
Abstract
In mice, naturally occurring and induced mutations in the suppressor of cytokine signaling-2 (Socs2) gene are associated with a high growth phenotype characterized by rapid post-weaning weight gain and 30–50% heavier mature body weight. In this work, we demonstrate an electroporation-based method of producing SOCS2 knock-out (KO) sheep. Electroporation of dual-guide CRISPR-Cas9 ribonucleoprotein complexes targeting SOCS2 was performed 6 h post-fertilization in sheep zygotes. Fifty-two blastocysts were transferred to 13 estrus-synchronized recipients, yielding five live lambs and one stillborn. These lambs all carried mutations predicted to result in SOCS2 KO. Three carried large deletion alleles which evaded detection in initial PCR screening. Off-target analysis using whole genome sequencing comparing the frequency of mutations in regions within 100 bp of possible sgRNA binding sites (up to 4 bp mismatches) and elsewhere in the genome showed no significant difference when comparing unedited control sheep to edited animals (p = 0.71). In conclusion, electroporation of zygotes with dual-guide CRISPR-Cas9 RNPs was effective at generating knock-out sheep with no substantial off-target activity.
Introduction
Conventionally, genome editing reagents are introduced to livestock oocytes and embryos via microinjection; however, this approach requires expensive equipment and skilled personnel and frequently results in mosaicism1–4 which can decrease the utility of producing genome-edited livestock by single-step approaches. Electroporation offers a rapid and efficient way to introduce editing reagents into mammalian zygotes. This is a commonly used method that can transfer different substances into a variety of cell types.5–10 The process of electroporation introduces short-term changes in plasma membrane permeability that allow for the transfer of CRISPR-Cas9 ribonucleoprotein (RNP) to the zygotic cytoplasm. Several reports have documented the production of gene-edited rats, 11 mice, 12 pigs, 13 and cattle 14 by electroporation of zygotes in a single-step approach, as reviewed by Lin and Van Eenennaam. 15
Suppressors of cytokine signaling (SOCS) are a family of eight proteins regulating cytokine intracellular signaling. One essential intracellular mediator of cytokine signaling is the Janus kinase and signal transducer and activator of transcription (JAK-STAT) pathway. 16 The SOCS2 gene encodes a 198 amino acid protein with two functional domains: the central SH2 and the highly conserved C-terminal SOCS box. When growth hormone (GH) binds to its cell surface receptor, the JAK-STAT pathway transduces the signal throughout the cell and activates transcription of growth-related genes and SOCS2. SOCS2 proteins exert negative feedback on the GH signaling pathway by binding to the growth hormone receptor (GHR) via the SH2 domain and blocking further phosphorylation and activation of STAT5 proteins. Additionally, SOCS2 functions as a substrate recognition subunit in the SOCS2 E3 ubiquitin ligase complex that tags the GHR for degradation.17,18 In Socs2 knock-out (KO) mice, an extended duration of STAT5 phosphorylation is observed in response to GH stimulation along with longer bones, 30–50% heavier mature body weight, and enlarged internal organs.19–21
Many of the effects of Socs2 KO in mice are also observed in sheep (Ovis aries). In a study investigating genetic susceptibility to mastitis in dairy ewes, a naturally occurring nonsynonymous point mutation in SOCS2 (p.R96C) was identified that disrupts the tyrosine binding pocket of the SH2 domain of SOCS2. 22 In vitro experiments showed a nearly complete lack of SOCS2 binding to its highest affinity phosphorylated residue on the GHR. This mutation was associated with increases of 24%, 18%, and 4.4% for height, weight, and milk yield, respectively, in SOCS2 p.R96C homozygotes. However, there was also a strong positive correlation with lifetime somatic cell count score (a proxy for genetic susceptibility to mastitis). 22 This mutation was subsequently found to result in undesirable structural and functional changes in mammary development in mice. 23 Recently, SOCS2 p.R96C sheep were produced by programmable base editing 24 ; however, the total rate of the intended substitution was below 45% for all animals.
Previously, we successfully targeted SOCS2 and PDX1 genes in sheep and OTX2 in goat embryos by electroporating zygotes with long-high/short-low voltage pulses 6 h after fertilization. 25 We found that this approach not only efficiently replaced microinjection but also increased the biallelic mutation rate in small ruminant embryos. In this study, we investigated the possibility of efficiently producing SOCS2 KO sheep by embryo electroporation of dual gRNA Cas9/sgRNA RNPs designed to target an 85 bp region of exon 1 of SOCS2 encompassing a part of the extended SH2 domain. We whole genome sequenced the genome-edited offspring and characterized the mutations present.
Materials and Methods
Experimental design
This work was conducted according to methodologies previously outlined in work by our group with respect to the production of electroporated small ruminant embryos. 25 In brief, two rounds of embryo production were used to generate SOCS2 KO animals. In each round, zygotes were produced by in vitro fertilization of in vitro matured cumulus-oocyte complexes (COCs). Then two sgRNA were introduced into the zygotes 6 h after fertilization by electroporation with short-high/long-low voltage parameters as previously described. 25 Embryos were cultured to blastocysts and transferred to synchronized recipients, then pregnancies were confirmed by ultrasound 30 days after fertilization. Lambs were genotyped with a combination of PCR-Sanger sequencing and whole genome sequencing (WGS). Western blotting was used to detect the presence of SOCS2 protein production in the live animals. The maternal breed used is unknown as the oocytes were collected from a mixed-breed slaughterhouse; the rams were a herd of primarily Rambouillet genetic background.
sgRNAs design
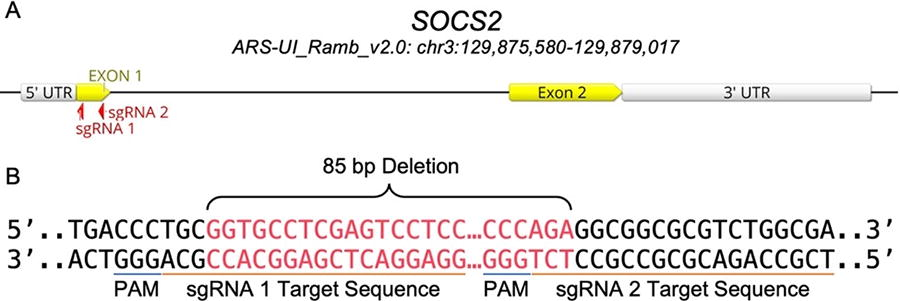
Two sgRNAs were designed using the online E-CRISP gRNA design tool 26 and ordered from Synthego (Synthego Corporation, Redwood City, CA) to target the beginning and end of the sheep SOCS2 exon 1 (Fig. 1; Supplementary Table S1); the expected deletion was 85 bp long. We used the Oar_v3.1 reference genome (GCA_000298735.1) 27 and strict search criteria specifically for KO.
Gametes
COCs: sheep ovaries were collected from a local abattoir (Superior Farms, CA) and transported to the laboratory in 37° C saline within 2 h of collection. COCs were aspirated with a 21 G needle under vacuum pressure.
In vitro embryo production
In vitro oocyte maturation: good quality COCs with several cumulus cell layers and homogenous cytoplasm were selected for maturation. COCs were washed five times with warm washing media and then transferred in groups of 50–400 µL of maturation medium (BO-IVM, IVF BIOSCIENCE) and incubated for 22–24 h at 38.5°C, 5% CO2, and humidified air.
In vitro fertilization
Matured COCs were washed three times with warm and pre-equilibrated fertilization media (BO-IVF, IVF BIOSCIENCE) and transferred in groups of 50–400 µL of fertilization media. Freshly collected semen was washed twice by centrifugation at 328 g for 5 min with 2 mL of semen preparation media (BO-Semen Prep, IVF BIOSCIENCE). The sperm concentration was adjusted with fertilization media to 2 × 106 spermatozoa/mL and 50 µL added to COCs for 6 h at 38.5°C, 5% CO2, and humidified air.
In vitro culture
Cumulus cells were stripped off presumptive zygotes by vortexing for 3 min in SOF-HEPES, then zygotes were washed five times, and transferred in groups of 50–500 µL of culture media (BO-IVC, IVF BIOSCIENCE) covered with 400 µL mineral oil and incubated for 7 days at 38.5°C, 5% CO2, 5% O2, 90% N2, and saturated humidity.
RNP preparation
All RNA work was conducted in an RNAse-free workstation at room temperature. A total of 40 ng/µL of each sgRNA was mixed with Cas9 protein (PNA Bio) in a mass ratio of 1:2 and incubated on ice for 10 min; RNPs were mixed immediately prior to electroporation.
Electroporation
Electroporation was performed using the NEPA21 Super Electroporator in 1 mm Nepa Electroporation Cuvettes (EC-001). All work was undertaken in an RNAse-free workstation. Denuded zygotes were washed three times with synthetic oviductal fluid-4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (SOF-HEPES) and three times with warm Opti-MEM (Thermo Fisher Scientific #31985062). Zygotes were moved to a new drop of Opti-MEM, mixed with RNPs to a total volume of 20 µL, and subsequently loaded into an electroporation cuvette. The poring parameters were set to four unipolar electric pulses of 40 volts of 3.5 ms each, and the transfer parameters were set to five bipolar electric pulses of 5 volts for 50 ms each. After delivery of electric pulses, the cuvette was washed several times with SOF-HEPES, and the recovered zygotes were washed three times with SOF-HEPES and one time with BO-IVC before being moved to pre-warmed and pre-equilibrated BO-IVC media and cultured for 7 days.
Recipient selection and estrus synchronization
All experiments involving animals were approved and performed in accordance with the University of California Davis Institutional Animal Care and Use Committee (Protocol # Protocol #: 20259). Healthy white-faced, multiparous, Corriedale-type ewes were estrus-synchronized using an intravaginal progesterone device (0.3 g of progesterone; CIDR-G; Zoetis). Controlled Internal Drug Release (CIDRs) were inserted for 6 days. On the day of CIDR removal, prostaglandin F2-alpha analog (10 mg dinoprost thrometamine; Zoetis) and PG600 (400 IU PMSG, 200 IU hCG; Intervet) were injected intramuscularly. Estrus was detected every 12 h after removing the CIDRs with the assistance of a vasectomized ram for heat marking. Thirteen ewes were selected for embryo transfer 6 days post-estrus.
Laparoscopic embryo transfer
Food and water were removed 24 and 12 h prior to surgery, respectively. Fifteen minutes before surgery, recipients were sedated by administration of 1.1–2.2 mg/kg of ketamine and 0.2–0.3 mg/kg of Midazolam. After preparing the surgery site, 2% lidocaine was injected at the incision site to induce local anesthesia. Following placement in Trendelenburg position, the abdomen was insufflated with CO2, and the uterus and ovaries were visualized with a 0-degree laparoscope (Karl Storz, Germany) introduced into the caudal abdomen. The horn ipsilateral to the corpus luteum was exteriorized via a small incision, and four blastocysts were transferred into the tip of the uterine horn.
Pregnancy diagnosis
Pregnancy diagnosis was performed 30 days post-estrus by abdominal ultrasonography. The frequency of the transducer was 3.5 MHz.
Genomic DNA extraction
Tissue/blood was collected from genome-edited lambs and two control rams, and DNA was extracted using DNeasy Blood & Tissue Kit (Qiagen Cat. No./ID: 69504).
In vitro on-target indel detection
PCR reactions were prepared with 10 μL of Promega GoTaq Green Master Mix (2×), 1 μL forward primer (10 μM), 1 μL reverse primer (10 μM), 100 ng of genomic DNA, and molecular grade nuclease-free water to a final volume of 20 μL. CRISPR-Cas9 target region was amplified by PCR; primer sequences and amplification conditions are available in Supplementary Table S2. PCR products were run on 1% or 2% agarose gels prepared with 60 or 120 mL of 1% tris base acetic acid and ethylenediaminetetraacetic acid (EDTA) (1% Tris base, boric acid, and EDTA [TBE]), and SYBRTM Safe gel stain (10,000×) diluted to 6×. Visualization of bands was performed under blue and UV light and compared with a control amplicon. DNA fragments were cut out of gels individually using a scalpel, and purified PCR products were gel-extracted either by the QIAquick Gel extraction kit (QIAGEN) or a freeze and squeeze method of placing the gel on top of a filter tip in a 1.5 mL conical tube, freezing the sample for 5′ at −80°C, then centrifuging for 3 min at 20,800 g. Gel-purified PCR products were outsourced to GeneWiz (Azenta, South Plainfield, New Jersey) for purified PCR product Sanger sequencing. To detect indels, first, each amplicon was aligned to a control amplicon using SnapGene® software (version 6.1.1) to visualize the modifications. The sequences were checked for insertions, deletions, and double peaks.
Allele analysis
The predicted amino acid sequences of each mutant allele were generated by SnapGene® software.
Whole genome sequencing
Genomic DNA from the six genome-edited lambs that were born and their two unedited sires (Ram 1 and Ram 2) were used for WGS with the Illumina NovaSeq platform following manufacturer instructions. DNA fragment sizes for library development were approximately 300 bp with 2 × 150 bp paired-end reads to an approximate depth of 13–45× per sample. Qualified reads were mapped to the ARS-UI_Ramb_V2.0 (GCA_016772045.1) 28 and Oar_v4.0 (GCA_000298735.2) reference genomes using Burrows-Wheeler Aligner-Maximum Exact Match and indexed with BWA-Index (BWA tools v7.1). Aligned WGS BAM files were visualized with Integrative Genome Viewer (v2.13.2).
Single nucleotide variants and small indel identification
For a detailed analysis, and genome-wide identification of single nucleotide variants (SNVs), the Illumina WGS data were mapped to the reference genome (ARS-UI_Ramb_V2.0) using CLC Genomic Workbench V11.0 (https://digitalinsights.qiagen.com). The reads were mapped to the reference by using conservative mapping parameters, which required that 75% of a read to map uniquely to the reference. Further affine gap cost penalty was used for insertions and deletions to the maximum cost allowed of 6 to open an insertion or deletion position and an extension cost of 1. Following mapping, all variants, including SNVs, small insertions, and deletions (indels <30 bp) were called using the basic variant detection tool using the following parameters: only reads mapped as intact pairs were included, with a minimum Phred quality score of 30 for the central variant, as well as the five adjoining bases on either side. The final step was to require a minimum variant frequency of 25% within a sample.
Structural variant analysis
Genome-wide structural variants (SVs) (deletions, inversions, intra-chromosomal translocations, tandem duplications), were identified by mapping reads to the reference with more liberal parameters than for SNV detection in order to minimize rejection of reads due to misalignment caused by large indels. CLC genomics software checks read mappings for evidence of breakpoints using “unaligned end” signatures. “Unaligned end” refers to the end of a read that does not map to the reference sequence at the positions presented in the read after the portion that does align to the reference. Thus, to maximize the discovery of SVs and minimize rejection of reads due to reference misalignment, a minimum of only 10% of a read was required to be mapped uniquely to the reference, allowing any unaligned ends to be incorporated in a stringent search for SVs. The algorithm first identifies positions in the mapping(s) with an excess of reads with left (or right) unaligned ends. Once these positions and the consensus sequences of the unaligned ends are determined, the algorithm maps the determined consensus sequences to the reference sequence around other positions with unaligned ends. If mappings are found that are in accordance with a “signature” of an SV, an SV is called.
On-target analysis
In addition to the SV analysis using CLC, a manual confirmation method was also used. For deletions that appeared to be within the span of overlapping paired reads the following method was used: (1) based on visual examination of the unaligned ends, haplotypes were identified and defined by a portion of the sequence in the unaligned ends plus adjoining sequences that aligned with the reference. These haplotypes were extracted from the raw fastq data using custom scripts and stored in separate files. (2) The haplotype-resolved reads were then aligned using the DeNovo aligner of CLC genomics. The resulting consensus alignment of each haplotype was compared with the Ovis aries Rambouillet reference genome (ARS-UI_Ramb_V2.0) using National Center for Biotechnology Information's (NCBI’s) basic local alignment search tool (BLAST). Alignments that were not contiguous with the reference were flagged as possible SVs. (3) The actual SV was identified by matching the contiguous parts of the BLAST with the reference sequence and counting the number of base pair (bp) deletions or insertions needed to enable alignments of the fragments to the reference sequence.
For deletions that were greater than the span of overlapping paired reads, an alternative manual method of verification was employed. This method utilized paid reads, which are fragments with ends that were sequenced as a physical unit but were mapped to the reference as broken reads, i.e., each end mapped to a different region. By visual examination, reads that were broken but at least partially mapped to the reference in the target region were extracted and their read IDs tabulated. The mates to the extracted reads were found by searching the raw fastq data file for each mate’s matching ID. The extracted reads from the target region along with their mates were then aligned using the DeNovo aligner and the process proceeded as previously described for short deletions.
Off-Target analysis
Potential off-target binding sites with up to 4 bp mismatches (Supplementary Table S3) were examined by mapping SNV and large SVs identified by CLC spanning ± 100 bp away from the putative cut site. To investigate whether endogenous variants at these sites could be associated with potential off-target activity, we used a custom R script, which detects if the possible binding site is either directly within or within an extended range spanning an additional 100 bp from either end of the mutated sequence (R version 4.3.2). Chi-square tests were conducted using Monte Carlo simulated p-values, comparing the frequency of mutated regions, which contained a putative binding site between edited samples and wild-type control rams.
Fibroblast cultures
Fibroblast cell lines from lambs were derived as previously described. 29 Briefly, 4-mm skin biopsies were taken from two different sites in each lamb. The skin was dissected, and small dermal pieces were cultured for 10 days in Dulbecco's Modified Eagle Medium high glucose media (Thermo Fisher, Cat#11995065) supplemented with 20% Fetal bovine serum (Gibco, Cat# 10100139), 1% GlutaMAX (Gibco, Cat#35050061), and 1% penicillin-streptomycin (Gibco, Cat#15140122). Cells were passaged using trypsin at least twice to obtain homogenous fibroblast cell lines.
Protein extraction and Western blotting
Proteins were extracted from fibroblast cells using RIPA Buffer (Sigma #R0278) with protease inhibitors (Roche #11836153001). Proteins were quantified using Pierce™ BCA assay (Thermo Fisher, Cat#23225). Western blotting was performed as previously described. 30 A total of 20 µg of proteins per sample were electrophoresed on 12% Mini-PROTEAN TGX precast protein gels (Bio-Rad) for 5 min at 50 volts followed by 115 min at 100 volts. Proteins were transferred to Immun-Blot PVDF membrane (Bio-Rad) for 120 min at 100 volts then blocked for 1 h in 3% BSA TBS-T buffer. The membrane was then incubated at 4°C overnight with the following primary antibodies: anti-SOCS2 antibody (Abcam#ab66733, 1:1000) and anti-β-Actin antibody (C4) (Santa Cruz#sc-47778, 1:1000). Then the membrane was washed three times in TBS-T and incubated at room temperature for 2 h with the following secondary antibodies goat anti-rabbit IgG H&L (HRP) (Abcam #ab205718, 1:2000) and m-IgG Fc BP-HRP (Santa Cruz, #sc-525409, 1:2000). The ChemiDoc-It imaging system was used to visualize the membrane.
Preliminary phenotype of homozygous SOCS2 F0 KO lambs
The growth variables of weight, height, and length were measured in the three surviving SOCS2(-/-) KO founder sheep (#7181 female, #7182 and #7183 males) through 20 weeks and compared with two phenotype-control ewes in the flock that were born at a similar time and raised in the same pen under commercial conditions.
Results
Pregnancies, abortions, lambing, and survival
Of the 13 recipients that received embryos, 6 were determined by ultrasound to be pregnant at day 30. Two recipients aborted at weeks 13–14 of pregnancy. One recipient delivered a stillborn male lamb (#7186), which may have died because of its large size (12.2 kg). Another recipient gave birth to triplets, two males and one female. The female (#7184) was euthanized at 2 days of age due to breathing difficulties, one male (#7185) died at 5 days of age while the other male was healthy (#7183). Two recipients gave birth to one healthy lamb each, one male (#7182) and one female (#7181), totaling three healthy lambs, which were followed for 20 weeks (Fig. 2).

SOCS2 genome-edited lambs female tag #7181 (front), male tag #7182 (left), and male tag #7183 (right).
Genotyping analysis of genome-edited lambs
Genotyping of the genome-edited offspring was initially performed by PCR amplification of a 479 bp region flanking the two target sites. All genome-edited lambs appeared to have deletion alleles via gel electrophoresis (Supplementary Fig. S1). Analysis of WGS data revealed that both sgRNA 1 and sgRNA 2 target sites were disrupted in each of the six genome-edited lambs (Fig. 3). Deletions varied in size from 30 bp in an allele of female tag #7184 to 2127 bp in a female tag #7181 allele. Three large deletions in the healthy lambs: female tag #7181, male tag #7182, and male tag #7183, evaded initial detection by PCR followed by Sanger sequencing due to the elimination of one or multiple primer binding sites. Additionally, three intermediate-sized deletions of 30 bp, 101 bp, and 109 bp were identified, with the 101 bp deletion present in male tag #7185 spanning both target sites and the other two initiating at target site 2. None of the mutations were the exact 85 bp deletion between the predicted cleavage sites, but five deletions were within a 5 bp window of both cut sites. Finally, four small insertions were observed, with a single base pair guanine insertion at cut site 1 occurring in allele 1 of the female tag #7181 and allele 1 of stillborn male tag #7186, as well as a “G” insertion and “TTT” insertion occurring in tag #7186 allele 2 at cut sites 1 and 2, respectively (Fig. 4).
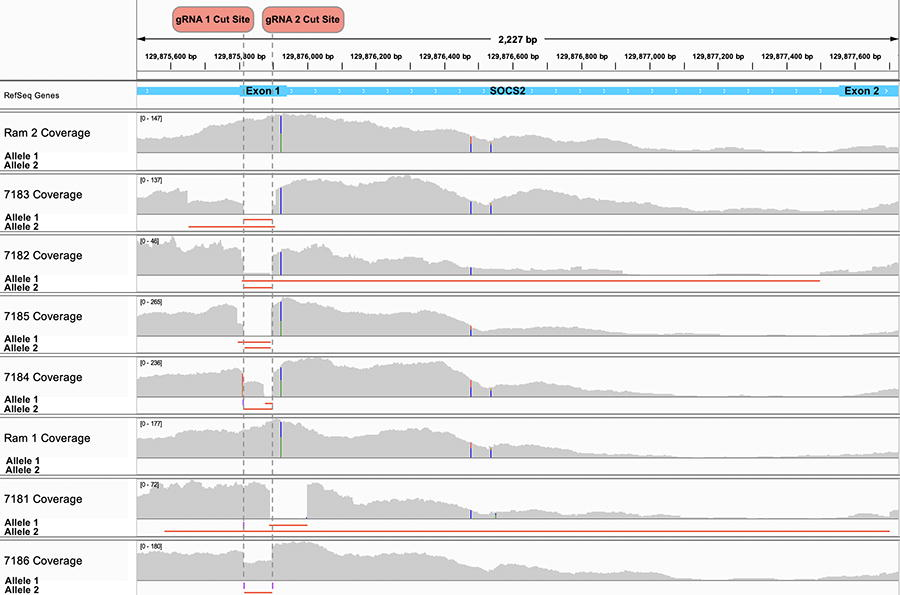
Read coverage tracks at the SOCS2 target locus for Ram 2, Ram 2’s genome-edited offspring tag #7183, #7182, #7185, and #7184; Ram 1, and Ram 1’s genome-edited offspring tag #7181 and #7186. The read depth is indicated in gray on the read coverage track with vertical dual color bars indicating a common SNP, the span of each deletion is indicated as a red horizontal line, and insertions are indicated as purple rectangles on the allele tracks.
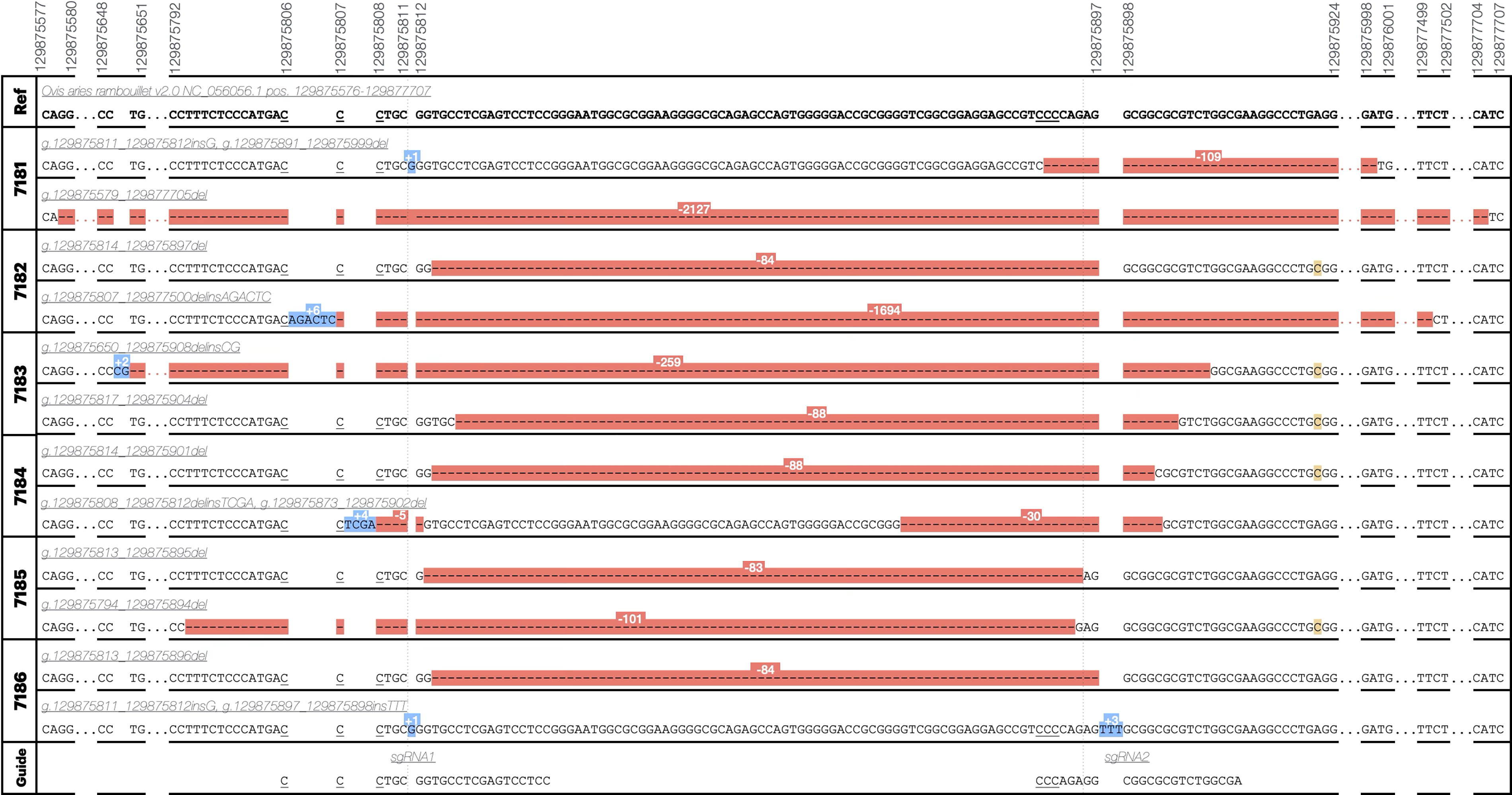
CRISPR-Cas9 genome-edited alleles. Variant names are according to human genome variation society nomenclature guidelines for genomic variants and indicate positions on chromosome 3 (NC_056056.1) in the ARS-UI_Ramb v2.0 reference genome. Red highlighting indicates a deletion, blue an insertion, yellow an endogenous SNP not considered to be a result of genome editing. Ranges not shown are indicated by ellipses and are either wild-type sequence or continued deletion sequence. PAM sites (NGG reverse-complement) are indicated by underlining.
The predicted effect of each of the major mutations in the gene was analyzed by prediction of the resulting SOCS2 translation (Table 1). All analyzed variants were predicted to result in KO of the SOCS2 gene either through deletion of a large section of the protein, deletion of the start site, or a frameshift and premature introduction of a stop codon.
Predicted amino acid sequences per edited allele and number of WGS reads (#)
Variant names indicate variant position on chromosome 3 (NC_056056.1) in the ARS-UI_ramb v2.0 reference genome. Predicted amino acid sequences were generated by in silico translation on SnapGene®: bold text indicates amino acid sequence from endogenous start site, an asterisk indicates stop codon.
SOCS2, suppressor of cytokine signaling-2; WGS, whole genome sequencing.
The frequency of each mutant allele, and wild-type reads in the WGS data are shown in Table 2. A small number of wild-type reads were observed for some samples in the WGS data (ranging from 0% to 12% of SOCS2 reads). This suggests that there was mosaicism in these samples.
Average sequence depth and relative proportions of each allele and wild-type sequence
Allele values are presented as discrete read count followed by the relative proportion of reads for that allele at the locus.
Off-target analysis
Genomic regions with up to 4 bp mismatches to the target sequence were found, revealing 133 possible off-target binding sites of the two guide RNAs (Supplementary Table S3). Cross-referencing these sites with genomic regions where a mutation was present (ranging ±100 bp from the mutated sequence) compared with the reference genome (ARS-UI Ramb v2.0) revealed a total of 40 mutations across all samples. Of these, six possible binding sites were directly within the mutated region (Supplementary Table S4). Chi-square analysis indicated that possible sgRNA binding sites were no more likely to exist within 100 bp of a mutation in edited sheep as compared with natural mutations that occurred at those same locations in wild-type control sheep (p = 0.7076) (Fig. 5). Notably, the number of naturally occurring mutations occurring outside of the 133 possible nonspecific sgRNA binding sites was several orders of magnitude higher in both edited animals and controls.
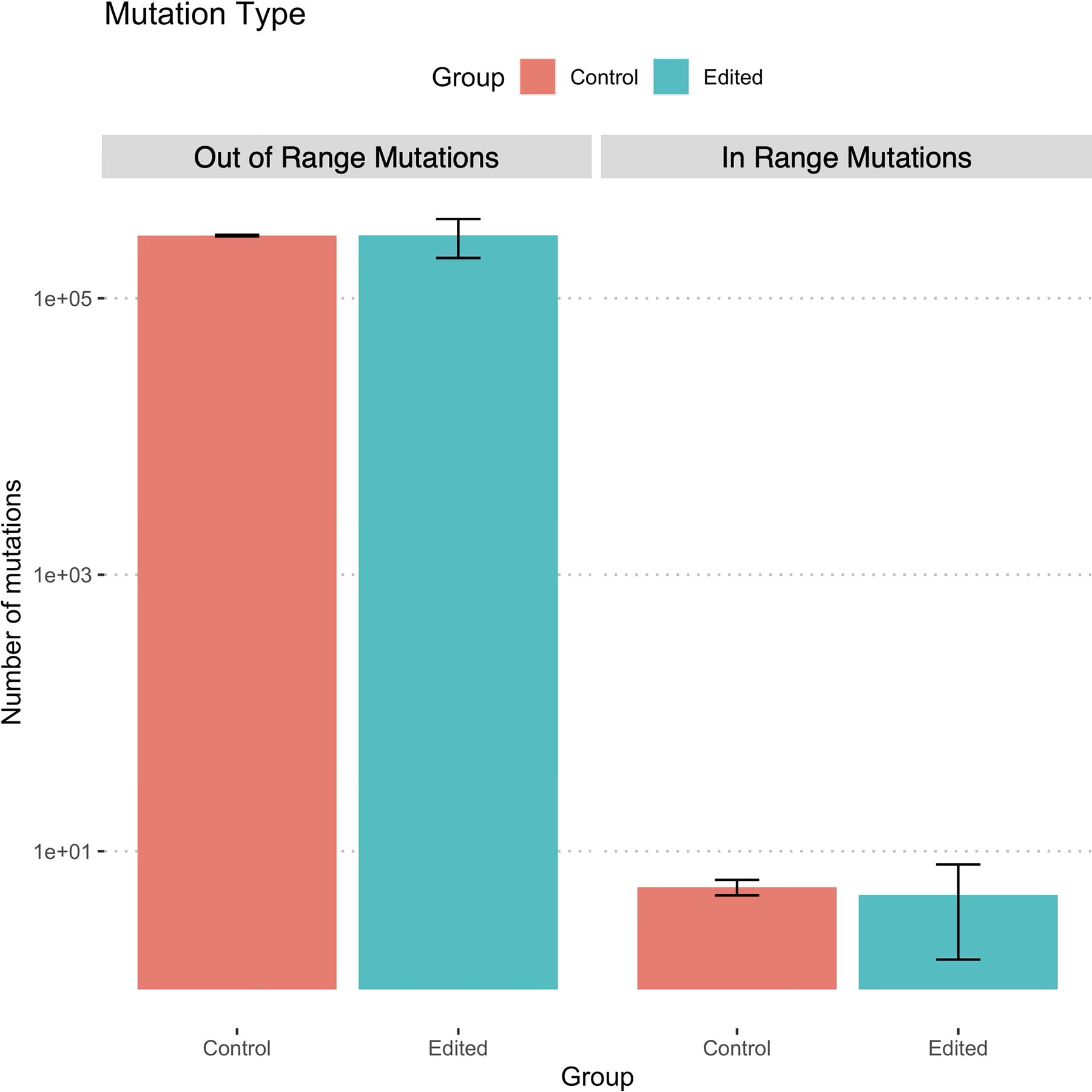
Comparison of the frequency of mutations in regions proximate (±100bp) to potential off-target sgRNA binding sites with up to four mismatches (in-range mutations) and elsewhere in the genome (out of range mutations) in edited animals (7181–7186; n = 6) as compared with unedited control animals (Ram 1, Ram 2; n = 2). A chi-square test did not show a significant difference between the frequency of mutations occurring in unedited control animals as compared with edited animals (p = 0.7076).
Protein expression results
Trace levels of SOCS2 protein expression were detected in males tag #7182 and #7183, and female tag #7181, respectively, compared with wild-type in a Western blot (Fig. 6).
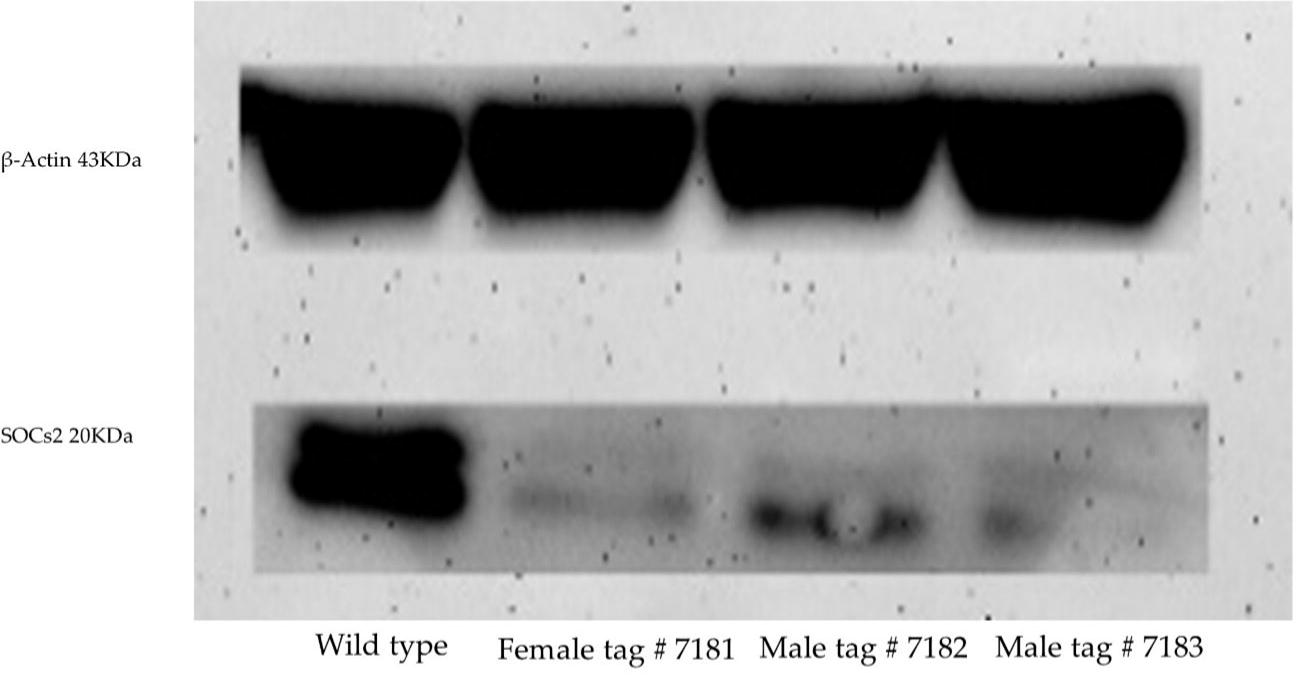
Western blot of protein extracted from fibroblast cell lines derived from three healthy SOCS2 knock-out lambs produced by zygote electroporation of CRISPR-Cas9.
Preliminary phenotype of homozygous SOCS2 F0 KO lambs
Results of this pilot study showed that the SOCS2(-/-) KO sheep had a faster growth rate (0.45 g/day vs. 0.31 g/day; p < 0.01), and a heavier average weight of 66.4 kg at 20 weeks of age as compared with 42.1 kg for the controls (Fig. 7). Furthermore, the SOCS2(-/-) KO sheep reached the final weight of the control sheep in approximately half the time. The increase in weight was accompanied by an approximately proportional increase in height and length. It is important to note that both phenotype-control sheep were female, the breed of the dams of the edited sheep was unknown, and #7183 was the only surviving lamb from a set of triplets. All of these factors are known to influence animal weight.
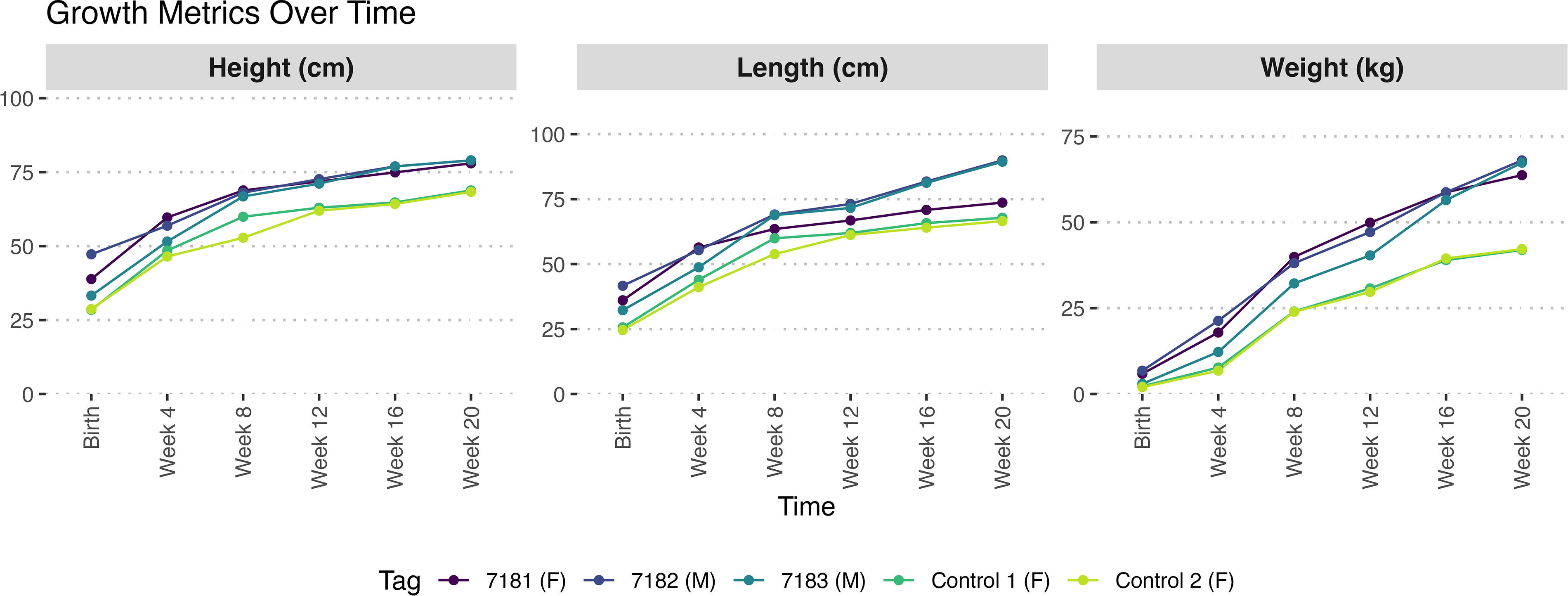
Preliminary growth results of the founder F0 SOCS2 genome-edited knock-out lambs (tag #7181 female, #7182 male, #7183 male) and two phenotype-control ewes over the first 20 weeks.
Discussion
In the current study, we report the generation of SOCS2 KO sheep by CRISPR-Cas9 electroporation of dual sgRNAs into in vitro produced zygotes. Of the 52 blastocysts transferred to 13 estrus-synchronized recipients (4 blastocysts per recipient), six ewes (46%) were determined by ultrasound to be pregnant at day 30. Two of the pregnant recipients aborted at weeks 13–14 of pregnancy, and of four that gave birth, one had triplets of which two died after birth, one delivered a large stillborn male, and the remaining two gave birth to one healthy lamb each. Of the six full-term lambs, three (50%) survived to 20 weeks. This is a relatively low survival rate, although it was not expected that all four blastocysts would reach term in every recipient. A U.S. study reported typical lamb mortality from birth to 4 weeks at 15.2%. 31 It is known that in vitro embryo production per se can generate larger-sized lambs with several abnormalities, usually termed “large offspring syndrome.” 32 Moreover, in vitro maturation and fertilization alone are known to compromise subsequent embryonic and fetal development in sheep, 33 irrespective of editing. It is not possible to extricate whether the high rate of morbidity observed in the current experiment was related to the edit per se, electroporation, or some other aspect of the in vitro embryo culture.
Electroporation of Cas9 RNP is a recent advancement in the genome engineering of livestock embryos that allows for the batched introduction of genome editing reagents for up to around 100 oocytes or embryos simultaneously, which offers several advantages over individual oocyte microinjection. 15 Early evidence suggests that electroporation is highly efficient at creating genome-edited embryos with a minor effect on embryo survival so long as electroporation is performed at the time of fertilization or at the pronuclear stage and electroporation settings are optimized for the species of interest.15,25 In mice, electroporation of gRNA into Cas9-expressing embryos was shown to result in a 100% or close to 100% mutation efficiency with no mosaicism. 34 Previous studies, using microinjection of CRISPR-Cas9 into sheep zygotes reported a lower mutation efficiency (10–38%).35–37
We electroporated the zygotes 6 h after in vitro fertilization to increase the chances of biallelic nonmosaic mutations by delivering the CRISPR-Cas9 reagents to the embryo before the DNA replication preceding the first embryonic cleavage. The method and the timing of editing reagent delivery are important, as electroporation of mouse zygotes before E0.5 has been shown to decrease mosaicism 1 and electroporation of sheep zygotes within 8 h post-fertilization decreases the frequency of biallelic mutation rate as compared with microinjection. 25
All the lambs generated from CRISPR-Cas9 electroporated embryos contained insertions or deletions at the targeted sites. This 100% mutation rate suggests the efficiency of electroporation as an effective zygote delivery method for CRISPR-Cas9 gene editing and has great potential for the generation of genome-edited sheep. Additionally, using this approach, all edited alleles present in the genome-edited lambs were predicted to result in a KO of the target SOCS2 gene (Table 1). However, the intended deletion of 85 bp did not occur in any of the founder animals, and there was a high degree of variability in the exact mutations that resulted in the following editing.
Of particular importance to livestock genome engineers and regulators is that 25% of all alleles characterized by WGS were undetectable by PCR and Sanger sequencing of a standard-length amplicon using PCR primers close to the target site. This is consistent with the results of a recent comprehensive analysis of CRISPR-Cas9 genome editing outcomes in human hematopoietic stem and progenitor cells using long-read Pacific Biosciences (PacBio) single-molecule real-time sequencing technology (SMRT-seq) that found large deletions (>200bp) occurred at a frequency of 11.7–35.4% depending on gene target. 38 Similar work undertaken in cattle using a dual-guide approach also resulted in large deletions. 39 The mechanism of long deletion is not well understood, but microhomologies are commonly associated with the repair of large deletions and were observed in our results. 40 Large deletions pose a major problem for the genotyping of genome-edited livestock embryos where small quantities of DNA limit the possibilities for WGS.
One concern of the public regarding CRISPR-Cas9 genome editing in food animals is the potential for off-target mutations, which we did not observe in our work. Off-target mutations could hypothetically result in unintended deleterious effects in sheep. The location of sites with 3 or 4 bp mismatches can be predicted effectively using web-based tools like CasOFFinder that search reference genomes for potential off-target sites.4,24,41 Our analysis of potential off-target binding sites with four mismatches or fewer from the target sequence identified a small number of mutations in edited samples which may be a result of off-target activity, but notably in-range naturally occurring mutations were also observed in unedited controls (Supplementary Table S4). Furthermore, we observed several orders of magnitude more mutations outside of this region in both edited animals and controls (Fig. 5). This highlights that natural mutations occur frequently in unedited food-producing animals (i.e., controls). In this case, the unedited rams had on average of 5.1 million naturally occurring SNP and indel mutations relative to the Rambouillet sheep reference genome. Similarly, a project that sequenced 2,703 cattle of different breeds found 84 million SNPs and 2.5 million naturally occurring indels. 42 Despite this known variation, animals produced by conventional breeding methods are not routinely evaluated for unintended mutations at the molecular level as genetic variation per se is not known to be associated with food safety hazards. 43 Increasing the search for binding sites from those with up to a 3 bp mismatch (n = 4) with the sequence of the two sgRNA guides, to those with up to a 4 bp mismatch (n = 133) resulted in a more than 30-fold increase in the number of potential off-target binding sites to be analyzed. Evidently, there are diminishing returns in performing such off-target analysis on the genomes of healthy gene-edited sheep. The food safety benefit resulting from such an exhaustive off-target analysis is unclear in the context of the abundance of naturally occurring genetic variation.
Several specialized programs for the detection of SVs have been developed, such as Manta, 44 DELLY, 45 Vg-Graph, 46 and Minigraph-Cactus, 47 along with commercial software such as CLC Genomics Workbench (QIAGEN, https://digitalinsights.qiagen.com) among others. However, the only accurate way to identify SVs is for species with defined haplotypes, such as with human populations. 47 This conclusion is based on the assumption that the SV only occurs on one chromosome. Unfortunately, if multiple SVs occur in the same region, as with CRISPR edits, preexisting haplotypes are irrelevant for that region. Even worse, if the SVs are at an intermediate frequency, i.e., a heterozygote, a consensus sequence for that region is not possible. A workaround was developed for our research whereby the haplotypes were resolved by manual inspection of the read overhangs, extraction of those reads, de novo realignment, and extraction of the consensus sequence, followed by a BLAST to the reference to reconstruct the SV. While this method is highly accurate, the procedure is laborious but tractable for examination of on-site edits. However, the method is not tractable for the assessment of off-site edits of four or more misalignments. Nevertheless, programs such as CLC genomics and perhaps others are accurate at finding where breakpoints have occurred, even if the programs are not accurate in reassembling the results into actual SVs. It may be that for the purposes of finding off-target effects, finding breakpoints is adequate for the purpose. However, as discussed previously the food safety value of these analyses is questionable given all animals that enter the food supply have millions of naturally occurring mutations relative to the reference genome. The results from this article support the mounting evidence that off-target editing is rare with well-designed gRNAs.
Another concern in genome editing is mosaicism, which can obscure the evaluation of founder animal phenotypes and result in a variety of offspring phenotypes. 4 Mosaicism that includes some remaining wild-type sequences is regularly observed with engineered nucleases and can diminish the value of single-step genome editing to produce gene-edited founders. 48 This could explain the low levels of SOCS2 mRNA and protein that were detected in the edited sheep. Given sequencing was performed on a mixed population of cells, it is not possible to differentiate between compound heterozygotes and mosaics containing two different subpopulations with biallelic edits.
The differences in growth between SOCS2(-/-) KO and wild-type sheep align well with the growth phenotype seen in the mice where Socs2(-/-) pups were indistinguishable from their littermates prior to weaning, but subsequently grew more rapidly. Male mice at 12 weeks of age were 40% heavier than controls, with heterozygous animals exhibiting an intermediate body weight. A similar and significant but less pronounced phenotype was seen in adult female Socs2(-/-) mice, which were only ∼30% heavier than sex-matched wild-type littermates. 19 The size, weight, and milk production of sheep with a naturally occurring homozygous point mutation in SOCS2 were significantly increased by 24%, 18%, and 4.4%, respectively, when compared with wild-type sheep. 22
While the preliminary growth results of this experiment are promising, there are caveats. The growth rate is affected by sex (intact rams grow faster), birth weight, ewe milk supply, health, and litter size. It is well known that triplets are significantly smaller than singletons, and female sheep are lighter than males.49,50 Additionally, the contemporary, control ewes that were available for this experiment were commercial sheep generated through natural mating and were not genetically related to the edited animals. As such their size differences may be due to factors other than the edit as it has been observed that in vitro embryo production per se can generate larger size lambs. 51 Moreover, there were no age-matched control rams. Ideally, controls would be matched for the known effects of breed, sex, litter size, and birthweight on growth and be produced and treated identically to the treated animals. This is particularly problematic given the unknown maternal genetic background of the gene-edited sheep as a result of the slaughterhouse ovary origin of the oocytes that were fertilized. The generation and maintenance of matched conventional comparators (e.g., nonedited animals of the same genotype, sex, litter size, and birthweight) is an expensive undertaking in large animal species, especially uniparous and biparous species. The Codex Guideline 52 for the conduct of food safety assessment of foods derived from recombinant-DNA animals highlights this problem stating that “it should be acknowledged that, particularly in the case of certain animal species, the available number of samples may be limited and there is likely to be large variation between animals, even those bred and raised under the same husbandry conditions.”
The high-growth phenotype could potentially be a valuable growth trait for introduction into sheep breeding programs. The greatly improved growth phenotype, without an associated increase in birth weight, could decrease the time to market weight, the amount of feed consumed, and reduce the carbon intensity of producing a unit of lamb. However, the negative effects that have been observed on reproduction in Socs2(-/-) mice including a failure to maintain pregnancy in certain lines, 53 reduced lifespan, 54 and undesirable structural and functional changes in mammary development 23 warrant caution. More data are required to determine how sheep carrying heterozygous mutations in the SOCS2 gene perform under commercial conditions, and if there are any negative phenotypic correlations or other pleiotropic effects on other traits.
Conclusions
This study documents a comprehensive genotypic analysis of CRISPR-Cas9 genome editing of six genome-edited sheep. Our findings demonstrate that electroporation with dual sgRNAs was highly efficient in generating SOCS2 KO sheep, with a high mutation rate, and the ability to process multiple embryos simultaneously. Notably, exhaustive analysis of mutations at sites with up to four mismatches to the guide RNA did not reveal concerning off-target activity with this approach, which will be highly applicable to other livestock editing endeavors. The growth phenotype of the SOCS2(-/-) sheep suggests there are likely benefits for productivity which will be investigated further in the next generation.
Footnotes
Acknowledgment
The authors would like to express their deep gratitude to Superior Farms (Dixon, CA) for providing sheep ovaries.
Authors’ Contributions
A.K.M., P.J.R., A.L.V.E., and J.F.M. designed the experiments. A.K.M. and D.S.F. carried out the experiment. B.R.M. and T.U.B. performed the surgical procedure of embryo transfer. D.E.H. performed the whole genomic sequencing analysis to visualize on-target edits and assist with primer design. W.M.M. and N.W. performed both on-target confirmation and potential off-target analysis using WGS data. A.K.M., D.S.F., A.L.V.E., and P.J.R. wrote the article. N.W. prepared figures, and all authors reviewed the final article.
Data Availability
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
UC Davis chancellor’s fellow award to P.J.R.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.