles, where recombinant PCR species in some cases may represent up to 12%. 24 Next-generation sequencing technologies offer some viable options in this respect. 38 Alternatively, targeted allele-specific sequence information may also be obtained through high-precision Illumina sequencing 39 and whole genome sequence information may be obtained by long-read sequencing technologies such as PacBio® amplicon and Nanopore® sequencing, 40 which can greatly aid mutant characterizations. Presently, Nanopore sequencing has a somewhat high random base calling error background (ca. 2%), which must be accounted for when assigning SNPs and length polymorphisms to the individual four alleles.
sgRNA and diagnostic editing scoring designs
Prediction servers for scoring sgRNA efficiency and off-target frequencies have been reviewed in studies by Chuai 41 and Alipanahi et al. 42 Recently, in vivo editing efficiencies of sgRNAs targeting the StGWD1 and StDMR6-1 genes, were compared to in silico predictions, using the servers CHOPCHOP (http://chopchop.cbu.uib.no/), CRISPRater (https://crispr.cos.uniheidelberg.de/), and SSC (http://crispr.dfci.harvard.edu/SSC/). Clear-cut correlations were, however, not found, 25 supporting the notion that in silico prediction at least for now is an initial supporting tool.
Protoplast transformation and explant regeneration optimizations
Current protocols for isolation of protoplast from leaves and PEG-mediated transformation of CRISPR-Cas vectors and RNPs can be found in14,15 with additional considerations and optimizations when using transient plasmid DNA or RNPs described in.24,25,43,44
Establishing and use of efficient editing screens
A number of editing scoring techniques have been devised and reviewed (e.g., summarized in studies by Bennett et al. 23 and Aoki et al. 45 ). One seemingly robust high-throughput editing scoring technique, the Indel Detection Amplicon Analysis (IDAA), is able to separate fluorescence-labeled PCR fragments down to ±1 bp in length 37 by means of capillary electrophoresis, and has proven highly valuable for editing scoring in organisms with high ploidy and complex genomes, such as potato. 23 Potato is rich in naturally occurring interallelic small insertions and deletions (Indels) in especially noncoding regions. This may be exploited and included in diagnostic IDAA PCR primer designs to ascertain that all four alleles are PCR amplified as demonstrated in previous work23–25 and shown in Figure 3. Reliable editing scoring of all four alleles enables fast assessment of sgRNA efficiency in the cell pool and identification of allele-specific (incl. full allelic) editing at the early callus/explant stage (Figs. 2 and 4).
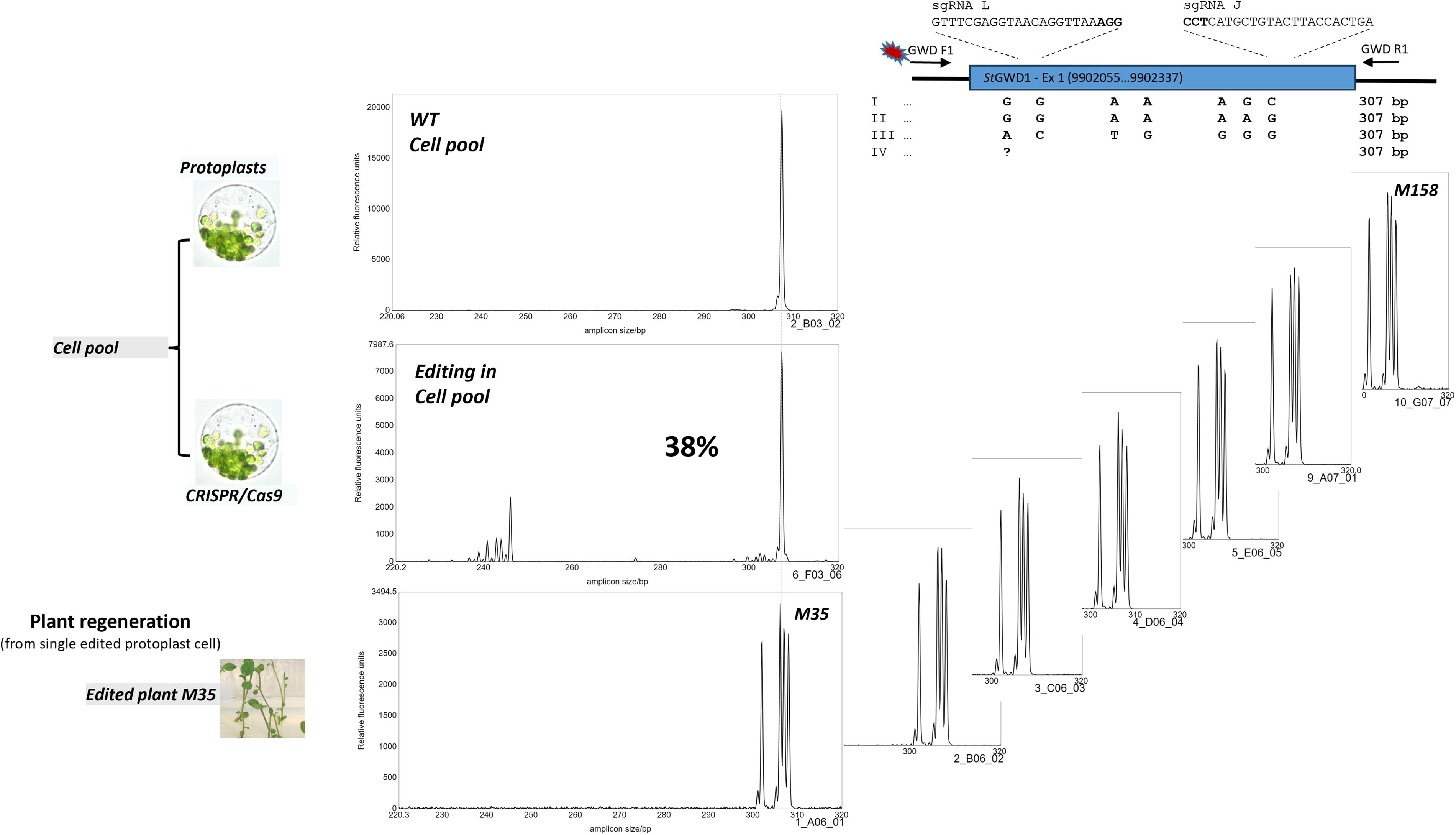
Protoplast density at alginate embedment: a balance between callus and shoot formation and non-chimerism. A high chloroplast cell density in the alginate lenses (embedment/casting) appears to favor callus- and later shoot formation, the so-called helper cell syndrome. A fine balance between protoplast density facilitating callus formation and growth and clonal integrity is desirable, as shown by IDAA of seven shoots from a single callus clone which was regenerated to explants, showing no chimerism. Target gene was the glucan water dikinase (GWD) 1 StGWD1, Soltu.DM.05G009520.2 in the Phureja DM1-3 v.61 reference genome, in cultivar Ydun, where exon 1, with the four alleles, SNPs and the PCR amplicon size indicated, is depicted in the upper right part of the figure. sgRNAs were sgRNA L (GTTTCGAGGTAACAGGTTAA
Gene editing optimizations
High editing efficiencies at the protoplast cell pool level are pivotal for achieving full allelic knock-out, reducing the downstream cumbersome and delicate explant regeneration, and enabling multiplexing (multiple targeting’s) in the same transformation event. This involves (i) in case of DNA-based expression of the CRISPR-Cas components: construct optimizations; (ii) considerations regarding multiple or single sgRNA/Cas targeting of a gene, and importantly (iii) design and test of several sgRNAs, either individually or in combination.
Previously, a 3- to 4-fold increase in editing efficiency at the protoplast level, typically with editing in >50% of alleles in the cell pool, was obtained by replacing the standard A. thaliana AtU6-1 promoter with endogenous StU6 promoters driving expression of sgRNAs here targeting the StGBSS 1 gene, and eventually resulting in 35% of explants having mutations in all four alleles. 24 Similar or even higher editing efficiencies (see Fig. 3) have been obtained when using DNA-free RNPs.25,43,44 We recommend the use of equimolar sgRNA and Cas9 components per RNP in pre-incubations in order to reduce the theoretical risk of RNP recomplexing in multiplex settings, and that the individual preincubated RNPs are mixed just prior to the transformation, accordingly 25 (see also the “Optimized Protocol for Genome Editing in Potato” section).
Considerations regarding gene/chromatin structure and sgRNA efficiency
Recently, we mapped the editing efficiency of RNPs targeting either the start (5′) or the latter third (3′) of the large starch biosynthetic StGWD 1 gene, which is encoded by 33 exons distributed over a 15.414 bp region on Chromosome 4 and the smaller S gene, StDMR6-1, which encoded by 4 exons distributed over a 6.398 bp region on Chromosome 5. In case of the GWD1, targeting the start compared to the end conferred higher editing, and the use of two RNPs (multiplexing) targeted at the end conferred a synergistic effect. Compared to the single RNP/gene targeting, no synergistic effects were found when simultaneously targeting both genes, residing on different chromosomes. 25 Again, >50% editing of all alleles at the cell pool level was obtained. Underlying mechanisms for these observations remain speculative and require larger and additional experimental set-ups. 25
Isolation of single edited protoplast cells—a balance between cell density and chimerism
Isolation and clonal propagation of individual single-edited protoplasts are delicate steps in the protoplast to explant regeneration. A high protoplast cell density in alginate lens embedment generally supports callus growth and later explant regeneration (reviewed in previous work 46 ). A cell density of ca. 5 × 10−4 protoplasts/mL in the alginate lens enables both efficient callus formation while still allowing for clonal isolation and explant development of seven shoots from a single callus clone which was separately developed into explants (see Fig. 4). Compared to Nicolia et al.,14,15 we have doubled the total volume to 4 mL in order to enable performing both IDAA editing analysis and protoplast alginate embedment in parallel.
Cultivar-dependent adjustments and starting material
Protoplast yield, transformation efficiency, and explant regeneration capacity are dependent on the quality of the plant material, requiring frequent generation of new plant material as outlined in. 13 Refreshment can be done ex vitro using material, for example, internodal stem sections taken from soil-grown plants or via mini tubers which significantly improve plant vigor typically yielding 4- to 10-fold increased transformation efficiencies. 47 Also, different cultivars may require adjustment of hormone composition and concentration in certain steps as evidenced by the special requirements for cultivars B101 (diploid) and table potato King Edward in comparison to the model cultivar Desiree13,47 and recently reported for the diploid tuber-bearing wild species Solanum bulbocastanum. 48
Somaclonal variation
Breeding strategies often involve targeting several genes and are, when comprising cell and tissue culturing, accompanied by generation of undesired random chromosomal mutations and rearrangements known as somaclonal variation during the regeneration process which may penetrate into phenotypic malformations at the explant level. Somaclonal variation derived from callus and shoot formation is a general concern, also in potato. 49 While much attention has been put on CRISPR-Cas-mediated off-target events, somaclonal variations from explant regeneration, where hormones are used to develop the whole explant from a single protoplast cell, have been shown to be significantly more prevalent than mutations arising from CRISPR off-target events.50,51 It is therefore desirable to optimize editing efficiencies to facilitate introduction of multi-gene targeting/multiplexing at the cell pool level in the same transformation event in order to avoid repeated rounds of tissue culturing. This also raises the question of how to distinguish somaclonal mutations from CRISPR-Cas9-derived off-target mutations in high-ploidy crops with complex genomes.
Streamlining protoplast transformation and explant regeneration and add-on’s
Basic protocols for PEG transformation of leaf-derived protoplasts with DNA constructs or RNPs, and regeneration of shoots from single protoplasts in potato have been established14,15 and recently summarized in. 13 However, streamlining and reduction of steps and time spans are important for reducing somaclonal variation52,53 and reducing contamination risks. We have further streamlined these protocols for PEG-mediated potato protoplast cell transformation and explant regeneration and identified light composition as a crucial factor in key steps of potato explant regeneration.
Protoplast isolation, transformation, and explant regeneration—key points
PEG-mediated potato protoplast transformation and their embedment into alginate were streamlined and reduced from originally 3 days14,15 to 2 days by omitting the ON incubation of the leaves in medium B and by—omitting the final wash step (Wash solution) before embedment (see below).
Explant regeneration was streamlined by omitting the medium G plate step, and root formation appeared feasible in hormone-free medium A instead of root induction medium I. Together, this shortens the entire regeneration time and reduces the time and number of steps the explant is exposed to hormones.
In our hands, the regeneration steps post-release and placement of calli on medium H plates may be cultivar, gene, and experiment (protoplast source and vigor) dependent and require close inspection of media renewal and hormone composition changes (callus, shoot, and root induction). These adjustments were tested for the cultivars Desiree and the in our hands more recalcitrant cultivar Ydun. The entire protocol, which is optimized from and based on the protocol by Nicolia et al. 14 is listed below.
Light conditions during explant regeneration
In vitro plant regeneration at the cellular, molecular, and physiology levels is reviewed in study by Bidabadi et al. 54 Plant cell culturing and explant regeneration in particular are generally tricky and delicate, often resulting in the accumulation of polyphenols at the callus forming step, termed oxidative browning, which may be attributed to light compositions/conditions and/or media composition with suggestive countermeasures outlined in previous work.55–57
Light conditions/composition after napkin removal have, in our hands, proven crucial for successful regeneration and are regularly not addressed in the literature including key regeneration protocols such as.14–16,58,59 Browning of callus was, in our hands, alleviated and shoot formation facilitated by applying a more red end light composition (e.g., Photosynthetic Photon Flux Density [PPFD]: 125.6 µmol m–2 × s–1 [Blue (400–500 nm); Green (500–600 nm); Red (600–700 nm); 20:34:46%], see also below). Light composition with a higher abundance of the blue end of the spectrum has, in our hands, for all cultivars tested conferred browning, often resulting in cell death of callus and blockage of shoot induction on medium H plates, underscoring the importance of light composition in the crucial callus and shoot induction steps.
Conclusions
Number and duration of potato protoplast to explant regeneration steps have been reduced, thus limiting exposure to hormones often linked to somaclonal variation.
Number and duration of steps in protoplast transformation have been reduced.
Considerations and a general scheme for identification of orthologous target genes in potato are presented.
Optimizations of loss-of-function gene editing strategies in potato, having a high ploidy and complex genome, are reviewed and summarized.
Optimized Protocol for Genome Editing in Potato
This protocol is optimized from and based on the protocol by Nicolia et al. 14 which was updated in 2021. 15 Optimizations and differences with regard to protoplast isolation, transformation and embedment, and explant regeneration, that is, omission of medium G and change to hormone-free medium A instead of root induction medium I, with additional tips not listed in Nicolia et al. 15 and including additional optimizations, are provided and detailed in the footnotes. Medium recipes are provided in the end. In cases of recalcitrance in steps post protoplast embedment, the earlier established protocols Nicolia et al.14,15 should be revisited.
Protoplast isolation, transformation, and embedment
Day 1—Cutting leaves and enzymatic digestion of cell walls A
Medium C B preparation
25 mL of medium C is prepared with vitamins and hormones, heated to 55°C for 10 min, and left too cool to room temperature, where after CaCl2 is added, pH is adjusted to 5.6 by KOH/HCl and the solution is filter sterilized.
Leaf slicing and enzymatic degradation of cell walls
During the cooldown of medium C, ca. 20–30 leaves (ca. 1 g) from 4–6 weeks old sterile in vitro plants are excised and placed in a glass petri dish containing 5–10 mL of medium B. Leaves are then cut in thin slices (1–2 mm wide) using a scalpel (e.g., BB521 scalpel [Aesculap®]) with a curved blade and collected in a plastic petri dish containing 10–20 mL medium B. When all leaves are sliced, medium B is removed using a sterile plastic Pasteur pipette, substituted with 25 mL plasmolysis solution, and incubated 15–30 min covered with tinfoil at RT. Plasmolysis solution is removed, and the room-tempered medium C is added, and the petri dish is sealed with parafilm, covered in tinfoil, and incubated overnight (ON) at 25°C with gentle shaking (60 rpm).
Prepare for day 2
RNP complex, for transfection of usually a 400 µL protoplast suspension, is assembled by mixing 1 µL 5 mg/mL TrueCut Cas9 v2 (Invitrogen A36499) and 0.375 µL 100 pmol/µL Trueguide sgRNA (Invitrogen) (1:1 molar ratio) in a PCR/Eppendorf tube and stored ON at 4°C. Place the sgRNA carefully into the small Cas9 droplet. For transfection of several RNPs, see footnote. C In case of plasmid-derived sgRNA/Cas expression, 10 µg sterile D pDNA, typically in concentrations of 1000 ng/µl, is used for transfections.
Day 2—Protoplast isolation, transformation, and initiation of plant regeneration
Isolation of protoplasts
Wash solution is heated in the microwave for 10–40 s to achieve room temperature. A 100 µm sterile filter mounted on a 100 mL beaker is prewetted with 5 mL Wash solution and leaf slices in medium C from Day 1 are filtered through and the petri dish is washed with 5 mL Wash solution and poured through the filter to obtain the isolated protoplasts including protoplasts sticking to petri dish 5 mL Wash solution is added to the filter to flush remaining protoplasts into the beaker. The solution is poured into a 50 mL Falcon tube and the beaker is washed with 5 mL Wash solution and added to the Falcon tube.
The Falcon tube is centrifuged at 50 × g with minimum acceleration and de-acceleration (this is crucial to ensure high protoplast viability) for 10 min, the supernatant is removed using a 10 mL Pasteur pipette. The green protoplast containing pellet is resuspended in 6 mL Wash solution. Two 15 mL Falcon tubes containing 6 mL Sucrose solution are briefly centrifuged to secure no droplets on the side of the tube. Using a sterile plastic pasture pipette the protoplast solution is very gently layered on top of the sucrose, 3 mL in each tube. The sucrose/protoplast tubes are centrifuged at 50 × g with minimum acceleration and de-acceleration for 15 min (while the protoplasts are centrifuging the polyethylene glycol [PEG] solution is prepared and filter sterilized E ).
After centrifugation of the sucrose/protoplast Falcon tubes, a thick green band will form in the middle of the gradient. The tube is gently tilted and this band is gently removed using a 1 mL pipette with the pipette tip cut in a corresponding angle.
The protoplasts obtained from the green band are added to a culture tube containing 3 mL sterile Transformation buffer 1. While estimating the cell density, the remaining protoplast solution is stored at 4°C.
Estimation of protoplast yield and adjusting cell density
Protoplast yield and density are estimated using a hemocytometer F . The protoplasts in Transformation buffer 1 are centrifuged for 5 min at 50 × g with minimum acceleration and de-acceleration and the supernatant is removed. The pellet is resuspended in Transformation buffer 2 to a density of ca. 1.6 × 106 protoplasts/mL.
PEG-mediated transformation
In total, 400 µL of the protoplast suspension at a density of ca. 1.6 × 106 protoplasts/mL is added to each pre-prepared culture tube containing RNP and gently mixed by pipetting. Of all, 400 µL of 25% PEG solution is added to each transformation tube, G gently mixed by pipetting, and left at RT for 3–5 min. Transfection is stopped by adding 5 mL of Wash solution and tubes are centrifuged 5 min at 50 × g with minimum acceleration and de-acceleration. Supernatant is removed and the pellet is resuspended in 1 mL medium E.
Editing analysis
0.5 mL of the protoplast suspension is transferred to new culture tubes containing 0.5 mL 0.4M Sorbitol solution and incubated at 25°C in the dark for 1–3 days. These samples are used for editing analysis, for example by IDAA.
Protoplast embedment in alginate lenses
To the remaining 0.5 mL protoplast solution 1.5 mL additional medium E and 2 mL Alginate solution are added yielding a total volume of 4 mL and a cell density of ca. 5 × 104 protoplasts/mL. The suspension is mixed by pipetting and 500 µL droplets, 4 on each plate, are placed on fridge cold (4°C) Setting agar. After approx. 1–1.5 h the formed lenses I are gently transferred with sterile spatulas to new petri dishes containing 10 mL medium E. Lenses can be lifted from the Setting agar by prewetting a spatula with liquid medium E and sliding it under the lenses. 9 Petri dishes are sealed with parafilm and wrapped in tinfoil and kept in darkness at 25°C for 5 days.
Explant regeneration
Light intensity, composition and regime and temperature from transfer of single calli to solid medium H plates and until transfer to soil were: PPFD: 125.6 µmol m−2 × s−1; (Blue (400–500 nm); Green (500–600 nm); Red (600–700 nm) 20:34:46%) (measured from the bottom of the shelf); 16 h/8 h (light/dark), 24°C/22°C.
In lenses calli formation and release
After 5 days in darkness, the tinfoil is removed, and the plates are placed under three layers of tissue paper (or napkins), one of which is removed each week the following 3 weeks (PPFD 4.26 (3 napkins), 6.5 (2 napkins) and 9.8 (1 napkins) µmol m−2 × s−1, respectively). After 3 weeks (start of week 4) small microcalli emerge (just visible to the naked eye) and medium E is changed to medium F and calli are subjected to continuous light (PPFD: 18.832 µmol m−2 × s−1 (no napkins); (R:G:B; 16:39:45) ([%], 24 h light/0 h dark; 24°C) and left for 1–3 weeks. J
Calli are released by removing liquid medium F and adding 8 mL of Releasing solution and incubating for up to 10 min. Released calli are washed in medium F with no hormones added. Calli, ≥2 mm K in diameter are transferred directly to shoot-promoting medium H plates. L Calli <2 mm can be cultivated for 1–2 weeks longer in medium F until they reach the ≥2 mm stage and can be transferred to solid medium H plates. M
Shoot induction on medium H solid plates
Plates are moved to a 16 h light/8 h dark, 24°C/22°C regime (PPFD: 125.6 µmol m−2 × s−1; [Blue (400–500 nm); Green (500–600 nm); Red (600–700 nm); 20:34:46%]) (measured from the bottom of the shelf), where calli are moved to fresh medium H plates every 10–14 day. After ca. 3 months shoots emerge from cali and when ≥1 cm, shoots are cut at the base and transferred to solid medium A boxes, where roots are formed typically within 2–6 weeks.
Root formation and transfer to soil
After rooting on medium A, plants are transferred to soil in 2 L pots and placed in greenhouse typically under a 16/8 h light/dark regime (typically, light: 06:00–22:00, light minimum of 250 µE, 15–18°C (light); 15°C (dark: 22:00 pm–06:00 am); Ventilation by +3c = 21c).
Media and hormone recipes
All media, materials, and tools listed in this procedure must be sterilized. All media except PEG and medium C may be kept for at least 3 months in the fridge as stock solutions without vitamins and hormones. Solutions with hormones and vitamins added can be kept for 1 week at 4°C before use, allowing for media preparation for multiple rounds of protoplast isolation to be prepared on the same day and used within a week.
*Medium A, B, C, E, F, H, Plasmolysis solution, Sorbitol solution, Alginate solution, Wash solution, Transformation buffer 1, Transformation buffer 2, and Setting agar are prepared according to Nicolia et al. 14 and references herein.
Medium A: 4.4 g/L Murashige and Skoog including vitamins (Duchefa M0222) 43.8217 mM sucrose, 8 g/L agar (Sigma-Aldrich A7921), pH. 5.6.
Medium B: 2.7 g/L Murashige and Skoog modified no. 4 (Duchefa M0238) 0.1 mL/L vitamin NN, 185.3225 μM casein hydrolysate (CAS 65072-00-6), 10.740
Medium C: 10 mL/L macrostock, 3 mL/of 6 mM CaCl2 stock(CAS 10043-52-4), 10 mL/L iron stock, 1 mL/L microstock, 5 mL/L vitamin stock 1, 2, and 3, 20/L mL organic acid stock, 926.6123
Medium E: 10 mL/L macro stock, 2.5 mM CaCl2 (CAS 10043-52-4) 10 mL/L of iron stock, 1 mL/L micro stock, 5 mL/L vitamin stock 1,2 and 3, 20 mL/L sugar stock, 10 mL/L organic acid stock, 926.61
Medium F: 2.7 g/L Murashige and Skoog modified no. 4 (Duchefa M0238), 2 mMNH4Cl (CAS 12125-02-9), 1 mL/L NN vitamin stock, 217.19
Medium H: 4.4 g/L Murashige and Skoog incl vitamins (duchefa M0222), 29.2 mM sucrose (CAS 57-50-1), 53.71 nM NAA (CAS86-87-3), 9.12
Plasmolysis solution: 91.1 g/L D-sorbitol (CAS 50-70-4).
Wash solution: 10 mL/L Macro stock, 3 mL 6 mM CaCl2 stock (CAS 10043-52-4), 10 mL/L iron stock, 1 mL/L microstock, 14.03 g/L NaCl (CAS 7647-14-5) 10.74
Sucrose solution: 0.43M sucrose (CAS 57-50-1).
PEG solution: 2.5 g/10 mL polyethylene glycol (PEG) (CAS 25322-68-3), 5 mL/10 mL of 0.8M mannitol stock (CAS 69-65-8), 500
Transformation buffer 1: 190 mM mannitol (CAS 69-65-8), 100 mM CaCl2*2H2O (CAS 10035-04-8) 0.5% w/v 2-(N-Morpholino)ethanesulfonic acid (MES) (CAS 145224-94-8), pH 5.6.
Transformation buffer 2: 499.53 mM mannitol (CAS 69-65-8), 14.95 mM MgCl2 *6H2O (CAS 7791-18-6), 0.1% w/v MES (CAS 145224-94-8), pH 5.6.
Alginate solution: 2.8% w/v alginate acid-Na salt (CAS 9005-38-3), 0.4M D-sorbitol (CAS 50-70-4).
Setting agar: 0.4M D-sorbitol (CAS 50-70-4), 50 mM CaCl2*2H2O (CAS 10035-04-8).
Releasing solution: 20 mM Na-citrate (CAS 6132-04-3), 0.5M D-sorbitol (CAS 50-70-4).
Macro stock: 0.73M KNO3 (CAS 7757-79-1), 0.24M MgSO4 *7H2O (CAS 10034-99-8), 24.98 mM KH2PO4 (CAS 7778-77-0).
Micro stock: 11.02 mM H3BO3 (CAS 10043-35-3), 33.11 mM MnSO4 *H2O (CAS 10034-96-5) 3.48 mM ZnSO4 *7H20 (CAS 7446-20-0), 582.75
Iron stock: 3.76 mM Na2EDTA (CAS 6381-92-6), 6.83 mM FeSO4*7H2O (CAS 7782-63-0).
Vitamin NN: 26.64 mM glycine (CAS 56-40-6), 0.555M myo-inositol (CAS 87-89-8), 1.48 mM thiamine-HCl (CAS 67-03-8), 2.43 mM pyridoxine-HCl (CAS 58-56-0), 40.61 mM nicotinic acid (CAS 59-67-6), 1.13 mM folic acid (CAS 59-30-3), 204.658
Vitamin stock 1: 9.12 mM pantothenic acid (CAS 79-83-4), 3.58 mM choline chloride (CAS 67-48-1) 5.68 mM ascorbic acid (CAS 50-81-7) 4.06 mM nicotinic acid (CAS 59-67-6), 72.92 mM p-aminobenzoic acid (CAS 150-13-0), 2.43 mM pyridoxine-HCl (CAS 58-56-0), 14.83 mM thiamine-HCl (CAS 67-03-8).
Vitamin stock 2: 453.104
Vitamin stock 3: 12.999
Sugar stock: 34.31 mM D-sorbitol (CAS 50-70-4), 18.26 mM sucrose (CAS 57-50-1) 34.69 mM D(-)fructose (CAS 57-48-7), D(-) 41.63 mM ribose (CAS 50-69-1), 41.63 mM D(+) xylose (CAS 58-86-6) 34.69 mM D(+) mannose (CAS 3458-28-4), 34.309 mM L(+) rhamnose monohydrate (CAS 10030-85-0), 18.26 mM D(+)cellobiose (CAS 528-50-7), 13.88 mM myo-inositol (CAS 87-89-8).
Organic acid stock: 11.36 mM pyruvic acid (CAS 127-17-3), 17.23 mM fumaric acid (CAS 110-17-8), 9.52 mM citric acid monohydrate (CAS 5949-29-1), 14.92 mM L-malic acid (CAS 97-67-6).
We found that incubation of leaves in medium B ON, prior to slicing and incubation with cell wall degrading enzymes as described in Nicolia et al.15 is not necessary and may be omitted.
Medium C including hormones should be fresh, prepared on day of use.
If, for example, two sgRNA/Cas9 complexes are combined in one transformation event, each Cas9/gRNA are mixed and preincubated separately to ensure complete (1:1 molar ratio) complex formation and avoidance of potential skewed complex formation between RNPs. RNP (assembled sgRNA/Cas9 complex [1.375 µL]) is then transferred to a 10 mL culture tube. In case of co-transfection of several RNPs, the RNPs should be mixed in the 10 mL culture tube just prior to transfection. We have successfully used the same molar amount as used in single RNP transfections, in cotransfections of two RNPs, that is, 2.75 µL in total.
Eluted in sterile nuclease-free ddH2O.
Polyethylene glycol (PEG) solution should be fresh and prepared on the day of use.
Yields may be cultivar dependent and are diluted in accordance to the hemocytometer in question.
Transformation tube is typically a 10 mL culture tube.
Protoplasts are fixed in the lenses which enables clonal development of eventually an explant from a single protoplast.
We found that no floating solution is needed.
We found 1–3 weeks of incubation on medium F, instead of the original up to 6 weeks, supports calli growth, big enough to be released. Also, in our hands, smaller calli size and younger age seem to improve shoot formation capacities in comparison to transfer to medium F after 6 weeks of callus growth.
In our hands, smaller calli sizes, not larger than 2 mm in diameter, spending a shorter time in Medium F, appear to support shoot formation (number of shoots) and regeneration.
Although included in Nicolia et al.,15 we found that Medium G is not necessary for regeneration for at least the cultivars Desiree, Saturna, and Ydun, and this step may thus be omitted, rendering a faster and overall more compact regeneration with less exposure to hormones.
We observed no difference in regeneration potential between early and later calli transfer to Medium H.
Footnotes
Acknowledgments
Professor Eva Rosenqvist, Department of Plant and Environmental Sciences, Crop Science, University of Copenhagen is acknowledged for helping with light measurements. Andreas Blennow is acknowledged for aiding in providing funding for and establishing gene editing of potato at UCPH.
Authors’ Contributions
F.M.C.: Conceptualization, methodology, validation investigation, visualization, and writing—review and editing. I.W.: Conceptualization, methodology, validation investigation, visualization, and writing—review and editing. I.E.J.: Conceptualization and methodology. E.A.: Conceptualization methodology, writing—review and editing, and funding acquisition. B.L.P.: Conceptualization, methodology, visualization, writing—original draft, and funding acquisition.
Author Disclosure Statement
The author(s) disclose no conflict of interests