
Abstract
Gene editing in human induced pluripotent stem (iPS) cells with programmable nucleases facilitates reliable disease models, but methods using double-strand break repair often produce random on-target by-products. Prime editing (PE) combines Cas9 nickase with reverse transcriptase and PE guide RNA (pegRNA) encoding a repair template to reduce by-products. We implemented a GMP-compatible protocol for transfecting Cas9- or PE-2A-mCherry plasmids to track and fractionate human iPS cells based on PE expression level. We compared the editing outcomes of Cas9- and PE-based methods in a GFP-to-BFP conversion assay at the HEK3 benchmark locus and at the APOE Alzheimer’s risk locus, revealing superior precision of PE at high expression levels. Moreover, sorting cells for PE expression level influenced allelic editing outcomes at the target loci. We expect that our findings will aid in the creation of gene-edited human iPS cells with intentional heterozygous and homozygous genotypes.
Introduction
Human induced pluripotent stem cells (iPS cells) are produced from somatic cells and have indefinite proliferative and differentiation potential.1–3 In addition, they can differentiate into multiple cell types while retaining their normal diploid karyotypes and genome from donors. Based on these characteristics, iPS cells are widely used as models to study human genetic diseases on a cellular level. Genetic disease modeling is generally achieved by correcting mutations in iPS cells derived from patients with hereditary diseases or by introducing mutations into iPS cells prepared from healthy donors without diseases. 4 In addition, modifying a target site by deleting, inserting, or replacing specific DNA sequences provides isogenic control cells to study pathogenic variants. 5
Point mutations represent almost 90% of the variants registered in dbSNP 6 and 58% of disease-related polymorphisms registered in the ClinVar database. 7 Precision gene editing technologies are required to reproduce these disease mutations in iPS cells at single nucleotide resolution. 8 The CRISPR-Cas9 system is conventionally used to generate targeted DNA double-strand breaks (DSBs) in the genome, which are subsequently repaired by cellular DNA repair pathways.9,10 Nonhomologous end-joining (NHEJ) results in insertion and deletion (indel) mutations, while microhomology-mediated end-joining (MMEJ) leads to predictable deletions. 11 Both NHEJ and MMEJ are collectively known as mutagenic end-joining (MutEJ), as both repair outcomes lead to the loss or gain of DNA sequence. 12 Typically, to produce specific changes, including single nucleotide variants, the homology-directed repair (HDR) pathway is exploited with custom repair templates containing the desired edit, such as double-stranded linear or plasmid DNA, or single-stranded donor oligonucleotides (ssODNs). However, various studies have shown a preference for MutEJ over precise repair by HDR when employing canonical Cas9.13–16 When applying ssODN editing to model dominant mutations causing Torsade de Pointes arrhythmias in iPSC-derived 3D cardiac tissue sheets, we also found that biallelic editing by HDR or a combination of HDR and on-target indels was more common than monoallelic editing. 17
Variations of Cas9, such as base editing (BE), combine Cas9 nickase (D10A) with a cytidine or adenine deaminase to directly convert specific DNA bases with reduced incidence of DSBs.7,18 Although BE can generate precise edits, it is limited to a specific editing window, and bases adjacent to the target may be simultaneously converted, resulting in bystander mutations. 7 In contrast, Prime Editing (PE) technology combines Cas9 nickase (H840A) with reverse transcriptase (RT) activity derived from the Moloney murine leukemia virus (M-MLV). 19 PE utilizes a PE guide RNA (pegRNA) with a 3′ extension that serves as a reverse transcription template and primer binding site to incorporate the desired edit. Since PE performs everything from single-stranded DNA cleavage to rewriting the genome, editing may be more intentional and is a breakthrough in genetic disease modeling. 20
This work demonstrates the optimization of PE applications in iPS cells using a GMP-grade electroporation platform. We established a fluorescence-based PE benchmark method by using GFP-to-BFP conversion in iPS cells. To maximize PE efficiency, we developed a strategy of FACS (Fluorescence-Activated Cell Sorting) enrichment in which the PE expression vector was modified with T2A-mCherry, allowing assessment of Prime Editor 2 (PE2) expression levels in cells and their fractionation. The efficiency of our method was benchmarked in iPS cells using HEK3 and the rs429358 (c.T388C) pathogenic risk variant in APOE. Our results demonstrate that the activity of PE increases with the expression level of mCherry, and PE is less mutagenic than the conventional genome-editing method using Cas9. Moreover, our results demonstrate that FACS enrichment can be used to control allelic editing outcomes.
Methods
Human iPS cell culture
409B2 (RIKENBRC #HPS0076), 317-A4 (GFP heterozygously targeted iPS cells), and 317-D6 (GFP homozygously targeted iPS cells)12,21 were maintained at 37°C and 5% CO2 in StemFit AK02N medium (Ajinomoto, Cat. No. RCAK02N) on 0.5 mg/mL silk laminin iMatrix-511 (Nippi, Cat. No. 892021) coated 6-well plates or 10 cm dishes. Cell passaging was performed every 7 days during maintenance. The cells were treated with 300 μL or 2 mL of Accumax (Innovative Cell Technologies, Cat. No. AM105-500) in 6-well plates and 10 cm dishes, respectively. The cells were incubated for 10 min at 37°C to dissociate the cells. Pipetting was performed to detach the cells from the surface and generate a single-cell suspension in 700 μL or 4 mL of medium containing 10 μM ROCK inhibitor, Y-27632 (Wako, Cat. No. 253–00513). Cells were seeded onto iMatrix511-coated plates at a density of 1 × 103 cells/cm2 in StemFit AK02N medium with 10 μM ROCK inhibitor for 24 h after seeding and then cultured without ROCK inhibitor. All the cell lines were routinely tested for mycoplasma contamination.
Cas9-gRNA vector cloning
The spacer sequence of GFP-targeting gRNA was designed based on a previous work. 12 pSpCas9(BB)−2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid # 48138; http://n2t.net/addgene:48138; RRID:Addgene_48138). PX458 was digested by EcoRI, and T2A-mCherry was inserted and ligated [KW1013: pSpCas9(BB)−2A-mCherry]. The gRNA construct was generated by Golden Gate assembly of annealed oligonucleotides into the BbsI-digested KW1013 plasmid. The oligonucleotides listed in Table 1 were used for gRNA cloning.
Cloning of PE-mCherry constructs
The T2A-mCherry fragment was PCR-amplified from the KW1013 plasmid. pCMV-PE2, a gift from David Liu (Addgene plasmid # 132775; http://n2t.net/addgene:132775; RRID; Addgene_132775), was digested by EcoRI and PmeI (Thermo Fisher Scientific). The digested product was gel-extracted using the Wizard SV Gel and PCR Clean-Up Kit (Promega). The two fragments were assembled in a single In-Fusion reaction (In-Fusion HD Cloning Kit, Takara, 639650), and the PCR-derived regions of the resulting plasmids were confirmed by sequencing.
pegRNA design and cloning
pU6-pegRNA-GG-acceptor was a gift from David Liu (Addgene plasmid # 132777; http://n2t.net/addgene:132777; RRID; Addgene_132777). pegRNA-GFP and pegRNA-APOE were designed using the PrimeDesign web platform version (https://drugthatgene.pinellolab.partners.org/). 22 The pegRNA construct was generated by Golden Gate assembly of annealed oligonucleotides into the BsaI-digested pU6-pegRNA-GG-acceptor plasmid and sequence verified, as previously reported. 19 The oligos used for the construction of vectors are listed in Table 1, and the plasmids used in this study are listed in Table 2.
Oligos used for vector construction in this study
Plasmids used in this study
PE, prime editing; pegRNA, PE guide RNA; ssODNs, single-stranded donor oligonucleotides.
Electroporation of plasmids and Ribonucleoprotein
For Cas9-based editing, all plasmids for electroporation were prepared using the HiSpeed Plasmid Maxi Kit (Cat. No. 12663), precipitated with ethanol, and dissolved in the MaxCyte electroporation buffer at a concentration of 2.5 μg/μL. For Cas9-based editing, 5 μg of plasmid encoding gRNA-GFP and Cas9 and 5 μg of ssODN repair template (IDT) were mixed in a total volume of 5 μL. The ssODNs used in this study are presented in Table 3. For PE-based editing, 5 μg of the KW1564 vector and 5 μg of the pegRNA-expressing plasmid were mixed in a total volume of 5 μL. Next, 5 × 106 cells resuspended in 50 μL of MaxCyte electroporation buffer were added to the DNA mixture. The suspension (50 μL) was electroporated into an OC-100 × 2 processing assembly (MaxCyte, Cat. No. SOC-1 × 2) using a MaxCyte STX electroporator (Opt 0-5 protocol). Electroporated cells were incubated at 37°C for 30 min and then transferred to an iMatrix511-coated 10 cm dish in StemFit AK02N medium supplemented with 10 μM ROCK inhibitor. Cell preparation for FACS analysis was performed 24 h after electroporation. Otherwise, medium exchange was performed 48 h after electroporation using StemFit AK02N without ROCK inhibitor, and cells were maintained until collection for genotyping on day five or flow cytometry (FC) analysis on day eight. We performed the APOE gene editing by ribonucleoprotein (RNP) of Cas9 with NEPA21 following the previously published protocol. 12 APOE gene editing was performed in either 317A4 or T8 iPS cells crRNA and ssODN are shown in Table 3. 23
Flow cytometry and cell sorting
In total, 5 × 105 cells were suspended in 1 mL FACS buffer (DPBS supplemented with 2% FBS), and GFP and mCherry fluorescence intensities were analyzed using a BD LSRFortessa Cell Analyzer or BD FACSAria II cell sorter (BD Biosciences) with BD FACSDiva software (BD Biosciences). After setting gates for the singlets, 10,000 events were measured for each population. For editing experiments in 317-A4 iPS cells, the cells were acquired using Pacific Blue (450/50 nm) and FITC (530/30 nm) filters. For cell sorting, cell suspensions were prepared in FACS buffer at a density of 1 × 106 cells/mL and filtered through a 35 μm nylon mesh cap of the tube (Corning, 352235) to remove clumps. Sorting gates were set for the singlet events. The desired population was collected using a BD FACSAria II cell sorter (BD Biosciences) in AK02N medium containing 20 μM Y-27632. Sorting efficiency was confirmed by re-analyzing 300 μL of the media. Raw data were analyzed using FlowJo 10 (FlowJo LLC). Rainbow Calibration Particles (6 peaks) and 3.0–3.4 μm (BD Biosciences) were utilized to calibrate the laser strength and determine the sorting gate. The 99 percentiles of the relative mCherry intensity of the cells electroporated without PE-expressing plasmid and 5 μg of pegRNA-expressing plasmid were used to define a negative threshold for mCherry in Figure 1. For FACS enrichment gating strategy, the 90 percentiles of relative mCherry intensity of un-electroporated cells were used as a threshold for the Low population. The mode of each peak of the ladder was first measured, and we defined the mode of the third and fourth peaks as the thresholds for medium and high populations, respectively.
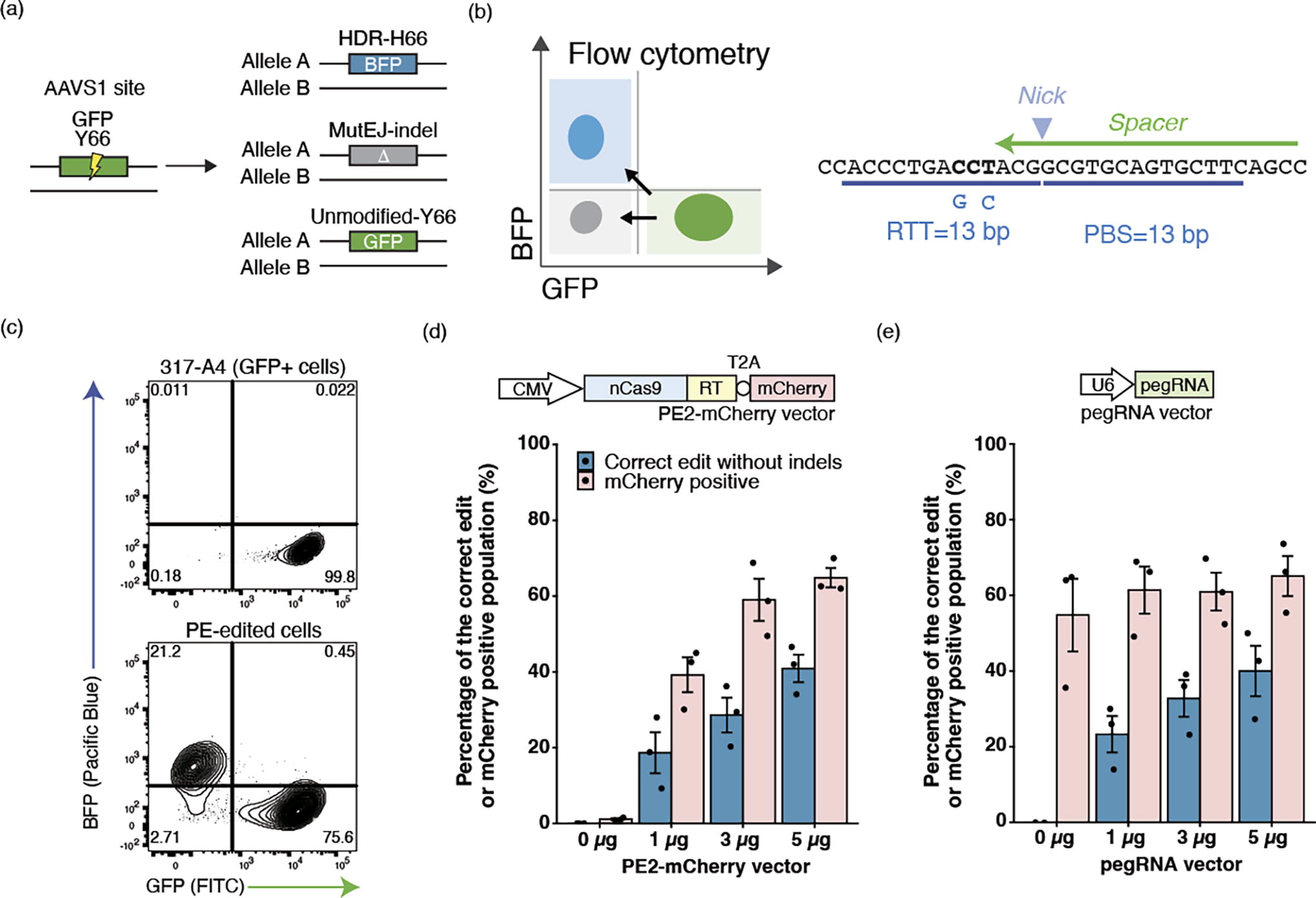
GFP-to-BFP conversion assay to optimize plasmid delivery of PE in human iPS cells using the MaxCyte platform.
Genotyping
For genomic DNA extraction, 0.5–1 × 106 cells were washed with 1X DPBS, and DNA was purified using the DNeasy Blood & Tissue Kit (Qiagen, Cat. No. 69506) following the manufacturer’s protocol. Purified DNA was eluted in 100 μL of AE buffer. Target sequences were amplified by PCR using the KAPA HiFi HS ReadyMix (Kapa Biosystems, Cat. No. KK2602). PCR product cleanup was performed using the ExoSAP-IT Express reagent (Cat. No. 75001) following the manufacturer’s protocol, and Sanger sequencing was performed using the BigDye Terminator v3.1 CS Kit (Thermo Fischer Scientific, Cat. No. 4337456). The final product was purified by ethanol precipitation and dissolved in HiDi formamide. Sequencing was performed on a 3500xl Genetic Analyzer (Applied Biosystems). Sequence alignments were analyzed with Snapgene (GSL Biotech LLC), and sequence trace files with low base-calling confidence were excluded manually. The primers used for genotyping are listed in Table 4. Sequencing analysis was performed on mixed sequences with ICE (https://ice.synthego.com/) 24 or DECODR (https://decodr.org/). 25 Sequence data from the unedited iPS cells were used as the reference genome. The parameters were kept at their default values.
Primers used for genotyping and sequence analysis
crRNA and ssODN Used in this study
Droplet digital PCR
To quantify the APOE 388 mutation created by PE, we prepared a mixture containing 10 μL of droplet digital PCR (ddPCR) Multiplex Supermix (Bio-Rad, Cat. No. 12005909), 1.8 μL of 20 μM forward and reverse primers each, 1.25 μL of 10 μM APOE-FAM and 0.5 μL of 10 μM hTFRC-HEX probes, and 30 ng of template DNA, adjusted to a final volume of 20 μL. Droplets were generated using a QX200 Automated Droplet Generator (Bio-Rad). The PCR amplification protocol was as follows: 95°C for 10 min, 40 cycles at 94°C for 30 s, and 58°C for 4 min, followed by 98°C for 10 min. The amplified droplets were then read with the QX200 Droplet Reader (Bio-Rad), and data were analyzed using QuantaSoft™ Analysis Pro v1.7.4.
Statistical analysis
The data are presented as the mean ± standard deviation from the indicated numbers of independent experiments and were processed using R 4.0.3, the R package tidyverse 1.2.0, 26 and ggprism. 27
Results
GFP-to-BFP conversion assay to benchmark editing with PE plasmids in iPS cells
First, we established conditions for PE expression from plasmids in iPS cells. We adopted a GFP-to-BFP conversion assay previously used to optimize ssODN editing, 12 where a single amino acid change from tyrosine to histidine (Y66H) in the fluorophore region of GFP is sufficient to convert fluorescent emission of GFP to BFP and can be quantified in single cells by flow cytometry (Fig. 1a). 28 The pegRNA-GFP was designed to create Y66H, along with a T65S mutation, which acts to stabilize BFP, increasing fluorescence by approximately two-fold,29,30 as well as block the PAM to prevent subsequent recleavage of the edited BFP allele (Fig. 1b). The spacer sequence is identical to that used in ssODN editing. 12 Conversion to BFP therefore represents the intended PE edit, while MutEJ is detected by loss of fluorescence and unmodified cells remain GFP-positive. These changes were quantified by flow cytometry, resulting in 21.2% editing to BFP in the 317-A4 iPS cell line (monoallelic AAVS1-targeted GFP) with 3 μg of each PE expression plasmid and pegRNAs expression plasmid (Fig. 1c).
The PE2 expression vector was modified to couple PE with mCherry using a T2A peptide that induces ribosomal skipping (PE2-mCherry), such that mCherry expression represents the level of PE expression. Previously, delivery of Cas9 ribonucleoprotein (RNP) and DNA plasmid to human iPS cells was established on the GMP-compliant MaxCyte platform.12,31,32 However, cotransfection of multiple plasmids in iPS cells has yet to be demonstrated, and we started by optimizing the amount of plasmids by titration. (Fig. 1d, e). We tested the editing efficiency based on the amount of PE-mCherry- or pegRNA-expressing plasmid (0, 1, 3, and 5 μg) while keeping the second component fixed (5 μg). The proportion of mCherry-positive cells detected at 24 h after electroporation indicated transfection efficiency. The proportion of BFP-positive cells on day 7 indicated the amount of correct editing. We confirmed that the increase in mCherry-positive cells correlated with the amount of PE plasmid, and the editing efficiency correlated with the amount of pegRNAs plasmid within the titration range. In addition, 5 μg of each plasmid showed a consistent transfection efficiency of ∼64.7% across multiple electroporations, and we decided to use this condition for the subsequent experiments. We also observed electroporation increased autofluorescence intensity. 317-A4 without electroporation exhibited a mean fluorescence intensity of 55.1 with a median of 51.3, whereas cells transfected without PE-mCherry and 5 μg of pegRNA showed a mean of 87.1 and a median of 84.9 (Supplementary Figure S1). We chose to maintain the fluorescence intensity of nonelectroporated cells for FACS enrichment because this approach allows us to include cells expressing low levels of PE without a negative effect on the analysis of editing in cells with high expression.
FACS enrichment of highly transfected iPS cells to maximize correct editing efficiencies
Next, we established parameters for FACS enrichment of cells expressing high levels of PE or Cas9. FACS enrichment for mCherry was performed 24 h after electroporation. Using fluorescent beads as a calibration ladder for consistency between experiments, the cells were divided into three groups: “Low,” “Medium” (or ‘Med’), and “High,” depending on the level of mCherry expression (Fig. 2a).
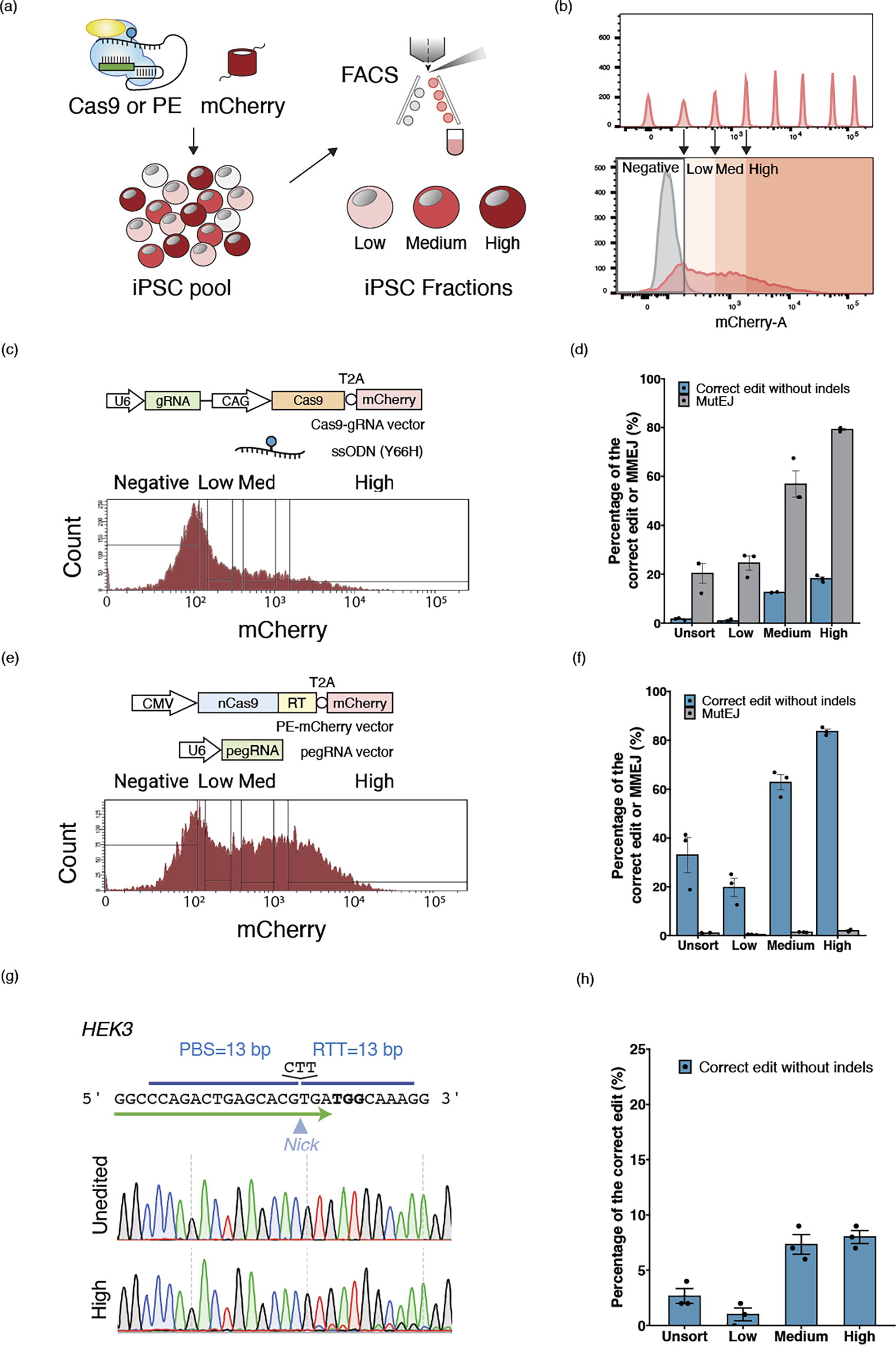
FACS enrichment to maximize PE or ssODN editing efficiency.
The effect of FACS enrichment on GFP editing outcomes was tested for both PE and ssODN editing (Supplementary Figure S2, Figure S3). In the total unsorted (“Unsort”) population, 33.0% of correct edits to BFP were observed for PE, whereas only 1.62% of the correct edits were observed in ssODN editing (Fig. 2c, d). MutEJ levels (fluorescence negative cells) in PE were found to be low (0.993%), while MutEJ was more prevalent than correct edits for ssODN editing (20.3%). In comparison, the ratio of correct edits to MutEJ was 0.08 in ssODN editing, as compared to 33.3 in PE (Fig. 2e, f). These data verify that PE with plasmids is more efficient and precise than editing with Cas9 and ssODNs in human iPS cells at the loci investigated in this study. In the High fraction, on average, 83.6% of iPS cells were converted to BFP by PE, while only 18.1% became BFP positive by ssODN. Compared with Unsort, the fold improvement of correct edits by PE were 0.60, 1.90, and 2.50 times in Low, Med, and High fractions, respectively. The fold improvement by ssODN editing was 0.53, 7.74, and 11.2 times for the respective fractions. Thus, for both PE and ssODN editing, the editing efficiency improved alongside mCherry intensity. Importantly for PE, the proportion of MutEJ only increased 0.43, 1.39, and 1.99 times in the Low, Medium, and High populations compared to Unsort, while in ssODN, MutEJ was increased 1.21, 2.80, and 3.90 times. In the High fractions, MutEJ reached only 18.1% for PE, but 79.2% for ssODN. These data demonstrate that FACS enrichment of PE increases the number of correct edits without a substantial increase in MutEJ, in stark contrast to ssODN editing. These data indicate that PE outcomes may be improved by enrichment using fluorescence without an increase in MutEJ.
Benchmarking editing efficiencies in iPS cells at endogenous loci
For benchmarking endogenous gene editing with PE and FACS enrichment, we selected the HEK3 locus (Fig. 2g, h) that has been used in diverse cell lines such as HEK293T, HeLa, K562, and human embryonic stem cells.33,34 The HEK3 benchmarking pegRNA inserts CTT and causes two detectable sequences. Overall, the combination of PE and FACS enrichment resulted in a 3-fold increase in correct edits, reaching 8.0% in the High fraction compared with Unsort population (2.67%). Med population showed a saturation in editing efficiency (7.33%) in HEK3 loci, with values comparable to those observed in the High fraction. MutEJ was undetectable across all fractions by ICE analysis. These data indicate that PE can be improved by fluorescent enrichment at endogenous loci without coenrichment of MutEJ.
Evaluation of allelic editing outcomes with FACS enrichment
Given the potential of iPS cells for modeling genetic diseases, it is important to determine the rate of mono- and biallelic editing at a cellular or clonal population level. We first evaluated allelic editing in 317-D6, a biallelic AAVS1-targeted GFP iPS cell line. In total, six editing patterns are expected (Fig. 3a). Since iPS cells with one or two active copies of GFP or BFP only double in their mean fluorescence intensities, 21 we recognize that distinguishing between biallelic editing and monoallelic editing with MutEJ is challenging by FACS. 12 However, considering the near-zero proportion of MutEJ generated by PE, we predicted that BFP single-positive iPS cells correspond to biallelic editing, whereas BFP/GFP double-positive iPS cells represent monoallelic editing (Fig. 3a). We also confirmed MutEJ population was not detectable in BFP positive population by Sanger sequencing (Supplementary Figure S4). The Unsort population exhibited nearly equal amounts of biallelic (29.4%) and monoallelic (23.1%) editing (Fig. 3b). Interestingly, the High population showed more than three-fold higher biallelic editing (72.6%) than monoallelic editing (22.4%). In the Med population, these proportions were more similar at 43.1% and 36.3% for biallelic and monoallelic edits, respectively. Finally, the Low population reversed this trend, showing nearly half the number of biallelic edits compared to monoallelic editing (7.65% and 14.7%). These results demonstrate that FACS enrichment for defined PE expression levels can skew the outcomes of mono- and biallelic editing at a cellular level.
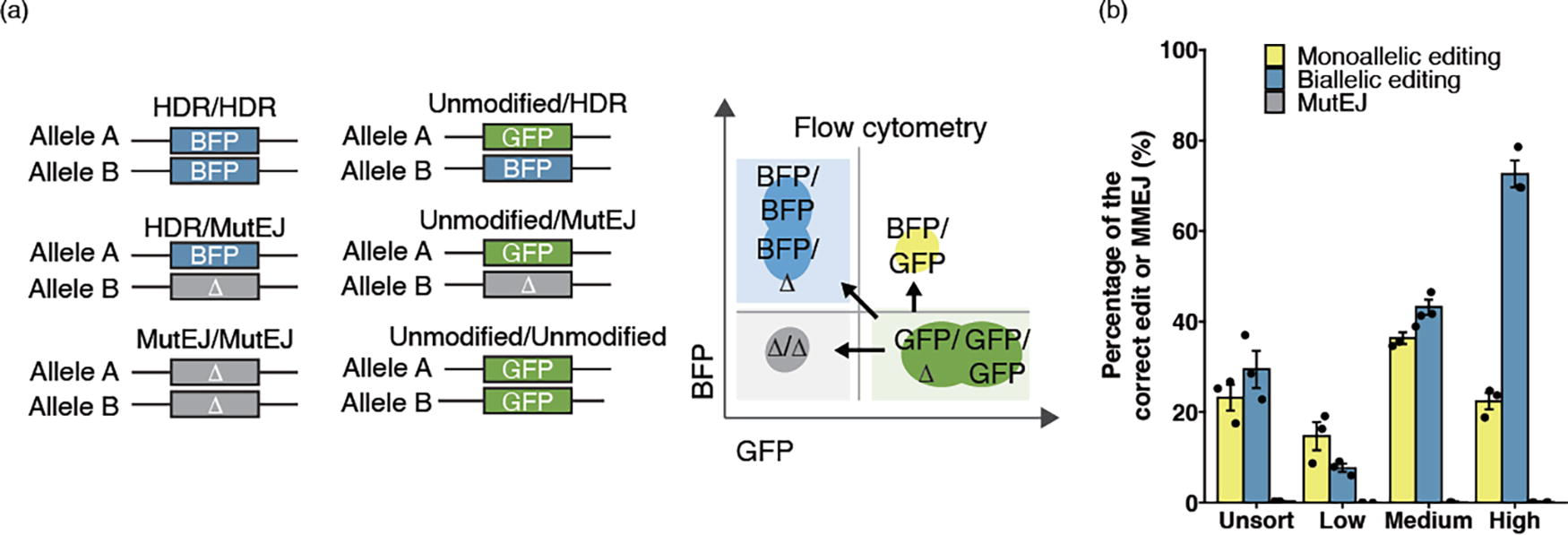
Modulating the allelic editing outcomes by FACS enrichment.
We then explored the clonal distribution of allelic editing by Cas9 and PE at an endogenous locus. Among the three variants of the apolipoprotein (APOE) gene, APOE2, APOE3 (c.C526T, rs7412), and APOE4 (c.T388C, rs429358), APOE4 is associated with the highest risk of Alzheimer’s disease (Fig. 4a). We therefore chose to engineer the rs429358 APOE3 variant to the APOE4 genotype by both ssODN editing and PE (Fig. 4b). Editing by ssODN and Cas9 RNP showed 11.3% correct edits (Fig. 4c). However, correct edits were outcompeted by 78.3% +1T insertions. We treated cells with NU7441 (a DNA-PKcs inhibitor), with the aim of depleting +1T insertions and increasing the correct edits (Fig. 4d). NU7441 treatment was able to maintain a similar level of correct edits (7.5%). While the decrease in +1T insertion by NU7441 was substantial (55.0%), it was not enough to eliminate it. Next, we used the same spacer sequence and designed APOEx386 pegRNA (Supplementary Figure S5). However, Sanger sequencing could not detect any activities from this pegRNA. Following this experiment, we designed APOEx398 pegRNA targeting the opposite strand while still creating the same variant (Fig. 4e, f). Sanger sequencing with DECODR software 25 estimated 23.6% APOE4 c.T388C correctly edited alleles in the High fraction by APOEx398 pegRNA, which was validated by droplet digital PCR (ddPCR, 21.3%) (Fig. 4f). In contrast, no edits were detected in the Low or Med fractions by Sanger sequencing and DECODR, whereas ddPCR identified 3% and 8% correct edits, respectively (Fig. 4f). MutEJ was not discernible in any of the fractions by Sanger sequencing (Fig. 4g).
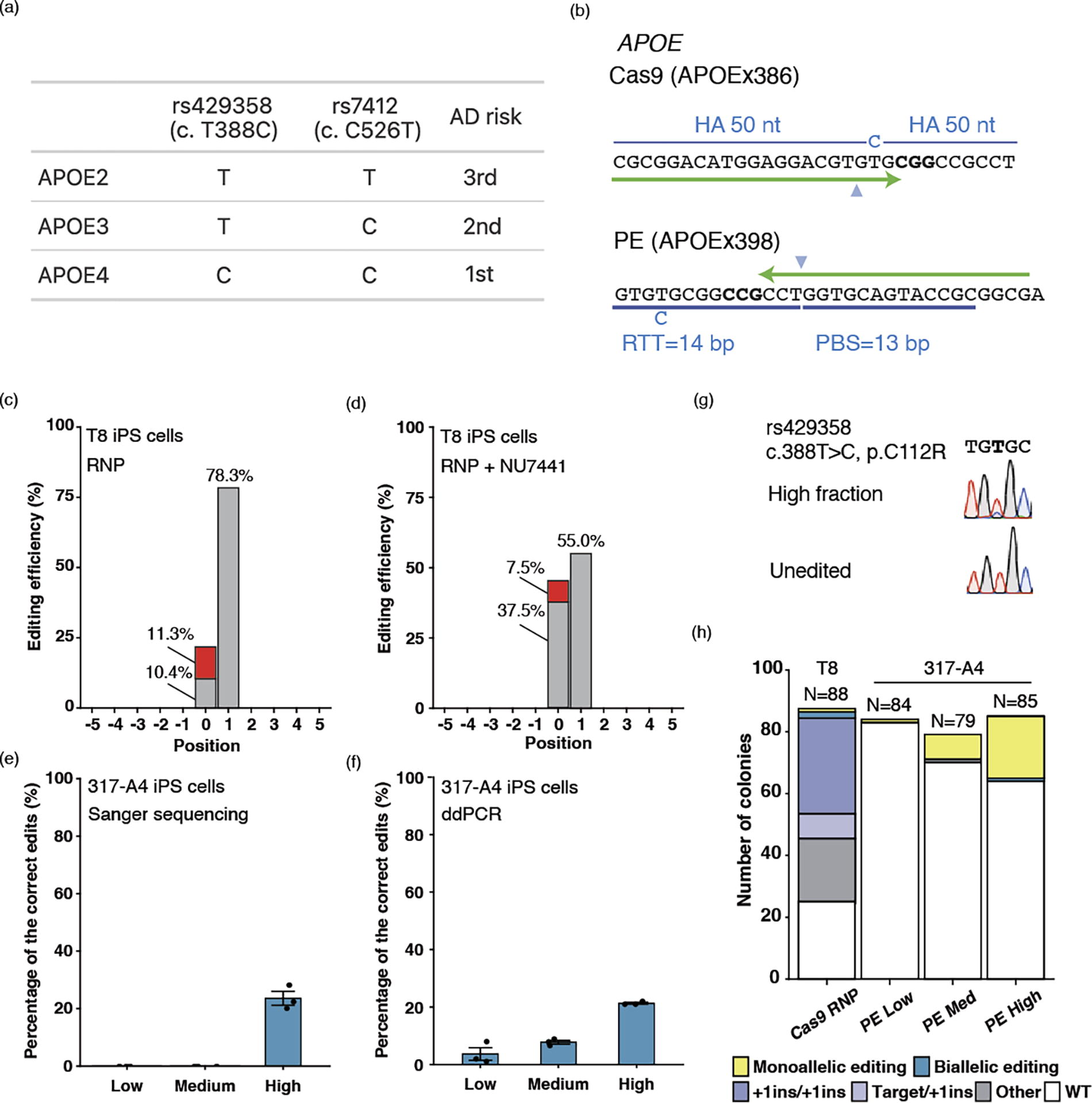
APOE gene editing to recreate a disease model for Alzheimer’s disease in iPS cells.
Finally, we determined the number of APOE4 biallelic and monoallelic edited iPS cells at a clonal level (Fig. 4h). Using Cas9 RNP, two bi- and nine monoallelically edited clones were obtained; however, eight of the monoallelically edited clones were accompanied by a +1 insertion in other alleles. Using PE, the Med and Low populations yielded only monoallelic clones (1 and 8, respectively), with many unedited clones. From the high PE condition, we isolated 24 mono- and one biallelically edited colony. Moreover, no unintended edits were confirmed by Sanger sequencing of clones. Collectively, these findings suggest that a combination of PE and FACS enrichment can fulfill a crucial need in establishing an allelic series of isogenic iPS cells for disease modeling.
Discussion
In this research, we demonstrate the superior efficiency and precision of PE over Cas9-based ssODN editing in iPS cells. Also, applying FACS, we successfully enriched cells, exhibiting varied editing efficiencies. Leveraging a GFP-to-BFP reporter assay with single-cell resolution, our findings revealed that PE generated notably fewer MutEJ events compared to ssODN editing. Finally, we show that FACS enrichment can modulate the frequency of mono- or biallelic editing, demonstrating its utility in controlling allelic editing outcomes.
The GMP-compliant MaxCyte platform has been used for plasmid transfection into T cells, HEK293T cells, and CHO cells.35,36 Previous research used the MaxCyte to successfully introduce plasmid, mRNA, and RNPs into iPS cells.12,31,32,37 As iPS cells hold significant promise not only for disease modeling but also for genome-edited cellular regenerative medicine anticipating ex vivo applications for cell therapy, our findings are of paramount importance.
Our protocol demonstrates a highly reproducible FACS gating strategy through the implementation of a fluorescent bead ladder. FACS provides advantages for PE editing using plasmid vectors by eliminating non-transfected cells and isolating cells based on their expression level. Our FACS enrichment data also indicate that PE activity was underestimated in the unsorted population, similar to a previous study on the combination of Cas9 and FACS enrichment reported. 38 Another study employed piggyBac transposition to introduce PE into iPS cells for long-term stable expression, and observed almost 50% of maximum editing after 8 days and a 2.5-fold increase in editing over an additional 32 days of continuous expression, 39 suggesting that the effects of PE are realized early after transfection. Using FACS enrichment after 24 hours, it is possible to select cells with modest PE expression for editing with high-efficiency pegRNAs (such as pegRNA-GFP) and high PE expression for editing with low-efficiency pegRNAs (such as pegRNA-APOE).
To bias the reaction of heteroduplex removal in the PE repair step, a second nicking guide RNA (ngRNA) can be used to nick ∼50 bases in the editing region. PE with the ngRNA used is called PE3.18,19,40 Although previous studies have shown that PE3 can be further improved by additional nicking, this also increases MutEJ. Also, using multiple gRNA including pegRNA in a single experiment increases the sites experimentalists require to confirm off-target activity. Thus, our study focused on PE2. Editing of GFP was found to be efficient; however, editing at endogenous loci was variable. PE3 can be a powerful method for improving editing efficiency. Furthermore, in PE, almost all edits were targeted edits. Previous studies have shown that PE is less likely to occur in cells with mature mismatch repair (MMR) such as iPS cells. 33 PE4 is a method in which the transient expression of a dominant-negative MMR protein (MLH1dn) is combined with PE2. This temporal inhibition of MMR showed the potential to increase editing efficiency in iPSCs. 41 Combined with FACS enrichment, this method may enable further flexible and accurate genome editing.
One of the bottlenecks of PE is the difficulty of pegRNA design. Although some webtools22,42 or prediction tools43,44 are available to design pegRNAs, it is difficult to accurately predict the function of a pegRNA until tested in cells. Despite a similar design scheme of pegRNAs, only 8% editing occurred in HEK3, while GFP reached 83% editing in the High fraction. It is also worth noting that our original GFP data (Fig. 3b) demonstrated a broader distribution of editing outcomes, including both mono- and biallelic editing events. However, this pattern was not observed in the APOE gene editing experiments (Fig. 4g), likely due to the lower activity of the pegRNA. We also attempted to create the same variant in APOE and designed pegRNAs with spacer sequences targeting complementary DNA strands. While one pegRNA showed activity, we could not detect any activity for the other. Previous research highlighted that cell type-specific DNA repair responses and chromatin contexts can affect PE efficiency. 43 To achieve predictable activities for pegRNAs, further research on design strategies and locus-dependent effects in multiple cell types will likely be necessary.
Even without FACS, we observed a significant disparity in by-products between ssODN editing and PE. Upon FACS enrichment, ssODN editing showed a marked increase in the proportion of MutEJ accompanied by HDR, whereas, in PE, only the proportion of correct edits surged. This disparity is caused by the increased likelihood of DSBs due to elevated Cas9 expression levels, whereas PE’s nick-based editing mechanism enhances mainly the rate of correct editing over MutEJ. While our observations were consistent for editing a single nucleotide variant in the APOE gene, we cannot guarantee that similar patterns would be observed for other genes or pegRNA and mutation designs without a genome-wide effort. In addition, pegRNA activity differs between GFP and APOE. This resulted in high biallelic editing in GFP, whereas high monoallelic editing was observed in APOE. Allelic editing may require a high level of PE activity.
Conclusion
A combination of PE and FACS enrichment allows highly efficient and accurate gene editing. The combined protocol is a useful method, as the accuracy of PE does not lead to a significant increase in by-products. Also, fractionation by FACS based on PE expression level can influence mono- or biallelic editing. Improving the reliability of gene editing outcomes will greatly facilitate the generation of genetic disease models and possibly therapies using human iPS cells.
Footnotes
Acknowledgment
The authors thank Suji Lee for technical assistance in cell culture and molecular biology and Dr. Kanae Mitsunaga for helping establish conditions for FACS analysis and cell sorting. The authors thank Dr. Ryo Yamada for his mentorship and guidance of R.N. as part of the Medical Innovation Program and for providing the inspiration for statistical methods applied to FACS.
Authors’ Contributions
R.N.: Conceptualization, formal analysis, investigation, resources, writing—original draft, writing—review and editing, and visualization. T.M.: Methodology, investigation, and resources. A.Y.L.: Investigation and resources. M.K.: Investigation. T.K.: Investigation and resources. K.T.: Resources. P.G.: Resources and writing—reviewing and editing. H.I.: Investigation and resources. T.L.M.: Conceptualization, investigation, and writing—reviewing and editing. K.W.: Conceptualization, formal analysis, investigation, resources, writing—original draft, writing—reviewing and editing, supervision, project administration, and funding acquisition.
Author Disclosure Statement
P.G. is an employee at MaxCyte Inc., providing electroporation systems used in this study. Other authors declare that they have no conflicts of interest.
Funding Information
This study was supported by the Japan Agency for Medical Research and Development (AMED grant numbers JP21bm0104001, JP23bm1323001, and JP23bm1223013), the COVID-19 Private Fund (Shinya Yamanaka, Center for iPS Cell Research and Application [CiRA], Kyoto University), and the National Research Council of Canada. R.N. is a trainee of the Medical Innovation Program managed by Kyoto University and supported by JST SPRING (grant number JPMJSP2110).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.