
Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein (Cas) technology has revolutionized genome editing across various biological systems, including the Apicomplexa phylum. This review describes the status, challenges, and applications of CRISPR–Cas9 editing technology in apicomplexan parasites, such as Plasmodium, Toxoplasma, Theileria, Babesia, and Cryptosporidium. The discussion encompasses successfully implemented CRISPR–Cas9-based techniques in these parasites, highlighting the achieved milestones, from precise gene modifications to genome-wide screening. In addition, the review addresses the challenges hampering efficient genome editing, including the parasites’ complex life cycles, multiple intracellular stages, and the lack of robust genetic tools. It further explores the ethical and policy considerations surrounding genome editing and the future perspectives of CRISPR–Cas applications in apicomplexan parasites.
Introduction
Gene editing changes DNA in an organism’s genome to modify its phenotype. The approaches include zinc finger nucleases (ZFNs), 1 transcription activator-like effector nuclease (TALEN), 2 meganucleases, 3 and the clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein (Cas) complex. 4 Meganucleases, ZFNs, and TALENs are old-generation techniques that laid the foundation for gene editing but are time-consuming and costly. Comprehensive reviews on meganucleases, ZFN, and TALEN approaches for gene editing can be found elsewhere.3,5 For a comparison of these gene editing methods, refer to Table 1.
Comparison of genome editing technologies
gRNA, guide RNA; RVD, Repeat variable di-residue; TAL, Transcription activator-like; eGFP, enhanced Green Fluorescent Protein.
CRISPR–Cas represents a newer, more advanced and precise genome editing technology.6,9 CRISPR is an RNA-mediated system that naturally forms an adaptive immune system in bacteria. It consists of two classes grouped into six significant types and at least 33 subtypes. 8 Class 1 (types I, III, and IV) uses multiple Cas effectors to cleave foreign DNA; due to this requirement, class 1 systems are challenging to program for genome editing. In contrast, class 2 (types II, V, and VI) uses a single Cas effector with multiple domains to cleave DNA or RNA, making them more adaptable to genome editing. Class 2 has several Cas proteins, including Cas9, Cas12, Cas13, and Cas14. 7
The type II CRISPR–Cas9 system, widely used in genome editing, recognizes the target sequence through a protospacer adjacent motif (PAM) downstream of the target. PAM allows the system to differentiate between the target DNA and the CRISPR RNA (crRNA) sequence.4,8 Cas9 cuts each target DNA strand, generating blunt-ended double-strand breaks (DSBs). 4 Various Cas9 nucleases recognize different PAMs. A catalog of 79 CRISPR–Cas9 orthologs is presented here. 10 The type V CRISPR–Cas12 system 11 has variants ranging from Cas12a to Cas12m, each recognizing unique PAM sequences. Cas12 DSB produces sticky ends with 4 or 5 nucleotide overhangs distal to the PAM site.12,13 This enhances efficiency during HDR repair. Cas12f’s small size makes it suitable for a single adeno-associated virus (AAV) vector, facilitating efficient in vivo gene editing.12,14 Cas12 can process pre-crRNA, enabling multiplex genome editing with multiple target sequences within a single transcript. Compared with Cas9, Cas12 demonstrates high specificity and low off-target frequencies, with proven efficacy in editing the genomes of apicomplexan parasites like Eimeria tenella and Plasmodium falciparum.12,15–17
Cas9 and Cas12 are designed to specifically recognize and cleave double-stranded DNA sequences, limiting their use in posttranscriptional regulations and studying noncoding RNAs (ncRNAs). However, type VI Cas13 can functionally characterize coding RNA (mRNA) and ncRNA and conduct posttranscriptional regulation without disrupting DNA. Cas13 binds and cleaves RNA, identifying target sequences with the aid of post-flanking sites.7,18 Cas13 has applications in transcriptome engineering, such as RNA editing, knockdown, and splicing manipulation.7,18,19 It exhibits high efficiency in mammalian and plant cells without causing off-target gene knockdowns, unlike the RNA interference (RNAi) system, making it a promising high-throughput genetic tool for apicomplexan parasites lacking RNAi mechanisms.20–22 DSBs triggered with Cas9 or Cas12 are repaired via nonhomologous end joining (NHEJ), homology directed repair (HDR), or microhomology-mediated end joining (MMEJ), as described later in the text.
NHEJ (Fig. 1a) begins when the Ku70–Ku80 heterodimer binds to a DSB, forming a Ku–DNA complex that recruits other NHEJ enzymes. DNA-dependent protein kinase catalytic subunit (DNA–PKcs) forms a synaptic complex, aligning the broken DNA ends, followed by DNA end processing. Diverse DSB ends, if incompatible, are trimmed or filled by nucleases and polymerases, causing indels. After processing, the DNA ligase complex (DNA ligase IV, X-ray repair cross-, complementing protein 4 (XRCC4), and XRCC4-like factor (XLF) ligates the DNA ends, and Ku is removed from the DNA.23,24 NHEJ occurs in all cell cycle phases. Apicomplexan parasites like Plasmodium, Cryptosporidium, and Theileria lack the classical NHEJ repair mechanism. 25
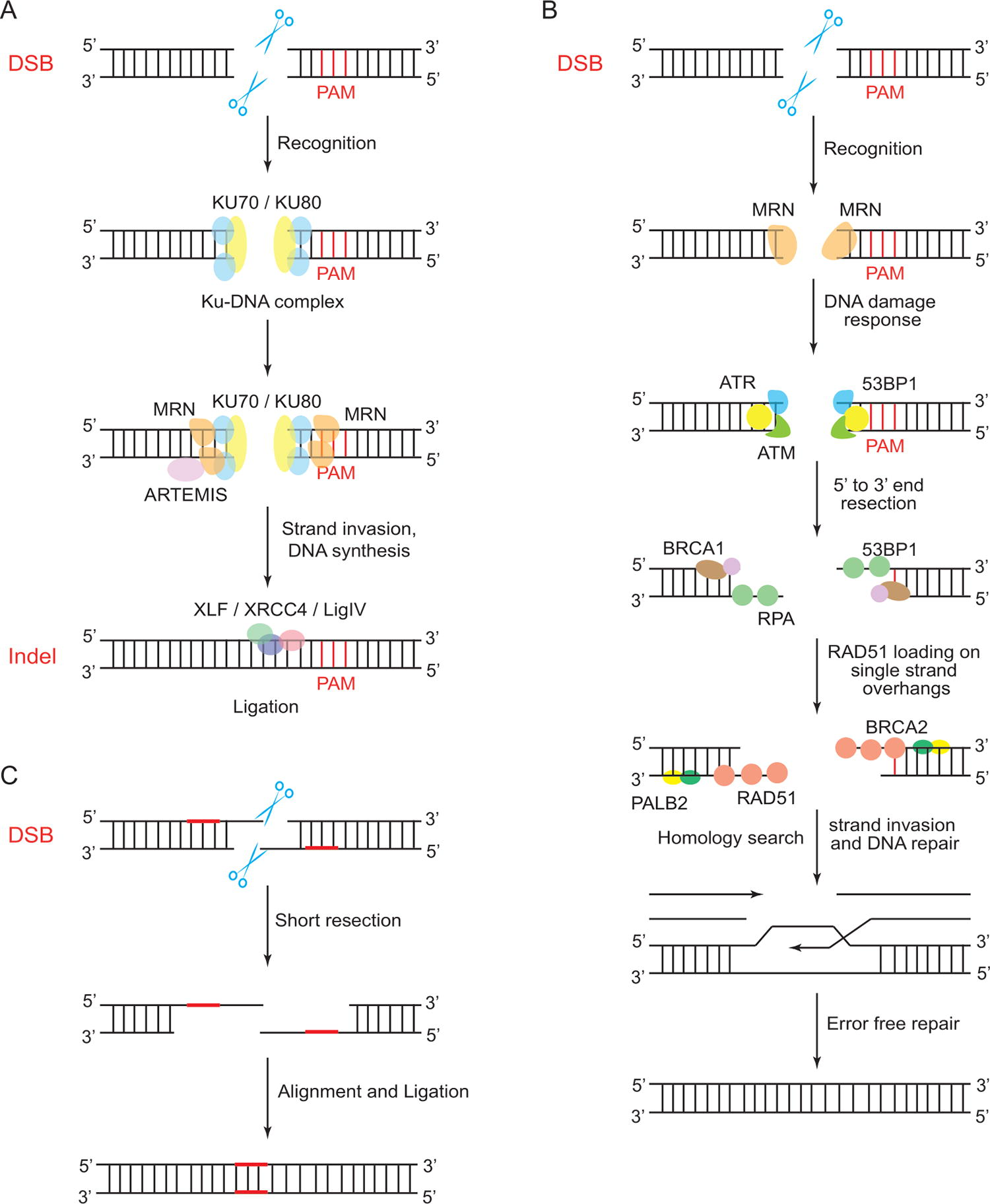
Comparison of NHEJ, HDR, and MMEJ pathways.
HDR is a precise DNA repair pathway requiring a homologous repair template (Fig. 1b). It is active in the late S and G2 phases when sister chromatids are available. An exogenous DNA template with homology arms (HAs) is introduced in HDR-based CRISPR gene editing. HDR begins with DSB end resection, creating 3ʹ single stranded DNA (ssDNA) overhangs that attract recombinases like Rad51 or Dmc1 to form a presynaptic filament. This filament binds the repair template to form a synaptic complex, and the ssDNA invades the homologous region, creating a displacement loop (D-loop). DNA polymerase extends the invading strand, capturing the second DSB end and forming double Holliday junctions (dHJs). dHJs can be resolved by endonuclease resolvase for crossover recombinants or by topoisomerase III for noncrossover products.26,27
In MMEJ (Fig. 1c), also known as alternative NHEJ (Alt-NHEJ), the repair process uses short homologous sequences known as microhomologous sequences near the DSB to guide the recombination of broken ends. The microhomologous regions anneal, and the overhanging 3ʹ ends are cleaved. DNA single-strand gaps are filled, and DNA ends are ligated. MMEJ is error-prone, resulting in deletions of various sizes. In specific contexts of immune system development, MMEJ is robust in mammals. Class switch recombination (CSR) transforms one antibody class into another in activated B lymphocytes and involves DSB generation in the immunoglobulin heavy chain region. XRCC4-deficient mouse B cells demonstrated that CSR proceeds through MMEJ. 28 To illustrate the functionality of MMEJ in Plasmodium yoelii, Xu et al., designed guide RNAs (gRNAs) targeting the P. yoelii circumsporozoite protein (PyCSP) and constructed plasmids, transcribing each gRNA and Cas9 enzyme without a homologous DNA template. 29 Without the homologous template and since Plasmodium spp. lacks the classical NHEJ pathway,25,30 overlapping microhomologous sequences anneal randomly, cleaving overhanging sequences and ligating gaps. Molecular characterization of edited parasites revealed seven unique clones with deletions ranging from 234 to 881 bp, showing that P. yoelii can repair CRISPR–Cas-generated DSB through MMEJ without a homologous template. 29
CRISPR–Cas has been applied to apicomplexan parasites such as Plasmodium, Toxoplasma, Theileria, Babesia, and Cryptosporidium, which are responsible for malaria, toxoplasmosis, theileriosis, babesiosis, and cryptosporidiosis, respectively. Notable applications of CRISPR–Cas technology in apicomplexan parasites include marker-free genome editing, genome-wide screening, and conditional gene expression. Applying CRISPR–Cas in apicomplexan parasites has challenges, including their complex life cycles and the lack of robust genetic tools such as known promoter and terminator sequences for certain parasites within the phylum. This review addresses the status of CRISPR–Cas applications in apicomplexan parasites, the milestones achieved, and the challenges faced. By examining the successes and limitations of CRISPR technology in this specific context, the review intends to pave the way for potential applications.
Gene editing in apicomplexan parasites
The phylum Apicomplexa consists of eukaryotic protozoa, most of which are obligate endoparasitic alveolates closely related to ciliates and dinoflagellates. They can be classified into Gregarines, Hemogregarines, Coccidia, and Hemosporids. 31 Apicomplexa life cycle involves sexual and asexual reproduction, often in different hosts; transmitted by vectors, ingested, inhaled, or transmitted sexually. They possess an apical complex with secretory organelles involved in host cell attachment. 32 However, the participation of the apical complex and secretory organelles during invasion varies among apicomplexans. Apart from Cryptosporidium and Gregarina, all apicomplexans possess an apicoplast, theorized to have arisen from an ancient secondary endosymbiosis with algae.
Antibiotics, vector control, vaccines, pasture management, hygiene, and sanitation are applied to control the spread of apicomplexan parasites. Many antiparasitic drugs exist, but there have been reports of the emergence of antibiotic-resistant parasites. Despite excellent progress in understanding the parasite developmental biology, the hope for effective control methods is yet to be realized. For example, developing a new vaccine takes ∼10–15 years, and one of the challenging processes is identifying candidate antigens in the preclinical phase 33 and for live modified vaccines to identify the genes to target for deletion and/or modification. The CRISPR–Cas system can hasten the identification of these candidate antigens, and it is also a valuable tool for validating gene functions, identifying drug-resistance genes, validating drug targets, and studying host–parasite interactions.34,35 Different CRISPR–Cas techniques are used for apicomplexan parasites depending on the species and the type of modification, which will be discussed later in the text.
Plasmodium parasites
Plasmodium spp. causes malaria in humans, rodents, reptiles, bats, nonhuman primates, and birds.31,36–38 Human malaria is caused by seven species, including P. falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi, but P. falciparum accounts for over 98% of malaria deaths.39–41 The female Anopheles spp. mosquito is the primary host and transmission vector for P. falciparum, while humans and other vertebrates are secondary hosts. An infected mosquito injects sporozoites during a blood meal; the sporozoites migrate to the liver, where they utilize proteins like circumsporozoite protein and thrombospodin-related anonymous protein (TRAP) to invade hepatocytes and mature into schizonts. Schizonts release merozoites into the bloodstream. Merozoites have an apex containing secretory organelles such as rhoptries, micronemes, and dense granules. These organelles invade erythrocytes in a multistage process involving adhesion, reorientation, junction formation, and entry. 42 The invasion process forms a parasitophorous vacuole (PV), protecting the parasite from host phagolysosomes. Clinical manifestations of malaria include fever, chills, headaches, muscle pain, nausea, vomiting, and diarrhea, progressing to brain swelling, anemia, and organ failure in severe cases. 43
Gene editing in human malaria parasites
A P. falciparum CRISPR–Cas9 gene editing system was established in 2014. 44 This approach employed a two-plasmid system (Fig. 2a) to demonstrate gene disruption and integration of a resistance gene through DSB-induced crossover recombination. One plasmid expressed Cas9 protein, and the other expressed single guide RNA (sgRNA) with the HDR donor DNA flanking a selectable marker human dihydrofolate reductase (hDHFR). Co-transfection of P. falciparum-infected erythrocytes with both plasmids was followed by drug selection and Polymerase Chain Reaction (PCR) analysis to check the integration of hDHFR in the target gene. 44 This system’s advantage is targeting multiple genes by introducing additional sgRNA sequences on the plasmids. However, high transfection efficiency is required for the successful co-transfection of the plasmids. This technique has been successfully applied in various apicomplexans.44,45
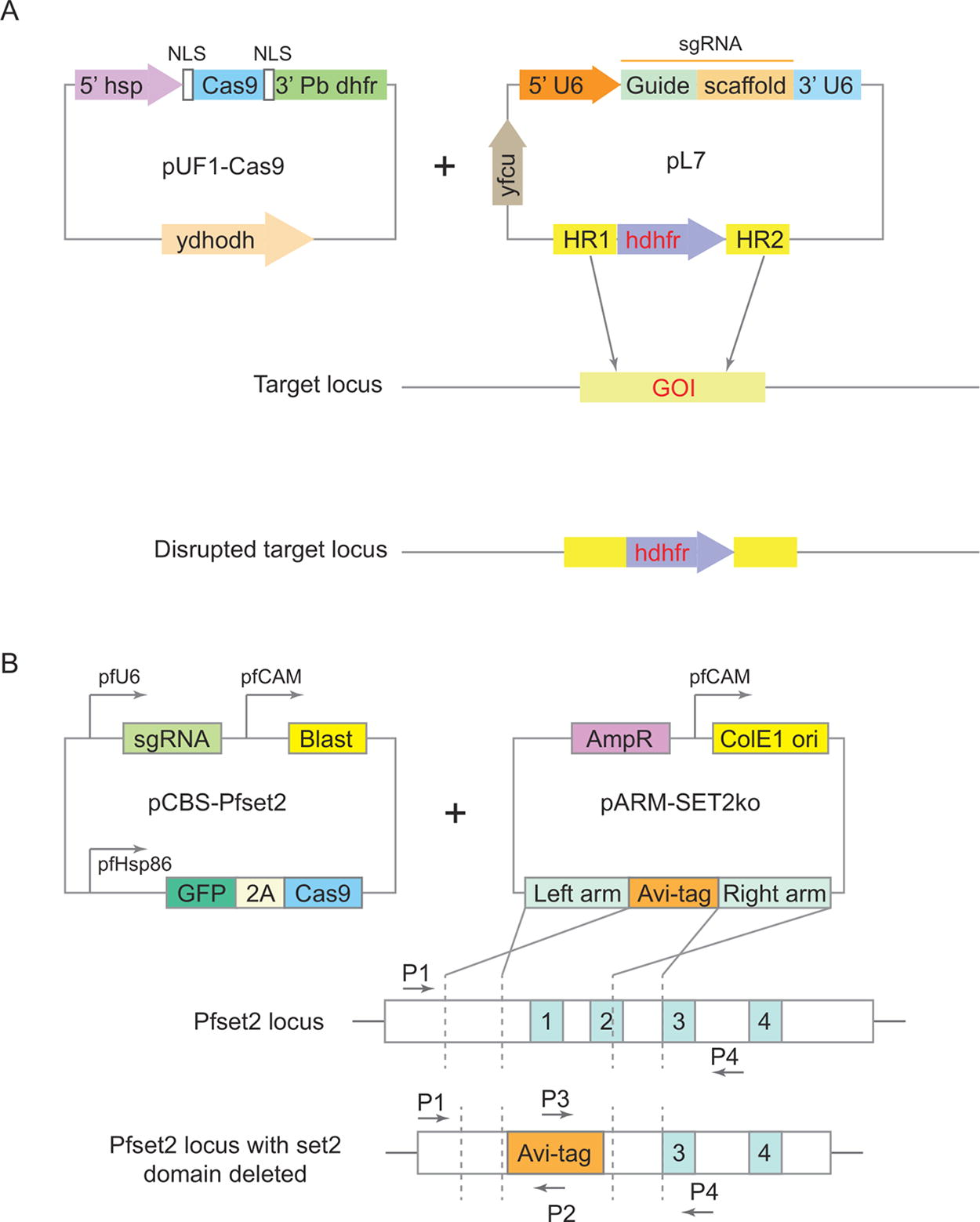
Illustrations of the two-plasmid and the suicide–rescue systems of CRISPR–Cas9 editing.
Marker-free genome editing system
A suicide rescue system for P. falciparum was designed with two plasmids: the suicide vector encoded Cas9 nuclease, sgRNA, and blasticidin selection marker, while the rescue vector contained HDR donor DNA. Parasites receiving only the suicide vector die due to inefficient DSB repair, while those with both vectors survive through HDR repair (Fig. 2b). Positive selection (blasticidin) identifies successfully transfected parasites, while negative selection is through self-destruction due to the lack of a rescue vector. A suicide vector, pCas9-BSD-sgRNA (pCBS), was constructed with expression cassettes for sgRNA, Cas9 nuclease, and blasticidin S deaminase. The PfSET2 gene, nonessential for in vitro blood-stage survival of Pf, was selected for disruption. The rescue plasmid, pARM-SET2ko, was the donor DNA template without a drug-selectable marker for Pf. It contained HAs flanking the Avi-tag. Co-transfection of pGFP-CBS-PfSET2 and pARM-SET2ko into Pf3D7, followed by blasticidin S selection, yielded drug-resistant parasites collected ∼4 weeks post-transfection. PCR confirmed Avi-tag integration, and over time, edited parasites dominated, with the WT (wild type) locus nearly undetectable after day 61. The suicide rescue system successfully generated a marker-free edited population, 46 avoiding the need for a selectable marker in the donor plasmid. The system has the potential to mediate consecutive gene manipulation; however, it requires a high transfection efficiency.
DiCre-mediated plasmepsin V gene disruption
Plasmodium falciparum plasmepsin V (PfPMV) is an aspartic protease that indirectly alters infected erythrocytes’ mechanical and adhesive properties, leading to vascular sequestration. PMV expression increases throughout schizogony. Boonyalai et al. used CRISPR–Cas9 and DiCre-mediated recombination for conditional gene disruption of PMV in the DiCre-expressing P. falciparum B11 clone. 47 A donor plasmid containing loxP sites flanking a synthetic PMV sequence also included 2A peptide sequences, a neomycin resistance gene, and a Green Fluorescent Protein (GFP) gene and was used to disrupt the PfPMV gene. Treatment of the B11 clone with rapamycin-induced excision of the floxed sequence led to a truncated form of PMV lacking a catalytic dyad residue. It also led to the excision of the neomycin resistance gene but induced the expression of GFP through a 2A peptide sequence. PCR confirmed successful PfPMV gene modification. Limiting dilution cloning yielded the PMV-C5 clone in which rapamycin treatment induced site-specific recombination confirmed by GFP-positive parasites. Microscopic observation of Giemsa-stained PfΔPMV mutant parasites showed severe growth defects hindering the ring-to-trophozoite transition, but the parasites appeared morphologically normal. The growth defect suggests that PfPMV is essential for this transition during intracellular growth. 47
Development and application of Cas9i and Cas12 systems in P. falciparum for gene editing
Zhao et al. developed the pUF1-DHODH-Cas9i plasmid to integrate Cas9 endonuclease into the P. falciparum genome. 17 Drug-resistant parasites were selected using DSM1, specific to the yDHODH marker post-transfection. Cas9 integration was screened through drug off/on cycles, and verification was achieved by PCR. Cas9 protein expression was confirmed by Western blotting, identifying a ∼156 kDa protein in the integrative clone F8. Limiting dilution isolated pure clones, with three clones (D3, D5, and F8) confirmed by PCR and Western blotting. The Cas9i system enabled multiplexing by using multiple gRNAs within the same construct, allowing simultaneous editing of K13 and Sir2B genes, verified by PCR, Western blot, and sequencing. In a separate experiment, Zhao et al. established a Cas12 editing system in P. falciparum with the pUF1-Cpf1 plasmid, incorporating AsCpf1 from Acidaminococcus. This plasmid featured a crRNA array targeting Sir2B and ARP6 genes. Mutations in Sir2B and C-terminal tagging of ARP6 were confirmed by PCR, Western blotting, and sequencing. The comparison between Cas9 and Cas12 systems showed that Cas12 could target multiple genes with greater specificity and more target sites due to AsCas12 5′-TTTV-3′ PAM, suited for the AT-rich P. falciparum genome. 17
Silencing Plasmodium virulent genes
Plasmodium falciparum relies on the exclusive expression of virulence genes like the 60-member var gene family encoding Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) for parasite survival in the host. PfEMP1 displayed on infected red blood cells (iRBCs) aids the parasite in immune evasion. The var genes found in specific chromosome regions are controlled epigenetically. One var gene is transcribed around 12 h postinvasion, silenced, and made ready for transcription later in the intraerythrocytic cycle. Another var gene is constantly active and euchromatic, while others are transcriptionally silenced throughout the cycle in repressive clusters. The activation mechanism for the active var gene expression has been unclear. CRISPR interference (CRISPRi) employs a catalytically dead Cas9 (dCas9), downregulating a 15-GC-rich member of a ncRNA gene family, revealing its role in mutually exclusive var gene activation. The sgRNA targeted all GC-rich family members in 3D7 G7 parasites expressing an active var gene.
Reverse transcription–quantitative PCR (RT-qPCR) on CRISPR clones and control groups linked GC-rich gene transcription to the chromosomal location of var genes. When there is one GC-rich gene predominantly transcribed and located adjacent to the 5ʹ region of an active central var gene, it stabilizes the expression of that var gene. The transcription of GC-rich genes is hypothesized to impact the local chromatin structure, making it more accessible to the transcription machinery of the adjacent var gene. This may help regulate var gene expression and, in turn, influence the conversion rate of individual var genes. 48
Host–parasite interactions and disease resistance
A genome-wide functional CRISPR screen of resistance genes in P. falciparum by introducing a genomic library of a drug-resistant strain into drug-sensitive strains, followed by drug screening-confirmed mutant parasites resistance. 35 Whole genome approaches for resistance screening include loss of function or gain of function screens. In loss of function, a gene confers resistance by eliminating its role, while in gain of function, a gene confers resistance by enhancing its function. Drug resistance gene pfmdr1 is a gain-of-function gene inducing resistance into drug-sensitive strains. In contrast, kelch13 is a loss-of-function gene; inserting it into a drug-sensitive strain does not induce resistance. pfmfdr7 was identified as a potential mefloquine-resistant gene through CRISPR knock-in. This approach helps determine the “gain of function” type of resistant genes but not the “loss of function” type of resistant genes. 35 Genes are manipulated to observe effects on disease-related phenotypes, pinpointing potential drug targets. Validation is done through CRISPR knockout or knockdown.
Optimizing CRISPR–Cas editing in Plasmodium knowlesi for use as a surrogate in Plasmodium vivax vaccine development
Host cell invasion by P. vivax and P. knowlesi depends on the Duffy binding proteins (DBP), specifically Plasmodium vivax DBP (PvDBP) and Plasmodium knowlesi DBPα (PkDBPα) which interact with the human RBC Duffy antigen/receptor for chemokines (DARC). The critical binding region of these proteins is the cysteine-rich region 2 (DBP-RII), which shows about 70% similarity between PkDBPα and PvDBP.49,50 PvDBP restricts P. vivax to reticulocytes and acts as a host specificity factor that prevents P. vivax from infecting macaques.51,52 PvDBP-RII is also the primary blood-stage vaccine candidate for P. vivax, with antibodies effectively blocking parasite invasion in ex vivo P. vivax assays.53–56
P. knowlesi possesses two PkDBPα paralogs, DBPβ and DBPϒ, which are highly similar to PkDBPα (68%–88% identity) but bind to different receptors via N-glycolylneuraminic acid (Neu5Gc), a sialic acid present on macaque RBCs but absent on human RBCs. Due to the lack of a long-term in vitro culture system for P. vivax, vaccine development currently relies on recombinant protein assays, low-throughput ex vivo studies, primate infections, or controlled human malaria infections.
Mohring et al. aimed to develop a P. knowlesi parasite line for P. vivax vaccine development. 57 CRISPR–Cas9 genome editing was established in P. knowlesi using an optimized PCR-based method for generating donor constructs. This method involved a three-step PCR process: amplifying an enhanced Green Fluorescent Protein (eGFP) cassette and 400 bp HAs with eGFP cassette adaptors, fusing each HA to the eGFP cassette, and using nested PCR to enhance background removal. The final amplicon was transfected with a plasmid containing Cas9, sgRNA (driven by PkU6 promoter), and a hDHFR-yFCU for negative selection. The sgRNA targeted the nonessential p230p locus. The resulting parasite lines demonstrated a 74% eGFP positivity rate.
Through this PCR approach, Mohring et al. identified critical parameters for effective genome editing including the length of HAs and their distance from the DSB. By varying the length of HAs targeting the p230p locus, they found that HA lengths ranging from 50 to 1600 bp produced integration events, but efficiency declined with HAs shorter than 400 bp. Parasites transfected with 50 and 100 bp HA constructs did not show eGFP positivity by microscopy though PCR confirmed integration. In contrast, constructs with HA lengths over 400 bp showed up to 81% eGFP positivity. They conclusively determined that increasing HA length proportionately enhanced integration efficiency. Additionally, they used the same p230p locus and moved the 400 bp HAs between 0 to 5 kb away from the DSB. They showed that integration efficiency declined with distance from the DSB. They also demonstrated that increasing the HA length from 400 to 800 bp at a 5 kb distance from the DSB improved integration efficiency from 14% to 54.8%.
Using these tools, they replaced the P. knowlesi PkDBPα gene with its PvDBP ortholog and deleted the P. knowlesi DBP paralogs, creating a transgenic P. knowlesi line (PkDBPαOR/Δ14Δϒ) that relies on the PvDBP protein for RBC invasion. Despite deleting the P. knowlesi DBP paralogs, the transgenic parasites multiplication rate in macaque RBCs did not decrease but slightly increased. This transgenic line represents a vital tool for P. vivax vaccine development. 57
Gene editing in rodent malaria parasites
Advancements in CRISPR–Cas9 genetic modification in Plasmodium berghei
Shinzawa et al. integrated the Cas9 nuclease gene into the P. berghei genome at the C-type subunit (CSSU) locus using a plasmid with positive (DHFR) and negative (5-FC) selection markers. 58 The resulting transgenic parasite, pbcas9, was verified to express Cas9 and showed average growth and development in various stages, indicating no significant disruption of the parasite’s life cycle. They tested a linear donor template to address technical challenges in genetic modification, particularly unexpected recombination issues with circular donor DNA. By introducing linear donor DNA and a plasmid containing guide RNA into pbcas9 parasites, they achieved successful site-directed mutagenesis and gene deletions with high accuracy. This method introduced a nonsense mutation and a 41-bp deletion in the IMC1I gene. Using linear donor DNA improved transfection efficiency and minimized unwanted recombination, thus ensuring precise genetic modifications. Shinzawa et al. further explored large-scale genome editing by utilizing donor templates with telomere sequences. This approach allowed the creation of new chromosome ends, facilitating extensive chromosomal deletions. Targeting the subtelomeric region containing the MFR1 gene demonstrated that large-scale deletions could be achieved without affecting parasite development. The results showed successful integration of telomere sequences and accurate chromosome modifications, confirming the effectiveness of this method for extensive genetic alterations. 58
Efficient gene editing in P. yoelii
Zhang et al. utilized three Cas9-mediated genetic modifications: gene deletion, reporter knock-in, and nucleotide replacement. For gene deletion, a plasmid containing Cas9, sgRNA, and a homologous repair template was used to target the P. yoelii SERA1 (PySERA1) gene, which encodes a nonessential serine protease. PCR and sequencing confirmed a complete 5 kb deletion in one of the edited clones, achieving 100% efficiency. In the reporter knock-in experiment, a separate plasmid with GFP in the donor template targeted the Py03652 locus. GFP expression was detected in 38% of the parasite clones via fluorescent activated cell sorting (FACS), with 22% showing successful replacement of the Py03652 locus after cloning, as confirmed by PCR and sequencing. The lower efficiency of knock-in (22%−38%) compared with gene deletion (100%) remains unexplained. In the nucleotide substitution experiment, a plasmid introduced a silent mutation in the PyHSP70 gene, incorporating an AatII site for restriction fragment lenght polymorphism (RFLP) analysis. To protect the donor DNA from Cas9, a three-nucleotide substitution was added at the sgRNA binding site. RFLP analysis showed a 25% editing efficiency. The lower efficiency of nucleotide replacement may be due to the functional constraints of the targeted genes. Despite these challenges, these modifications provide valuable insights for functional studies by enabling precise DNA alterations and gene expression regulation. 59
Toxoplasma gondii
Toxoplasma gondii, a zoonotic parasite, infects various nucleated mammalian and avian cells. 60 Highly adaptable, it is a model organism amongst apicomplexans, the most experimentally amenable, and was the first transfected apicomplexan 61 and notably the first apicomplexan with a CRISPR–Cas9-based genome-wide loss of function screening. 62 Felids, acting as definitive hosts, produce fecal oocysts that contaminate soil, water, and food. 60 Toxoplasma gondii has three infectious stages: tachyzoite, bradyzoite, and sporozoite. Host cell invasion involves attachment, penetration, moving junction formation, PV creation, and host cell membrane closure. 63 Invasion involves the consecutive release of three secretory organelles, namely, micronemes, rhoptries, and dense granules, playing critical roles in parasite motility, adhesion to the host cell, PV protection from lysosomal destruction, and tachyzoite to bradyzoite differentiation.64,65 Clinical symptoms in felids range from pneumonia to encephalitis, particularly in felids with immune deficiency syndrome. 60 In immunocompromised humans, severe symptoms like ataxia, confusion, and coma can occur. Prenatal infection may lead to congenital disorders like hydrocephalus and blindness, with potential abortion.60,66,67
Attenuation of T. gondii strains
Despite 98% genetic similarity, T. gondii strains have significant differences in virulence. Type I strains are lethal to mice at low doses, while types II and III are less virulent. Type II strains are commonly associated with toxoplasmosis, while type III strains are not. Type I strain causes severe infections in neonates, eye infections, and AIDS-related encephalitis. ROP18, a rhoptry-secreted protein, shows significant genetic variation among strains. Its expression level in type I is high, and in type III, it is almost undetectable, suggesting a pivotal role in acute virulence variation. 68 Shen et al. used CRISPR–Cas9 to validate ROP18’s role. 69 A sgRNA targeting the 5ʹ coding region of ROP18 in GT1 and RH type I strains and a donor template with a DHFR-selectable marker were used. Transfected parasites were selected with pyrimethamine, and Western blot analysis confirmed the loss of ROP18 expression. When RHΔROP18 mutant parasites were injected into mice at various dosages and monitored for 30 days, their survival was prolonged for 2–3 days, compared with wild-type tachyzoites, meaning the mutants were slightly attenuated. Conversely, 75% of mice injected with GT1Δrop18 parasites survived the challenge with 300 tachyzoites, but the challenge with 3,000 tachyzoites led to 60% mortality. 69 ROP18’s role in type 1 strain virulence was confirmed. Further, using CRISPR, GRA12, and TGME49_289150 genes were knocked out. Mice infected with GRA12 mutants had a complete absence of cysts in the brain, and those infected with TGME49_289150 mutants had overall fewer cysts than the control infections, suggesting the potential for live attenuated vaccines. 70
A stable expression system for genome-wide CRISPR screening
In 2016, a stable expression system was established in the T. gondii RH strain. 34 This technique utilizes constitutive Cas9 expression, which was detrimental to the parasite; therefore, a nonfunctional (decoy) sgRNA was introduced to prevent unintended Cas9 activity, successfully yielding Cas9-expressing parasites. A genome-wide genetic screen utilized a library of sgRNAs with 10 guides against each of the 8,158 predicted T. gondii protein-coding genes. The sgRNA library incorporated into an expression vector served as barcodes to assess the impact of each gene on the parasite’s fitness during human fibroblast infection. The study identified 200 indispensable conserved apicomplexan proteins (ICAPs) lacking functional annotation. CRISPR-mediated endogenous tagging of 28 ICAPs identified claudin-like apicomplexan microneme protein (CLAMP) as an essential invasion factor localized to micronemes. The essentiality of the CLAMP homolog, PfCLAMP, was confirmed in P. falciparum during the asexual cycle. 34
Efficient gene knock-ins in T. gondii
Toxoplasma gondii, easily cultured with high transfection rates and genetic amenability due to its balanced nucleotide composition, 34 faces challenges in efficient gene replacements, targeted knockouts, and knock-ins due to its preference for NHEJ. NHEJ is initiated when Ku70 and Ku80 proteins form a heterodimer tightly binding DSB DNA ends. Deleting the Ku80 gene significantly decreased NHEJ activity, enhancing HDR efficiency to nearly 100%. In Ku80 mutants, gene replacement efficiency increased by up to 400-fold compared with wild-type strains. 71 CRISPR–Cas9 effectively drove HDR in Ku80 mutants, introducing the Ty tag to the Calcium-dependent protein kinase 3 (CDPK3) protein in T. gondii. Sidik et al. designed a plasmid pU6-CDPK3-Ct to target the CDPK3 gene. The donor template contained the Ty epitope tag sequence and flanking homologous regions for in-frame integration within CDPK3. Both KU80 and wild-type parasites were transfected with pU6-CDPK3-Ct and the donor template. Quantification on the third day post-transfection showed ∼5% endogenous tagging in wild-type parasites and 15% in ΔKU80 parasites. The successful CDPK3 tagging demonstrated efficient gene knock-ins without NHEJ. 72
Babesia bovis
Babesia parasites are intraerythrocytic apicomplexan parasites that induce babesiosis after transmission by ticks. They have two classes of hosts: an invertebrate and a vertebrate host. Invertebrate hosts are ixodid ticks except for Ornithodoros erraticus a non-ixodid tick that is a reservoir for Babesia meri. Vertebrate hosts include mammals and birds. 73 Babesia bovis transmitted by Rhipicephalus microplus, causes babesiosis in cattle. Once a tick feeds on an infected animal, the parasite begins to develop new organelles, most notably the development of arrowhead-shaped organelles called ray bodies. 73 These ray bodies are involved in the fusion of gametes to form zygotes, which use the arrowhead to penetrate the epithelial cells of the tick gut. From the gut, the parasites migrate to the salivary gland. Sporozoite development takes place within the salivary gland, and they mature only after the tick starts to feed again. Once in the bloodstream, Babesia sporozoites directly invade the RBCs.73,74 Inside the RBC, the parasite rapidly escapes from the PV that is formed by the invagination of the RBC membrane during the invasion. The parasites divide by binary fission and then lyse the RBC, releasing more parasites into the circulation. These parasites rapidly re-invade RBCs, continuing the infection cycle. Some parasitized RBCs (pRBCs) differentiate into gametocytes which are ingested by blood-feeding ticks, completing the sexual stages of the parasites’ life cycle.73,75 Infected animals suffer acute and chronic hemolytic anemia as the parasite replicates in RBCs, disrupting their structure and function. B. bovis infections can lead to severe complications, including cerebral babesiosis, respiratory distress, and multi-organ failure, due to pRBC accumulation in the microvasculature. 75
CRISPR/Cas9-mediated epitope tagging, point mutation and gene replacement in Babesia bovis
Babesia bovis was first edited in 2019, where Hakimi et al. demonstrated the use of the CRIPSR–Cas9 system in epitope tagging, introducing a point mutation and gene replacement. 76 Individual plasmids were designed for all three processes. First, they constructed a plasmid to insert a sequence encoding a 2-myc tag epitope at the 3ʹ end of B. bovis spherical body protein 3 (SBP3) open reading frame (ORF). Cas9 and hDHFR were simultaneously driven by the ef-1α intergenic region (ef1-α IG), which functions as a bidirectional promoter. In contrast, a 20-nucleotide sgRNA was driven by the B. bovis U6 spliceosomal RNA promoter. SBP3 was selected due to its expression in the cytoplasm of parasite-infected RBCs (iRBCs) and its characteristic staining pattern. 77 The iRBCs were transfected with a single circular plasmid, and transgenic parasites were observed 10 days after selection with the DHFR inhibitor WR99210. PCR confirmed insertion of the myc-tag. An indirect immunofluorescence antibody test performed using an anti-myc antibody confirmed that the myc-tag was fused to SBP3. A survey of 100 parasites revealed that all were fluorescence-positive, indicating efficient tagging and a minimal presence of wild-type parasites. Secondly, to introduce a point mutation, they created a plasmid with Cas9, hDHFR, and a donor DNA from a thioredoxin peroxidase 1 (TPX1) ORF containing a mutation that changes the peroxidatic Cys (Cys49) to Ser. TPX1 is a cytoplasmic peroxiredoxin with an essential peroxidatic Cys at its catalytic site functioning to reduce the peroxide substrate. 78 After the appearance of transgenic parasites, the tpx-1 ORF was amplified and sequenced, confirming the presence of the desired mutation. Transfectants from two different sgRNAs showed varying efficiencies from mixed wild-type and mutant populations to nearly pure mutants. Finally, to validate the CRISPR–Cas9 system for gene knockout in B. bovis, they targeted TPX1 gene, nonessential in the erythrocytic stage. They designed a single plasmid to replace TPX1 ORF with gfp ORF. The presence or absence of the TPX1WT locus was evaluated by PCR showing a 1.6 kb DNA fragment for the WT parasites and no amplified product for the transgenic parasites. Their results indicated a pure transgenic parasite population. Southern blot analysis evaluated whether the transfected plasmid was episomally maintained or integrated into the genome. The results indicated the integration of the CRISPR–Cas9 plasmid into the genome. Fluorescent microscopy of over 100 parasites showed GFP expression from all the parasites. 76
Cryptosporidium parvum
Cryptosporidium, a zoonotic enteric parasite, globally causes gastroenteritis in humans and domesticated animals, leading to potentially fatal cryptosporidiosis in immunocompromised individuals.79,80 Cryptosporidium parvum is not an obligatory intracellular parasite because it does not require host cell invasion to complete its life cycle. 81 The life cycle involves both sexual and asexual development stages, initiated when a host ingests or inhales sporulated oocysts. After surviving the stomach’s acidic environment, oocysts release sporozoites in the small intestine. These sporozoites, structurally similar to merozoites, possess an apical complex with organelles like rhoptries, micronemes, conoids, and dense granules. 82 Apical complex proteins such as circumsporozoite-like antigen, glycoprotein 900 (GP 900), glycoprotein 40 (GP40), and thrombospondin-related adhesive protein facilitate sporozoite attachment through ligand–host receptor interactions. The PV of C. parvum is unique, located at the host cell’s surface rather than deep within its cytoplasm. Sporozoites reside in an extracytoplasmic compartment, separated from the enterocyte’s cytoplasm by an electron-dense band formed through host actin accumulation during the initial invasion. Cryptosporidium is thus described as an intracellular but extracytoplasmic parasite.83,84
Cryptosporidium parvum was first edited in 2015 when thymidine kinase (TK) gene was knocked out, and luciferase and neomycin were introduced through HDR in sporozoites. 85 Modified parasites, tested in mice treated with paromomycin, showed detectable luciferase 6 days after infection. This study established a CRISPR–Cas editing system while demonstrating the nonessential nature of the TK gene. Lack of the TK gene increased susceptibility to antifolates. 85
A protein degradation system in C. parvum
The effect of calcium-dependent protein kinase 1 (CDPK1) downregulation was demonstrated in multiple stages of the C. parvum life cycle. CRISPR–Cas9 was used to attach a 3× hemagglutinin epitope (3×HA) tag and an E. coli DHFR degradation domain (DDD) at the C-terminus of CDPK1 forming the unstable fusion protein CDPK1-HA-DDD. 86 Cryptosporidium parvum proteasome can degrade this fusion protein. Trimethoprim was introduced to regulate CDPK1 expression in vivo and in vitro due to its affinity to DDD. In its presence, the fusion protein stabilized, promoting parasite growth, while its absence led to degradation and growth inhibition. Fluorescence-labeled antibodies specific to the hemagglutinin tag visualized the presence and localization of CDPK1. CDPK1 was found to be expressed during merozoite development in the asexual proliferation of C. parvum. 86
Theileria parva
Theileria parva, an obligatory intracellular parasite causing East Coast fever in cattle, is transmitted by the brown ear tick, Rhipicephalus appendiculatus. It infects and transforms bovine mononuclear cells, causing severe uncontrolled lymphocyte proliferation. African buffalos, also affected, serve as asymptomatic carriers, transmitting the parasite to cattle. 87
Sporozoites enter the host through tick saliva, rapidly invading lymphoid cells. They lack motility and an apical complex; therefore, they enter the host cell in any orientation after attachment. The invasion involves continuously zippering closely opposed parasite and host membranes, shedding the sporozoite surface coat, and internalizing within the host cell.88,89 Theileria parva does not form a PV; instead, tightly apposed host and parasite membranes separate after complete internalization. The host membrane dissolves, releasing the parasite into the host cell cytoplasm, where host cell-derived microtubules form around the parasite. Secretory organelles, including rhoptries and micronemes, play a significant role in establishing T. parva within the host cytoplasm, distinguishing its entry process from other apicomplexans.88,89
Ongoing research on T. parva has not yet yielded publications on gene editing. De Goeyse et al. performed the first successful transient transfection of T. parva sporozoites using a 4D-Nucleofection™ System. 90 Traditionally, transfection in Theileria has been challenging due to the need for plasmids to cross two membranes (host lymphocyte and parasite membrane). Here a novel protein expression vector, pMotif-EF-1–100, which is based on the mammalian vector pRNAi-GL but modified with the T. parva elongation factor 1 alpha (EF-1α) promoter was used. This plasmid includes a transmembrane Azami Green sequence for fluorescence visualization. The sporozoites were freshly prepared from salivary glands of ticks, as cryopreserved sporozoites were not effective. The successful transfection was determined by observing fluorescence using a GFP filter, which corresponded with 4′,6-diamidino-2-phenylindole and Polymorphic Immunodominant Molecule staining. Transfection efficiency was low, with results comparable to those seen in T. annulata (0.3–0.9%).90,91 Similarly, efforts in our labs have yielded transient transfection of fresh sporozoites using electroporation. These studies mark the initial step in the genetic manipulation of T. parva. The successful development of a stable transfection method could significantly advance gene editing research.
Editing in other apicomplexan parasites
Other apicomplexan parasites successfully edited through CRISPR–Cas include Neospora caninum 92 and Eimeria tenella. 16 Challenges like low transfection efficiency, common in most apicomplexans compared with other eukaryotic organisms,90,93 affect high-throughput screening due to reduced coverage. They also introduce variability in experimental outcomes resulting from challenges in accurately interpreting the results. Developing methods to enhance transfection efficiency would be highly advantageous. Other challenges are described in the next section.
Challenges of editing apicomplexan parasites
Delivery of editing components
Efficient delivery of CRISPR components into the target nucleus is crucial in gene editing. These components can be delivered physically, chemically, or by viral vectors. Physical methods applicable to apicomplexa parasites include electroporation and microinjection.94,95 This review focuses on electroporation, microinjection, and viral vectors. Electroporation involves applying an electrical pulse to cell membranes, creating temporary pores for molecule transport. The main disadvantage is the loss of cell viability. 94 Apicomplexan parasites reside within the host cells, rendering them inaccessible except for a transient sporozoite stage. Identifying ideal electroporation conditions to facilitate payload transfer through host cells into the parasite nucleus is crucial. The schizont stage of P. falciparum and the tachyzoite stage of T. gondii have been electroporated successfully.96,97
Microinjection, a physical method for introducing molecules into cells, is based on Barber’s technique for inoculating bacteria into living cells. 95 The micropipette’s tip is injected through the cell membrane, delivering contents into the cytoplasm under a microscope. While theoretically simple, injecting one cell at a time is laborious, substantially limiting the number of transfected cells per experiment. This is technically challenging for apicomplexans due to their small size, e.g., 1 µm T. parva sporozoites. 98 Skilled handling is vital to avoid cell damage. In addition, microinjection apparatuses are expensive, making them hard to implement in many laboratories. However, it is ideal for manipulating the intracellular parasite stages because it enables manipulation of the cellular internal environment.
Viral-based deliveries use retroviruses, adenoviruses, and AAVs to carry Cas9 and guide RNA into a cell. Retroviruses and AAVs ensure long-term transgene expression, while adenoviruses provide transient expression.99,100 Attempts to identify protozoan viruses are promising. Cryspovirus from Cryptosporidium species belongs to the Partitiviridae family and is linked to plants and fungi. 101 Matryoshka RNA virus 1 (MaRNAV-1) from P. vivax shares high sequence similarity to narnaviruses found in fungi, plants, and other protists. Leptomonas seymouri, harbors a symbiotic narna-like virus.102,103 Viral vectors are valuable for genetic manipulation, and exploring virus-like particles from apicomplexan-affecting viruses may enhance gene delivery to parasites, though further research is needed.
In vitro cultivation
In vitro cultivation is essential for the genetic manipulation of organisms due to the difficulty of studying genes in vivo. Cell culture is complicated for apicomplexan parasites because one must mimic the host’s nutritional and environmental conditions. Cryptosporidium, Eimeria, Sarcocystis, Neospora, Toxoplasma, Plasmodium, Babesia, and Theileria have known cell culture techniques but are limited to specific parasite stages. Theileria parva can only be cultured in the schizont stage, employing the immortalization of the lymphocytes that the sporozoites are infecting. 104 Manipulating the schizont is difficult due to their sequestration within host cells and the complexity of targeting multiple nuclei in a macro schizont. There are no known tissue culture procedures for maintaining other life cycle stages in culture. Plasmodium spp. benefit from robust in vitro cultivation, 105 allowing the availability of various genetic tools, including stable transfection methods. 106 It is possible to culture all life cycle stages of P. berghei. 107 Advancements in in vitro culture technologies for other apicomplexan parasites will facilitate broader biological studies and genetic manipulation for controlling their pathology.
Incomplete gene annotations
While genome projects to sequence apicomplexan parasites have identified an extensive catalog of genes, many of their functions remain unknown and are listed as hypothetical proteins [VEuPathDB]. This makes it difficult to identify suitable gene editing targets.
Complex parasite life cycle
Apicomplexan parasites have a complex life cycle comprising multiple stages, each with unique biological and physiological characteristics. The diversity of life stages complicates the development of universal transfection techniques, requiring tailored strategies for each stage. The parasites often reside within host cells; thus, manipulating them is technically challenging, as the interactions between the parasite and host cell can affect the efficiency of genetic delivery and expression. Additionally, some stages, such as Plasmodium and Theileria schizonts, possess multiple nuclei within a single cell.89,108 This multinucleate condition complicates the delivery and integration of transgenes, as ensuring that genetic material reaches and integrates into all nuclei simultaneously is technically demanding. Protective barriers, like the thick oocyst wall in Toxoplasma, impede the efficient introduction of genetic constructs and must be overcome for successful transfection. Developing stage-specific selectable markers is challenging, yet it is crucial for precise selection and maintenance of transgenic lines. 62
Ethical and policy considerations in gene editing
The desire for live attenuated vaccines has led to exploring the potential of using CRISPR–Cas for live attenuated vaccine development. One candidate is the protozoan parasite Leishmania, transmitted by sandflies. Before the extensive use of CRISPR–Cas technology, conventional methods were used to knock out the centrin-1 gene in Leishmania donovani. Centrin-1 is crucial for the parasite’s growth and differentiation. This knockout affected duplication and cell division in amastigotes, leading to apoptosis and cell arrest in the G2/M phase, resulting in easy clearance by the immune system. In animal models, the mutant parasites did not survive beyond 5 weeks, and the animals developed long-lasting Th1 protective immune responses against L. donovani and other Leishmania species.109,110 Although these centrin-1 deficient parasites (LdCen−/−) showed potential as live attenuated vaccines, the use of an antibiotic-resistance marker prevented progression to human trials. 111 A subsequent study revealed that LdCen−/− had unexpected deletions ranging from 350 to 6900 bp in various chromosomal regions. 112 These findings underscore the need for thorough product characterization, especially when developing CRISPR-based vaccines. CRISPR technology, while powerful, can induce unintended on- and off-target effects, including unwanted indels or large structural variants such as translocations, inversions, and duplications impacting cellular health and function. 113 In further efforts, CRISPR–Cas9 was employed to achieve a centrin-1 gene knockout in Leishmania donovani, L. mexicana, and L. major, aiming to create a live attenuated vaccine without antibiotic resistance markers. While the knockout was unsuccessful in L. donovani, it was achieved in L. mexicana and L. major. Leishmania mexicana showed in vitro attenuation but not ex vivo, whereas L. major elicited protective immunity in animal models and reported no detectable unintended off-target effects.114,115
Various insect species are vectors for apicomplexan parasites. One proposition for controlling these diseases is eradicating the vectors or making them refractory to parasites. CRISPR–Cas9 has enabled gene-drive technology, successfully eradicating the Anopheles gambiae mosquito population in a laboratory within a year. 116 Gene drive is a genetic engineering technique that modifies genes to alter the chances of inheritance. Ordinarily, there is a 50% chance that any gene will be passed from parent to offspring; however, gene drive increases this chance to almost 100% in favor of a particular gene. In this case, when a mosquito carrying the gene drive mates with one that does not, CRISPR–Cas in the gene drive is triggered. It recognizes the natural gene copy in the opposite chromosome and cuts it out before embryonic development begins. This technology can potentially drive a species to extinction. However, several concerns are raised about gene drives. Is it ethical to deliberately drive a species to extinction? What are the implications for the ecosystem? Which body will decide whether to release a gene drive that can surpass geographical barriers? On the surface, gene drives appear to have significant potential for humans, but eradicating an entire species will require the organized effort of all governments, scientists, and the public. The potential for resistance development in gene drives, as demonstrated by CRISPR-based drives in mosquitoes, raises ethical concerns. Resistant mutations can eventually replace the drive in a population, undermining the intervention’s effectiveness and potentially causing unforeseen ecological consequences. 117 Similarly, a CRISPR-based live attenuated parasite can undergo mutations in the environment. Although the initial genetic modification may be stable, natural selection, environmental pressures, and random genetic drift can accumulate additional mutations over time. These mutations could potentially restore virulence or cause other unintended consequences. Alternatively, gene drives can be engineered to include an effector molecule 118 that makes mosquitoes refractory to the malaria parasite, thereby reducing disease transmission in the target population. 119 This gene drive is designed to eliminate the malaria parasite without eliminating the mosquito.
CRISPR has extensive potential for beneficial use, but like most other technologies, it also has potential for misuse, hence its regulation. Before genome editing techniques, genetically modified organisms (GMOs) involved introducing foreign genetic material into an organism, usually from the genome of another organism. In contrast, gene editing typically involves making precise changes to an organism’s existing DNA without necessarily adding genetic material from another species. Some countries regulate gene editing products as GMOs; however, the main contention with GMOs is DNA transfer between genetically unrelated organisms, which consumers often perceive as unnatural. Scientists and regulators proposed terminologies to distinguish GMO products based on the origin of the introduced DNA. Transgenics involve DNA from a sexually incompatible organism, while cisgenics involve DNA from a sexually compatible organism. The idea was that cisgenic organisms should attract less regulation. Similarly, gene editing has been categorized into three types: Site Directed Nucleases (SDN) -1, SDN-2, and SDN-3. In SDN-1, the organism relies on its own cellular repair mechanisms. In SDN-2 and SDN-3, a template is constructed instructing the organism on how to repair. Minimal repairs fall under SDN-2, while introducing long genetic sequences from a sexually incompatible organism falls under SDN-3. Scientists argue that SDN-1 and SDN-2 processes resemble natural mutations or those occurring through breeding and thus should face less strict regulations. 120 In Kenya, for example, gene-edited products are considered GMOs if a foreign gene or DNA, such as reporter genes or selectable markers is inserted in the end product. Knockouts without inserting foreign DNA are not regulated under the Biosafety Act, 121 meaning a gene-deleted parasite without foreign DNA can be used and regulated as an ordinary vaccine.
Conclusion and future perspectives
The future of genome editing in apicomplexans looks promising, particularly with the diverse applications of CRISPR-associated proteins like Cas9, Cas12, and Cas13. The widely used Cas9 from Streptococcus pyogenes recognizes the 5′-NGG-3′ PAM, which may be more effective in GC-rich genomes and less so in AT-rich ones. Bioinformatics analyses have revealed new Cas9 orthologs with different PAMs, sizes, and Trans-activating CRISPR RNA (tracrRNAs), such as the 5′-NAAN-3′ PAM from Streptococcus macacae, which is beneficial for targeting the AT-rich genome of Plasmodium. Additionally, near-PAM-less Cas9 variants, which rely mainly on target-spacer complementarity rather than strict PAM sequences, can increase target accessibility and overcome the complex apicomplexan genome. 122
Cas12, a smaller nuclease, offers advantages for editing apicomplexan genomes due to easier delivery into smaller parasites and the potential for AAV-mediated delivery. Smaller Cas12 proteins provide more space in AAV vectors for additional elements, making the gene editing system more functional. With ongoing efforts to identify protozoan AAVs, Cas12 will be valuable for delivering CRISPR components into these parasites. Cas12a’s ability to recognize the PAM sequence 5′-TTTV-3′ allows for more target sites than SpCas9. More Cas12 effector variants are being discovered to improve cleavage accuracy and increase PAM flexibility for editing. Cas12 also supports multiplexing, enabling simultaneous targeting of multiple genes, which is useful for complex modifications in apicomplexans.13,122
Despite advances, many genes in apicomplexans remain functionally unexplored. RNAi has limitations in some apicomplexans due to the absence of RNAi-inducing genes.20–22 Cas13, which targets RNA, presents new opportunities for studying gene expression and regulation. The newly discovered Cas13d improves RNA silencing efficacy. 122 Conditional tools are needed in apicomplexans to regulate gene expression at the genome, transcriptome, or protein levels. Additionally, the absence of NHEJ in some apicomplexans limits the application of high-throughput CRISPR–Cas9 screens. Cas13 could enhance the functional characterization of multigene families and accelerate drug discovery.
An effective gRNA must maximize its activity on the intended target while minimizing potential off-target effects. Achieving this balance has led to creating computational tools designed to aid in gRNA development. However, accurately predicting gRNA performance regarding both activity and specificity remains challenging. One reason is that gRNA activity is significantly affected by the context of the target sequence, including the surrounding PAM site sequence and the local structure of the target site. 122 Additionally, the binding and activity of CRISPR complexes at endogenous targets can be obstructed by other DNA-binding proteins that regulate transcription or chromatin accessibility. Another crucial factor affecting gRNA activity is its secondary structure. Many existing models have concentrated mainly on the thermodynamics of interactions between the spacer and the DNA target, overlooking other aspects of gRNA folding. Advances in developing models that integrate the biophysical aspects of gRNA folding and target sequence context, based on empirical data from extensive experiments, are expected to enhance gRNA design. These models should accurately reflect how modifications to gRNA design—like shortening the spacer or adding aptamers—impact gRNA function. 122 Such progress could reduce or eliminate the need for experimental testing of new gRNAs and speed up the development of CRISPR tools for precise gene editing or regulation in apicomplexans and beyond.”
Footnotes
Authors’ Contributions
E.W., H.A., G.O., and L.S. conceived and designed the article. E.W. did literature search and writing. H.A., G.O., P.N., V.N., and L.S. critically revised the article for important intellectual content. S.X. and S.Z. prepared the figures. L.S. took care of funding acquisition.
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
This work was funded by the United States Agency for International Development (USAID) Bureau for Resilience and Food Security under Agreement # 7200AA20CA00022 as part of Feed the Future Innovation Lab for Animal Health.