
Abstract
CRISPR-Cas systems have proven effective in a variety of applications due to their ease of use and relatively high editing efficiency. Yet, any individual CRISPR-Cas system has inherent limitations, necessitating a diversity of RNA-guided nucleases to suit applications with distinct needs. We searched through metagenomic sequences to identify RNA-guided nucleases and found enzymes from diverse CRISPR-Cas types and subtypes, the most promising of which we developed into gene-editing platforms. Based on prior annotations of the metagenomic sequences, we establish the likely taxa and sampling locations where Class 2 CRISPR-Cas systems active in eukaryotes may be found. The newly discovered systems show robust capabilities as gene editors and base editors.
Introduction
CRISPR systems have consistently proven to be robust gene-editing tools for both research and therapeutic applications. CRISPR-associated (Cas) proteins are categorized into two major classes and further divided into different types and subtypes. Those most useful for gene editing are found in Class 2 systems of Types II, V, and VI, with the most frequently used enzymes being SpCas9 (Type II) and AsCas12a (Type V-A).1–3 Cas9 recognizes a unique Protospacer Adjacent Motif (PAM) sequence at the 3′ end of the protospacer (5′-NGG-3′ PAM for SpCas9) for recognition and creates a double-strand DNA (dsDNA) break in the targeted DNA region. 4
In contrast to the Cas9 enzyme, the Cas12a enzyme does not need a trans-activating crRNA (tracrRNA) to process crRNAs, and it recognizes a T-rich PAM in the targeted DNA sequence (5′-TTTV-3′ PAM for AsCas12a), although activity has been observed at noncanonical PAMs for AsCas12a. 5 Furthermore, Cas12a can produce “a staggered DNA double-strand break with a 4 or 5 nucleotide 5′ overhang.”6,7 Recently, the development of base editing has enabled the creation of mutations beyond random insertions and deletions (INDELs) resulting from dsDNA breaks. By fusing CRISPR-Cas enzymes with a cytidine deaminase and two uracil glycosylase inhibitors (UGIs), a C → T (or G→A) substitution can be achieved without generating dsDNA breaks.8,9 However, base editing using any specific Cas protein encounters limitations because it can only target bases adjacent to specific PAM sequences, which can be resolved by utilizing a variety of Cas proteins. 10
The CRISPR-Cas toolbox has been expanded with additional effector proteins,11–13 which have higher gene-editing efficiency and expanded PAM sequence preferences, allowing them to outperform in multiple applications, such as gene activation and base editing. 14
To develop effective treatment strategies, it is crucial to have access to a diverse assortment of CRISPR-Cas systems that are compact, devoid of off-target effects, and jointly targetable to diverse sequences. To partially meet this demand, we utilized existing public tools for CRISPR identification and applied them to a large public metagenomic data repository to discover RNA-guided nucleases (RGNs). We were then able to systematically test 91 newly identified systems for bacterial activity and subsequent eukaryotic activity to create a large collection of active Class 2 systems suitable for human genome editing. By searching public metagenomic data repositories and applying sequence-based prediction techniques, we pinpointed candidates encompassing a variety of PAM sequences. We demonstrated activity of multiple Types II, V-A, and V-B systems in both bacterial and eukaryotic cellular environments. We show how existing methodologies can be successfully applied to create a large genome-editing toolbox.
Methods
Metagenomic CRISPR-Cas identification
CRISPRCasTyper was run on 52,515 metagenome-assembled genomes (MAGs) downloaded from https://portal.nersc.gov/GEM/genomes/, and the corresponding metadata about sampling locations and taxa was used to create summary for the discovery of diverse, active CRISPR systems in metagenomic sequencing data.15,16 Enrichment of classes and types of CRISPR systems among bacterial taxa was tested by Fisher's exact test. Sequences were curated by the presence of intact catalytic residues, length, and diversity and aligned to known reference sequences11,13,17 using a MAFFT E-INS-I 18 alignment and the neighbor-joining method and Jukes-Cantor genetic distance model using Geneious Tree Builder in Geneious Prime (Biomatters Ltd.). Divergence from known systems was determined by a search of the GenomeQuest database (GQLifeSciences, accessed February 2021). Sequences where the taxonomy was not determined by Nayfach et al. 15 were here designated as Environmental Genomic Sequence (EGS). The proportions of active CRISPR systems by sampling sources were compared by Z-test for two independent proportions.
Single guide RNA computational identification and crRNA identification
For Type II systems, tracrRNA and single guide RNA (sgRNA) were predicted using https://github.com/skDooley/TRACR_RNA. 19 For Type V-A systems, repeat-arrays identified were searched for the consensus crRNA sequence UCUAC[N3–5]GUAGAU and the appropriate crRNA region was extracted. 11
Determination of PAM requirements for each RGN through bacterial PAM depletion
For Types II and V-A systems, the identified Cas protein was codon-optimized for expression in Escherichia coli via the GenScript Codon optimization tool 20 and synthesized with a SV40 NLS on the 5′ end and a nucleoplasmin NLS on the 3′ end under control of the T7 promoter in pET28. The identified targeting RNA with an appropriate targeting sequence was synthesized under control of a separate T7 promoter on the same vector. All syntheses were carried out by Twist Biosciences. For Type V-B systems, the native CRISPR operon was synthesized under control of the T7 promoter in pET28 without any optimization. Three repeats of the CRISPR array were maintained, but the spacer sequence was replaced with a specific target sequence. Synthesis was performed by GenScript.
PAM identification was performed similarly to previously established methods. 21 In brief, a known target sequence was cloned into a pUC19 vector flanked on one side by a random octamer. Then, T7 Express Competent E. coli (New England Biolabs) containing the Cas/targeting RNA expression plasmid were cotransformed with the random octamer library and grown to saturation in double selective media. The cells were then transferred to double selective Overnight Express™ Instant TB Medium (Novagen) supplemented with 2% glycerol at 5% v/v and allowed to grow at 37°C for ∼4 h before being incubated at 30°C overnight. The following day, the plasmids were extracted with the MagJET Plasmid DNA kit (Thermo) according to the manufacturer's instructions. The resulting plasmid library was then amplified by PCR using Q5® High-Fidelity DNA Polymerase (New England Biolabs) using barcoded primers for the random PAM library followed by an Agencourt AMPure XP clean-up step (Beckman Coulter Genomics).
Deep sequencing (55bp paired end reads) was performed on a NextSeq (Illumina) using custom primers, to obtain typically 1–4 M reads per amplicon. PAM regions were extracted, counted, and normalized to total reads for each sample.
PAMs that lead to plasmid cleavage were identified by being underrepresented when compared to controls (i.e., when the library is transformed into E. coli containing the RGN but lacking an appropriate sgRNA). To identify the PAM requirements for a RGN, an enrichment value was computed for each kmer as the ratio between the read counts, normalized to library size, in the control sample and in the targeting sample. To reduce noise, only ratios of at least 4 were retained. This was achieved by subtracting 3 from each ratio and rounding to the nearest integer.
Sequence logos were generated for each PAM from a list of enriched kmers, where each kmer in the list was added in proportions commensurate with the score described above, and were visualized using the command line utility weblogo. RGNs were considered to have bona fide PAMs if the same logo could be observed across multiple windows, especially the window that includes the full PAM and the adjacent non-PAM base on one or both sides. If multiple guides were used, the PAM was required to be consistent across guides.
tracrRNA identification for type V-B1 systems
Active Type V-B systems underwent bacterial RNA sequencing to identify their tracrRNA similar to established methods. 17 In brief, the same expression plasmids used for PAM determination were expressed under identical conditions and RNA was prepared from E. coli lysates using TRIzol followed by isolation with the mirVana miRNA Isolation Kit (Invitrogen) and total RNA cleanup with the RNA Clean and Concentrator kit (Zymo). Libraries were prepared using the NEBNext Small RNA Library Kit for Illumina (NEB). Libraries were sequenced with a 2 × 150 paired-end NextSeq run (Illumina). Adapters were removed from reads using Trim Galore, and paired-end reads were merged using flash. 22 Both merged reads and reads that failed to merge (due to insufficient overlap) were aligned to plasmid sequences using Burrows-Wheeler Aligner. 23 Unmerged read pairs were pseudo-merged based on their aligned positions. Strand-specific pileups for both merged and pseudomerged reads were created using samtools. 24
Uracil protecting peptide identification
Public genome collections were searched for sequences of less than 200 amino acids with a total of at least 10 aspartic acid and/or glutamic acid residues on the predicted protein surface and a negative charge on at least 10% of the predicted surface residues.25,26 Twenty-one candidate uracil protecting peptides (UPPs) were selected for testing and codon optimized for expression in human cells. Selected UPPs were synthesized fused to the 3′ end of a known eukaryotic cytosine deaminase (APOBEC3A) and a known Cas9 Enzyme (Nme2Cas9)27,28 with a 5′ SV40 NLS and a 3′ nucleoplasmin NLS in the pTwist CMV plasmid (Twist Biosciences).
Dual-plasmid Lipofectamine eukaryotic activity assay
Expression plasmids were synthesized for each selected Cas enzyme with dual NLS tags after codon optimization for human expression (GenScript) into the pTwist CMV plasmid (Twist Biosciences). 20 Targeted genomic sequences adjacent to the relevant PAM were selected and used to test the identified Cas enzyme/PAM. Respective targeting RNA was then synthesized under the U6 promoter on the pTwist CMV plasmid (Twist Biosciences) expressing a codon optimized Green Fluorescent Protein. The eukaryotic activity assay was conducted following the method outlined in a previous study. 29 293T cells were seeded in 24-well plates at 1.3 × 105 cells per well in 500 μL growth medium (Dulbecco's modified Eagle's medium, 10% fetal bovine serum, 1% Pen-Strep) and incubated overnight at 37°C, 5% CO2.
In accordance with the manufacturer's instructions, the cells were transfected with 500 ng of plasmid containing targeting RNA and 500 ng of plasmid containing the effector protein using Lipofectamine 3000 (Invitrogen). The cells were then incubated at 37°C, 5% CO2 for 48 h.
Following the 48 h incubation, genomic DNA extraction was performed using the Nucleospin-96 Tissue Kit (Machary Nagel) as per the manufacturer's instructions. Gene-specific primer pairs incorporating Nextera amplification regions were used to amplify targets using the Platinum SuperFi PCR Master Mix (Invitrogen) on the ProFlex 2 × 96-well PCR System (Invitrogen). Amplicons were visualized using the Flash Gel System (Lonza Bioscience) and subsequently purified with the NucleoSpin 96 PCR Cleanup Kit (Machary Nagel) following the manufacturer's protocol.
Nextera DNA Indexes (IDT) compatible with Illumina sequencing were added to the purified amplicons using the Phusion Hot Start II High-Fidelity PCR Master Mix (Thermo Fisher) on the ProFlex 2 × 96-well PCR System (Invitrogen). Barcoded amplicons were visualized on the Flash Gel System (Lonza Bioscience) and equal amounts of these amplicons were pooled together. The pooled barcoded amplicons were subjected to double size selection using Agencourt AMPure XP beads (Beckman Coulter). The Qubit 1X dsDNA assay (Invitrogen) was used to determine the concentration of the pooled sample. The sample was prepared for next-generation sequencing on a Miniseq or NextSeq (Illumina) following the manufacturer's instructions for 2 × 150 reads. The reads were analyzed with CRISPResso2, and statistical comparisons were done in GraphPad Prism 9.2.0 (GraphPad Software, LLC) using a paired t-test. 30
Results
The discovery of diverse, active CRISPR systems in metagenomic sequencing data
We searched for CRISPR effector proteins in 52,515 MAGs that were sampled from diverse habitats collected from all continents and oceans, human and animal hosts, engineered environments, and natural and agricultural soils. 15 In total, 8904 Class 1 systems and 4227 Class 2 systems were identified. As previously reported, Types I and III systems are most abundant across bacterial phyla. 31 When considering phyla with over 100 putative CRISPR systems, Type II systems were most abundant in Bacteroidota, and Type V systems were most abundant in Cyanobacteria as previously reported. 31 The Deinococcota phylum was most significantly enriched for Class 1 systems [Fig. 1a; Fisher's Exact Test (FET), p < 10−100]. The classes Bacilli and Clostridia were most enriched in Type II-A systems (Fig. 1b; FET, p < 10−100 for both), and the class Bacteroidia was most enriched for Type II-C systems (Fig. 1b; FET, p < 10−100).

Distribution of CRISPR-Cas system in major archaeal and bacterial phyla. Distribution of the six types of CRISPR-Cas systems identified from metagenomic samples in major archaeal and bacterial phyla
Clostridia and Desulfomonilia were most enriched in Types V-A and V-B1 systems, respectively (Fig. 1c; FET, p < 10−100, p = 2.6 × 10−27, respectively). Among the identified systems, 166 Type II, 58 Type V-A, and 4 Type V-B1 systems were assessed to be diverse, with lower than 80% sequence identity to existing systems, as determined by phylogenetic analyses and a BLAST search to known systems, and likely to be functional, as determined by the presence of intact catalytic residues. Of those, the 29 Type II systems shorter than 1300 amino acids and all the Type V systems were selected for bacterial PAM determination, using either computationally predicted sgRNA (for Type II systems), the consensus UCUAC[N3–5]GUAGAU crRNA motif (for Type V-A systems), or transferring the entire CRISPR operon into E. coli (Type V-B1 systems).
These systems were tested against an exhaustive 8-mer library positioned in the appropriate PAM location with respect to the guide sequence(s). We identified the PAM for 18 Type II systems (62%), 37 Type V-A systems (62.7%), and three Type V-B1 (75%; Supplementary Fig. S1). The vast majority of Type II-A systems from Bacilli (82%) and Type V-A systems from Clostridia (100%) were active in our bacterial PAM assay (Fig. 1d), consistent with the enrichment of these systems in their respective bacterial classes. Small RNA-seq was performed on active Type V-B1 systems to identify their tracrRNA (Supplementary Fig. S2). Selected systems were then tested against multiple targets to determine active DNA editing via dual plasmid lipofectamine delivery in eukaryotic cells with a nuclear localization sequence at both termini (Supplementary Tables S1–S4).
Only Type II systems from Bacilli were found to be active for eukaryotic editing (Fig. 1e), whereas Type V-A systems from multiple bacterial classes were found to be active for eukaryotic editing, most abundantly from Bacteroidia and Clostridia (Fig. 1e). Comparing sampling sources across CRISPR systems, Class 2 systems were most enriched in deep subsurface among environmental sample categories (FET, p = 4.34 × 10−8) and were depleted in plants compared to all categories of multicellular animals among host-associated samples (Fig. 2a, b; Z-test, p < 5.25 × 10−7 for all comparisons). Systems derived from mammalian host-associated samples were significantly more likely to be capable of active editing in E. coli than systems derived from environmental or engineered samples (Fig. 2c; Z-test, p = 4.3 × 10−3, p = 0.041, respectively).
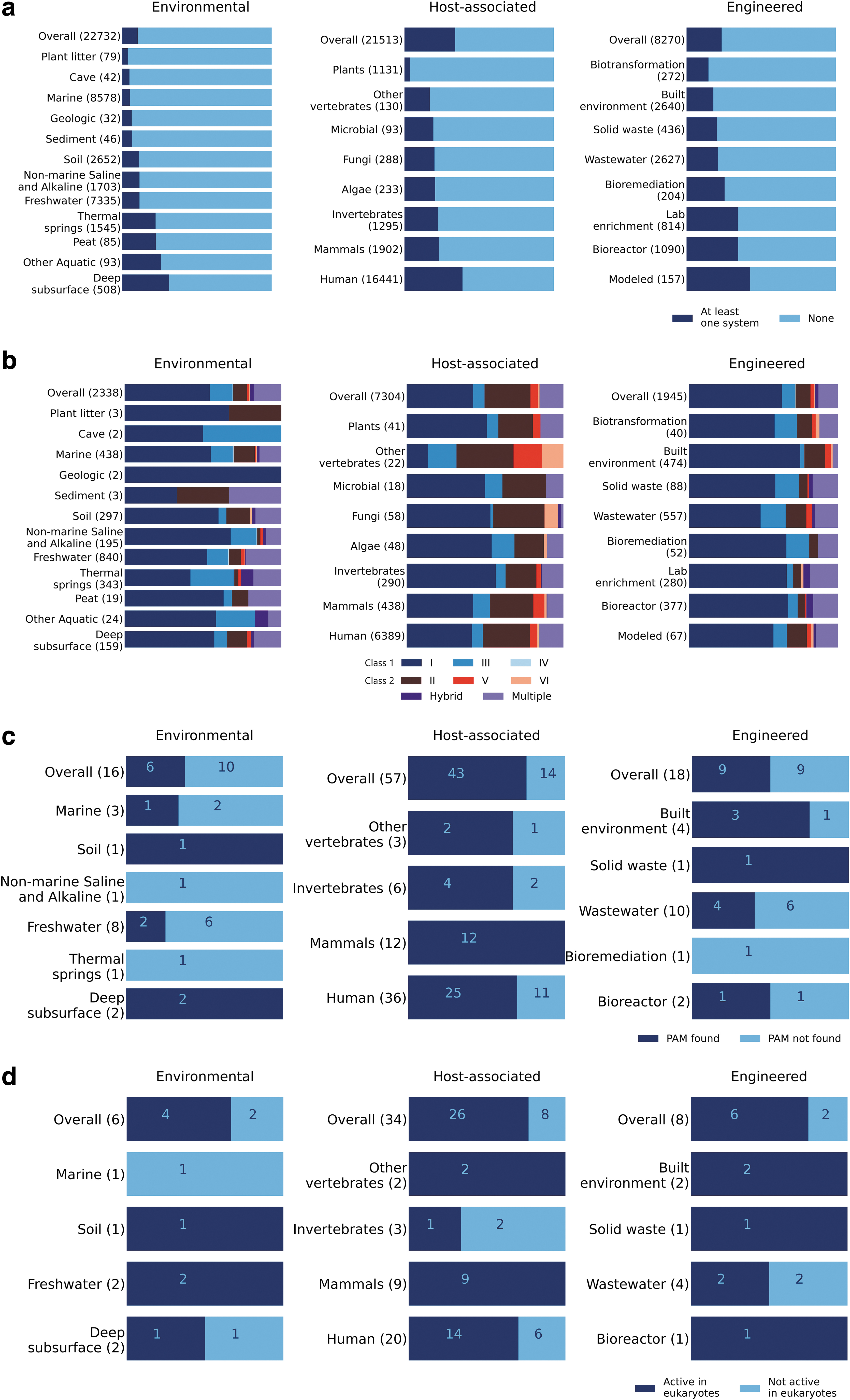
Abundance of CRISPR-Cas systems by sampling environment. Distribution of the six types of CRISPR-Cas systems according to sampling environment of metagenome-assembled genomes
A higher proportion of systems derived from mammalian host-associated samples were capable of active editing in human cells compared to other sources, but this difference was not significant, likely due to the small number of systems tested from environmental or engineered sources (Fig. 2d).
Selected diverse CRISPR effector proteins display genome-editing efficiencies comparable to leading literature systems
Of the CRISPR systems tested in eukaryotes, one Type II-A system inferred to originate from the genus Holdemanella, here designated HoCas9, and three Type V-A systems inferred to originate from Eubacterium brachy, the genus Treponema, and the family Saccharimonadaceae, here designated EuCas12a, TrCas12a, and EGS0068, respectively, were further characterized as genome editors due to their compact PAMs (Fig. 3) and editing performance across a range of eukaryotic targets (Fig. 4a). Compared to Nme2Cas9, a Type II-C effector protein of similar length but only 20% sequence identity, HoCas9 shows comparable performance across a range of targets while also possessing a desirably compact PAM. 27 EGS0068, EuCas12a, and TrCas12a share low sequence identity to AsCas12a (27%, 29%, and 35% respectively) and to each other, but EuCas12a and TrCas12a both showed significantly higher levels of eukaryotic editing across a range of targets than AsCas12a, whereas EGS0068 showed comparable activity (Fig. 4b).
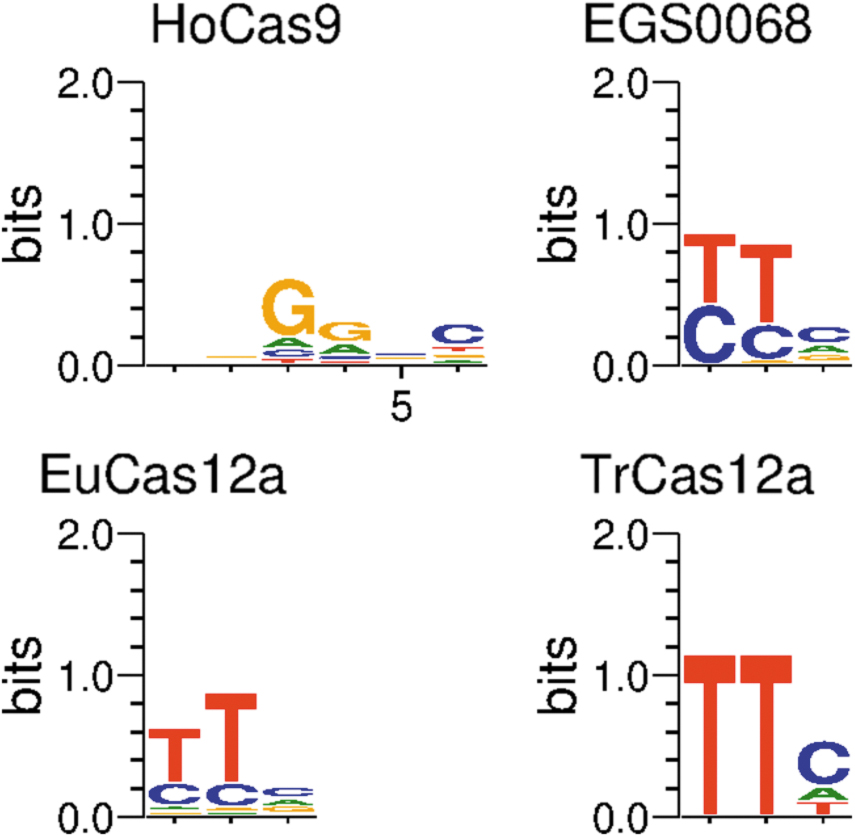
PAM logos of highest performing Class 2 CRISPR systems. PAMs of highly performing Class 2 effector proteins. HoCas9 has a 3′ PAM; EGS0068, EuCas12a, and TrCas12a all have 5′ PAMs. PAM, Protospacer Adjacent Motifs.
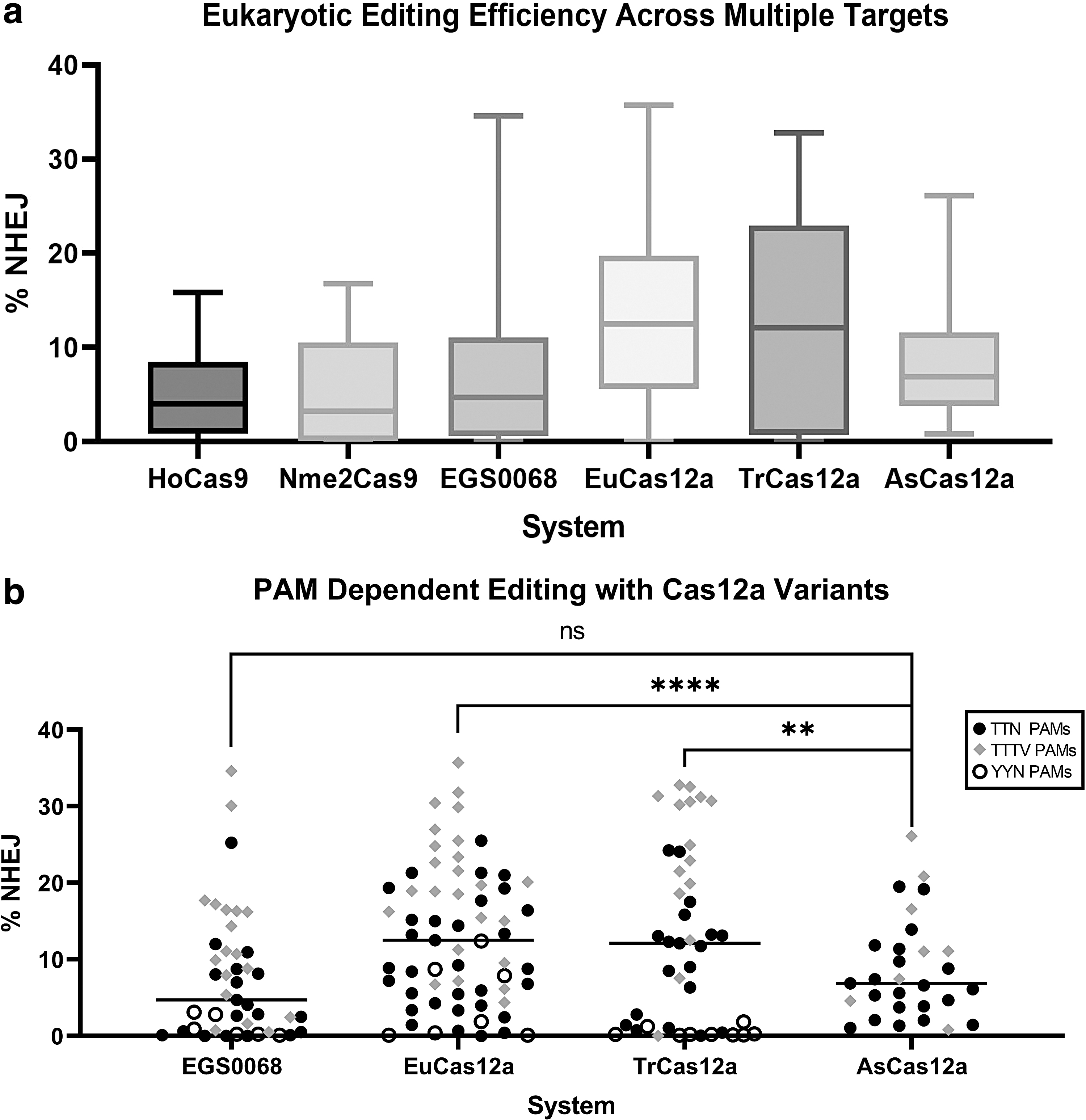
Eukaryotic activity of lead candidates. Average
While the bacterial PAM depletion assay of ESG0068 and EuCas12a indicated a higher tolerance for cytosines in the PAM than AsCas12a, editing performance was still below average for YYN PAMs compared to TTN or TTTV PAMs. We additionally observed activity of AsCas12a at PAMs that did not fit the canonical TTTV PAM of AsCas12a, similar to reports in previous studies. 5 To test if the performance of these three Cas12a candidates could be further improved, residues homologous to those previously introduced to create an enhanced version of AsCas12 (enAsCas12a; E174R_S542R_K548R) were identified via multiple sequence alignments and introduced into EuCas12a (Q158R_S528R_K535R), TrCas12a (E165R_D584R_K590R), and EGS0068 (Q167R_N575R_K581R). 14 Multiple targets were then tested with a variety of PAMs in HEK293T cells and performance was improved for TrCas12a and EGS0068, but, surprisingly, the mutations impaired activity in EuCas12a (Supplementary Fig. S3).
Initially, EGS0068 and TrCas12a showed higher performance on targets with a YTTN PAM compared to RTTN PAM, but with the enhanced versions, we saw improved activity at both RTTN and YTTN PAMs, indicating improved performance across both preferred and nonpreferred targets. The leading systems were then compared against enAsCas12 with a variety of PAMs across a range of targets, and all showed comparable editing (Fig. 5).
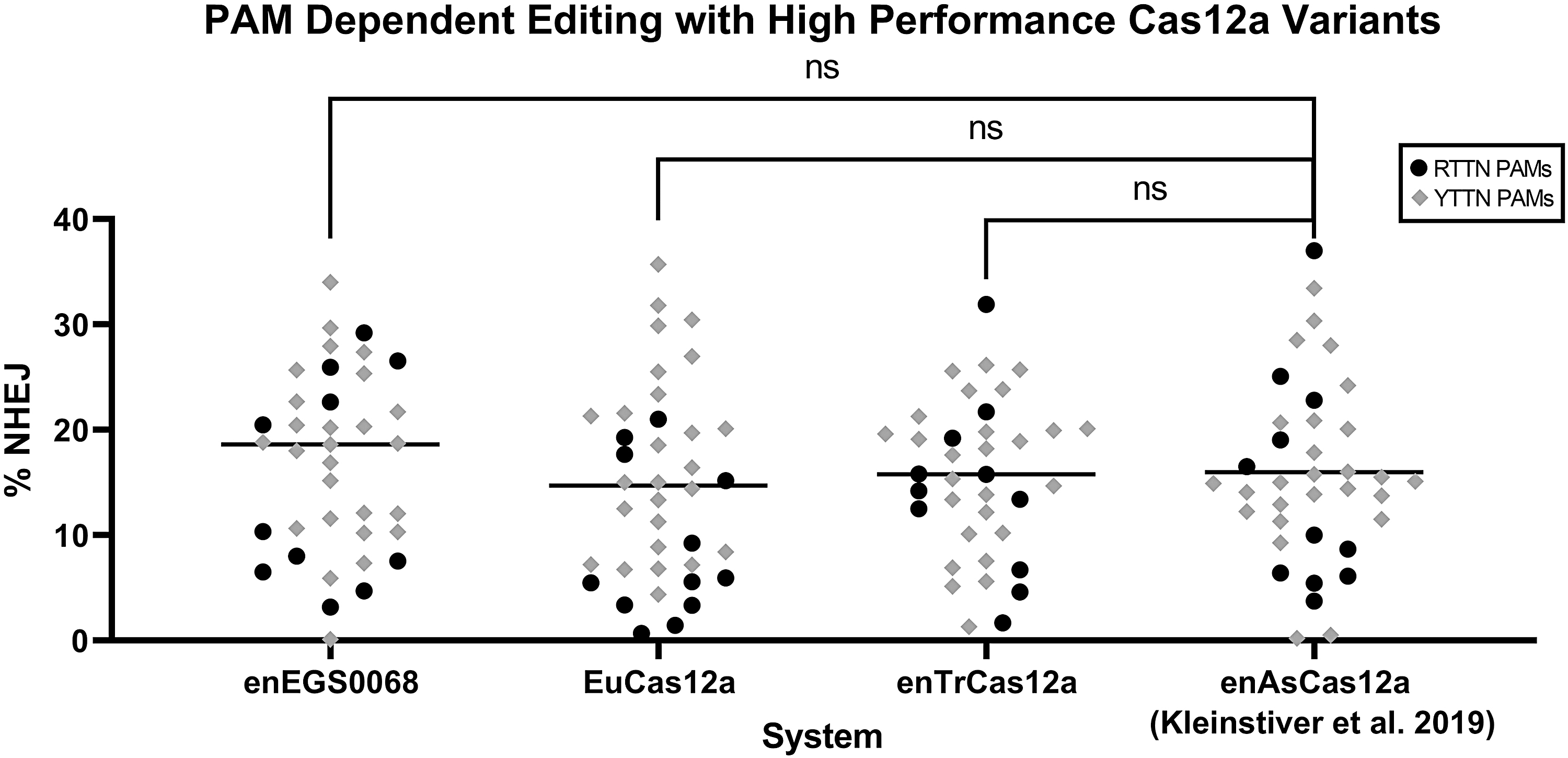
Multiple target activity of high-performing variants compared to enAsCas12a. Indel activity in 293T cells of enEGS0068 at 37 targets, EuCas12a at 38 targets, enTrCas12a at 37 targets, or enAsCas12a at 37 targets with the indicated PAM sequence. Each dot represents a single target site, averaged from replicates. Paired t-test comparisons were performed considering just identical target sites across systems. Median performance of all sites in the system is indicated with a horizontal bar.
Use as a base editing system
Cytosine base editors (CBEs) encourage fixing of deamination events by fusion with UGI(s). 9 We filtered public genome collections by custom criteria for protein length and protein surface amino acid identity and charge (see Methods section) to identify a novel putative uracil-protecting protein in Bacillus phage vB_BpuM-BpSp (BpSpUPP). BpSpUPP, when fused to APOBEC3A and a nickase-mutated Nme2Cas9, increased the rate of on-target C > T mutations, while reducing off-target mutations compared to a fusion lacking the UPP (Supplementary Fig. S4), presumably by protecting the uracil generated by cytidine deamination. BpSpUPP is 121 amino acids, more than 50 amino acids shorter than the dual UGI used in the well-characterized BE4 design. 9
To generate a wholly novel CBE, EuCas12a was rendered catalytically dead by mutating all three catalytic RuvC residues into alanine (D9556A_E1050A_D1291A, dEuCas12a), fused to APOBEC3A at the amino terminus and to BpSpUPP at the carboxy terminus with flexible linkers (Supplementary Table S2). Productive CBE was observed across several sites (Fig. 6a), but at a lower frequency than nonhomologous end joining with the wild-type enzyme. Nevertheless, on-target C > T mutations were the predominant editing outcome for the base editor (Fig. 6b,c). In addition, the editing window appears to be rather broad, with high levels of base editing occurring from positions 11–23 downstream of the PAM at the preferred TC motif.
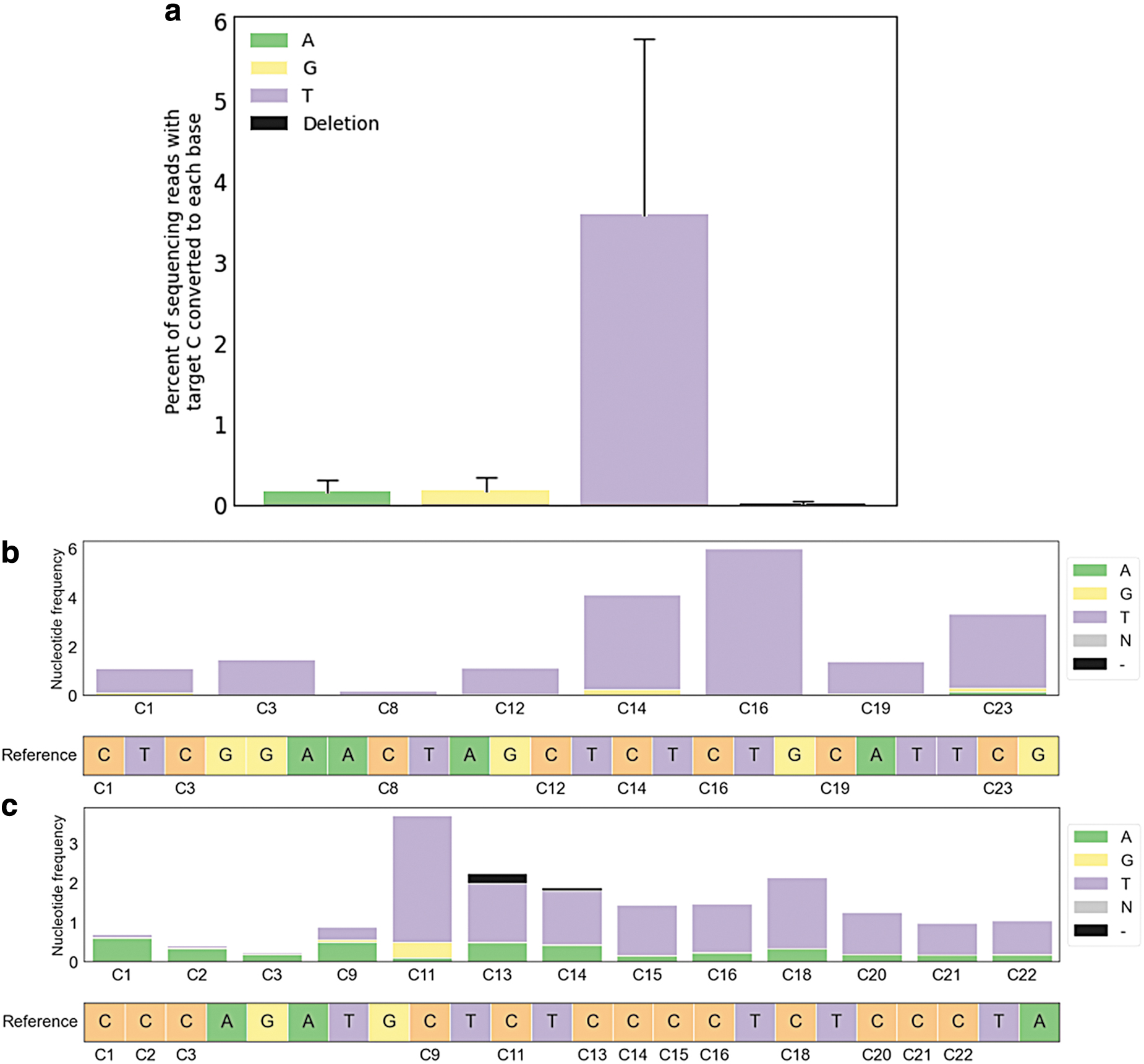
Cytosine base editing with EuCas12a. Performance of base editing on cytosines within the target window
Discussion
We conducted a broad analysis of diverse Class 2 CRISPR families based on analysis of metagenomes collected from a variety of environments. By performing a large-scale exploration on diverse metagenomes, we were able to identify Firmicutes (Cas9 and Cas12a) and Bacteroidota (Cas12a) as the bacterial phyla most likely to contain systems capable of performing eukaryotic gene editing in human cells. Nonplant host-associated environments appear to be the prime sampling location for discovering Class 2 CRISPR systems in general, potentially by providing a favorable environment for Firmicutes and Bacteroidota. The greater frequency of CRISPR systems in either nonplant host-associated or fluid environments may imply greater competition between bacteria and phages in these environments.
The observed enrichment may be attributable to increased interactions caused by the mobility of the host, resource density, and/or enhanced diffusion, as sessile plants, solid soil, and resource-poor air, are much less likely to contain a CRISPR phage defense system of any type. In addition, Class 1 CRISPR systems are more common in environmental and engineered samples than Class 2 systems. Notably, systems from environments where extremophiles are found, such as thermal springs and deep subsurface, were highly enriched in Class 1 systems as well, with a few Class 2 systems as well, but the cause is not immediately apparent. The above observations may be used to guide metagenomic sample site selection when such efforts are motivated by CRISPR system discovery.
As expected, systems taken from environments most similar to assay environmental conditions, such as host-associated, were more likely to demonstrate activity in E. coli and 293T cells, potentially reflecting an overlapping range of active temperatures or other environmental conditions common across systems useful for human genome editing. These discoveries can be used to guide future discovery work for systems suitable for eukaryotic gene editing.
We discovered thousands of CRISPR-Cas nucleases, diverse within and across families. For a subset of these, we establish guide RNA requirements (or a single crRNA for Cas12a enzymes) and PAM requirements. Collectively, the diversity of the discovered PAMs enables diverse targeting capabilities. By design, these effectors are not close homologs to previously reported enzymes and, together, greatly expand the diversity of Class 2 nucleases. When testing nucleases for in vitro editing activity with plasmid transfections, we found a Cas9 enzyme, HoCas9, small enough to be packaged into an adeno-associated virus (AAV) vector that performs as well as Nme2Cas9 and with a unique PAM sequence, which broadens the addressable target space for AAV-based delivery of gene editing. 27
In addition, we identified three Cas12a enzymes that edit as well or better than AsCas12a at TTN PAMs. In the case of EGS0068 and EuCas12a, we also demonstrated activity at PAMs not preferred by well-characterized Cas12a systems in addition to typical TTN PAMs. We confirmed the reports that AsCas12a exhibits broader targeting specificity than the canonical TTTV PAM and performs equally well at TTN PAMs. 5 When attempting to enhance the activity and broaden the targeting range of these Cas12a variants, we introduced mutations homologous to those reported elsewhere to create enAsCas12a. 14 We observed significant improvement with one of them (EGS0068), moderate improvement with another (TrCas12a), and a decrease in activity with the third mutant (EuCas12a).
Notably, when compared to enAsCas12a directly, we saw comparable activity with enEGS0068, enTrCas12a, and EuCas12a, the last of which—as a native sequence with high baseline performance and a more flexible PAM than enAsCas12a—shows promise as an object of subsequent, targeted engineering efforts. We developed an active CBE from EuCas12a and a newly discovered peptide from Bacillus phage vB_BpuM-BpSp, capable of fixing C > T mutations. This fusion protein showed a broad base editing window. Taken together, these nucleases demonstrate highly efficient gene editing and expand the genome-editing toolbox.
Conclusion
The CRISPR systems discovered here increase the known diversity of Class 2 single-effector CRISPR nucleases and demonstrate the genome-editing potential of diverse enzymes from a variety of sources. Moreover, we show that Class 2 systems are more common in nonplant host associated environments than other environments. Thirty-six diverse nucleases showed in vitro activity with varied PAM requirements, and four showed extremely high levels of editing, comparable to leading Class 2 enzymes. They show utility as base editors and utilize a novel uracil protecting protein for improved CBEs. These nucleases expand the toolkit of CRISPR-associated enzymes with diverse genome-engineering applications.
Footnotes
Acknowledgments
The authors thank Lauren Drowley for helpful discussions and everyone at UCB for creating a productive and stimulating scientific environment. The metagenomic data were produced by the U.S. Department of Energy Joint Genome Institute (![]() operated under Contract No. DE-AC02-05CH11231) in collaboration with the user community.
operated under Contract No. DE-AC02-05CH11231) in collaboration with the user community.
Authors' Contributions
It is the authors' opinion that M.W. and L.R. contributed equally to this work. M.W.: investigation, writing-original draft, and validation. L.R.: uracil protecting protein data curation, formal analysis, methodology, software, visualization, and writing–original draft. J.v.B.: investigation, methodology, and writing–review and editing. M.M.: investigation and writing–review and editing. S.M.: investigation, methodology, and writing–review and editing. I.M.: conceptualization, formal analysis, software, supervision, and writing–review and editing. T.B.: conceptualization, formal analysis, investigation, methodology, project administration, supervision, visualization, and writing–original draft.
Author Disclosure Statement
All authors are/were employees of UCB Biosciences Inc., receive salary from the company, and might own equity in the company. Patent applications related to the article have been filed.
Funding Information
This work was funded entirely by UCB Biosciences Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.