
Abstract
CRISPR-based genome-editing technologies, including nuclease editing, base editing, and prime editing, have recently revolutionized the development of therapeutics targeting disease-causing mutations. To advance the assessment and development of genome editing tools, a robust mouse model is valuable, particularly for evaluating in vivo activity and delivery strategies. In this study, we successfully generated a knock-in mouse line carrying the Traffic Light Reporter design known as TLR-multi-Cas variant 1 (TLR-MCV1). We comprehensively validated the functionality of this mouse model for both in vitro and in vivo nuclease and prime editing. The TLR-MCV1 reporter mouse represents a versatile and powerful tool for expediting the development of editing technologies and their therapeutic applications.
Introduction
Recent advancements in CRISPR-based genome-editing technologies have transformed both basic research and therapeutic applications.1,2 These technologies have expanded to encompass a wide range of approaches, such as nuclease editing,3–7 base editing,8–10 and prime editing. 11 One of the main objectives in gene editing is to correct or eliminate disease-causing mutations in human patients, 2 and advancing toward this goal is the subject of intense research. 12 However, before these approaches can progress to clinical trials and become approved drugs, extensive research and development are necessary to establish compelling evidence of the safety and efficacy of gene-editing treatments.13,14 To expedite and streamline this process, a good reporter mouse can be valuable for facilitating comprehensive investigations into the in vivo activity and delivery of gene-editing approaches.
Several fluorescent reporter mouse models exist for this purpose. One example is the mT/mG mouse, 15 where the membrane-targeted tdTomato (mT) is initially expressed, flanked by loxP sites. Upon Cre-mediated recombination, the mT sequence is excised, leading to the expression of downstream membrane-targeted EGFP (mG). Another notable example is the Ai9 mouse, 16 which will exhibit tdTomato expression following the removal of a loxP-flanked stop cassette by Cre recombinase. Both of these models, while originally developed as Cre reporters, have also proven useful as reporters of CRISPR-Cas-mediated genome editing when guides are designed to target the appropriate site(s).17–21 However, while these models are useful for assessing nuclease-mediated excision, they have limitations when applied to precision editing approaches, such as homology-directed repair (HDR), base editing, and prime editing.
Recently, a mouse model with a disrupted EGFP gene (mouse lines with 1 or 13 bp deletion available) has been developed to evaluate CRISPR-Cas-based cleavage and HDR. 22 While these reporters are designed specifically for HDR (precise repair using a donor template), in some cases indels may also restore the reading frame, complicating the interpretation and comparison of different editing outcomes.
By contrast, Traffic Light Reporter (TLR) systems offer a unique advantage by enabling the simultaneous tracking of different repair and editing outcomes using distinct fluorescent markers.23–25 Typically, the TLR design involves a broken Marker-X, such as GFP, followed by a frameshifted Marker-Y, such as mCherry. Before gene editing, no signal is detected. However, in the presence of nuclease-mediated indels, frame-restoring mutations can occur, leading to the correction of mCherry reading frame and its subsequent expression. Alternatively, when a donor template is provided alongside the nuclease, successful HDR events can restore the functionality of GFP. Recently, we have developed a versatile traffic light reporter construct called TLR-multi-Cas variant 1 (TLR-MCV1), 26 which enables the comparison of different nuclease systems (SpyCas9, Nme1Cas9, Nme2Cas9, SauCas9, CjeCas9, Geo1Cas9, Geo2Cas9, AspCas12a, LbaCas12a, FnoCas12a, etc.) in cell lines.
In this study, we generated a knock-in mouse line carrying the TLR-MCV1 reporter integrated into the Rosa26 locus, a safe harbor site (Fig. 1A, B). First, we demonstrated in vitro and in vivo nuclease-mediated editing for this reporter mouse. Moreover, using different prime-editing guide RNAs (pegRNAs), we successfully applied this reporter to prime-editing-mediated restoration of either mCherry or GFP expression in vitro and in vivo. Overall, our findings highlight the TLR-MCV1 mouse as a versatile reporter system for various nuclease editors and prime editors, particularly for assessing in vivo activity and delivery strategies; this model promises to facilitate genome-editing research and its applications.
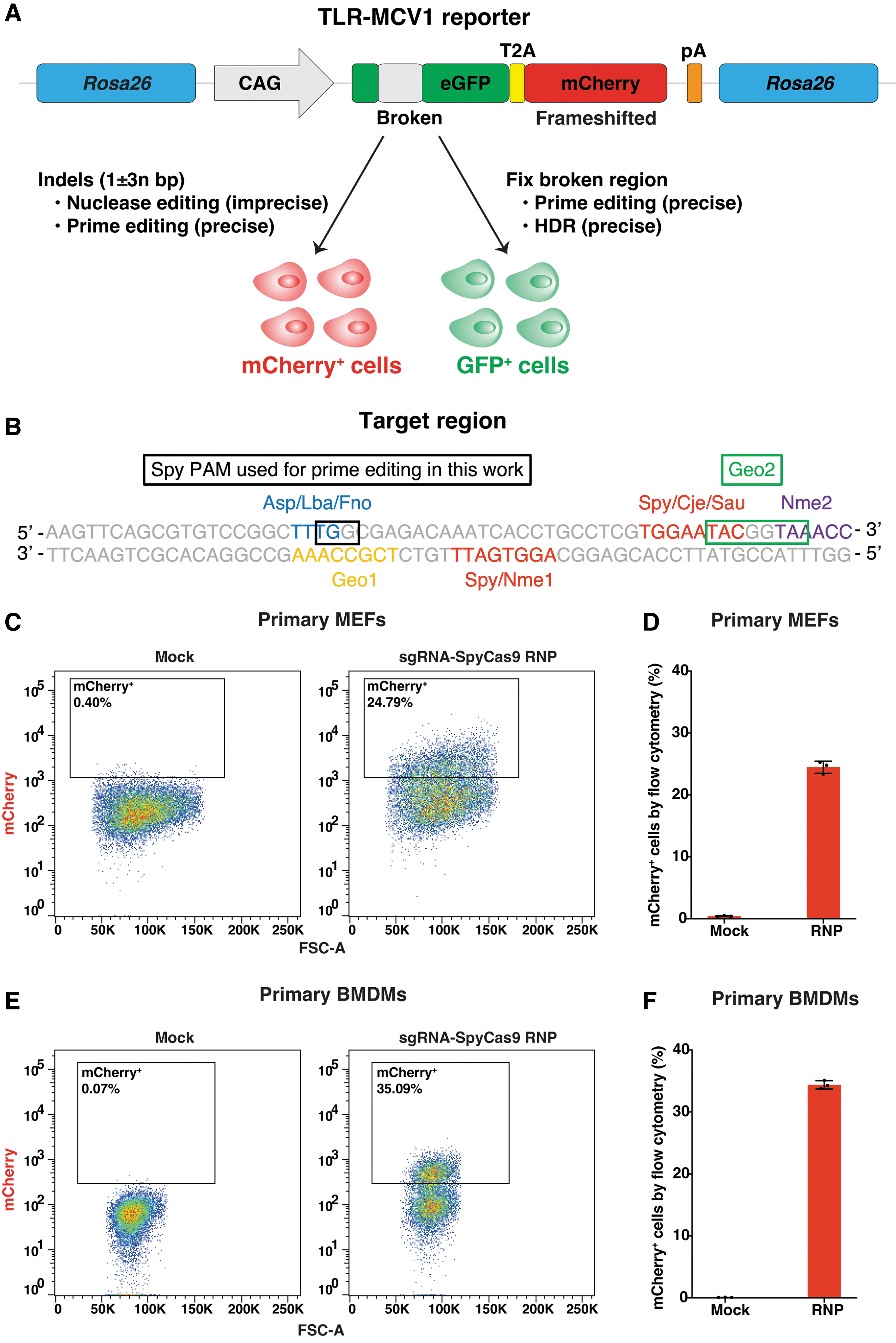
Nuclease editing in primary cells from the TLR-MCV1 reporter mouse.
Methods
Generation of plasmids
Donor plasmid for pronuclear microinjection was made by an assembly of TLR-MCV1 transgene into the vector (Addgene plasmid 74286; http://n2t.net/addgene:74286; RRID:Addgene_74286). 27 pegRNAs and nicking single-guide RNAs (sgRNAs) were designed using PrimeDesign software. 28 pegRNA or sgRNA plasmids were assembled between U6-promoter-contained vector and pegRNA or sgRNA sequence insert using HiFi DNA assembly master mix (NEB). pCMV-PE2 plasmid was a gift from David Liu (Addgene plasmid 132775; http://n2t.net/addgene:132775; RRID:Addgene_132775). 11 pAAV.pU1a-SpCas9 plasmid was the Addgene plasmid 121507 (http://n2t.net/addgene:121507; RRID:Addgene_121507). 29 pAAV.sgRNA (for SpyCas9) plasmids were assembled between the vector containing AAV2-ITR and the U6 promoter, and the insert including the sgRNA sequence. pAAV.Nme2Cas9.sgRNA was constructed by inserting a pair of annealed oligonucleotides corresponding to the spacer region into the vector (Addgene plasmid 119924, All-in-one AAV plasmid expressing Nme2Cas9 with sgRNA cloning cassette). 30
Plasmids were confirmed by Sanger sequencing, and purified using a miniprep kit (Promega) for in vitro experiments, or an endo-free maxiprep kit (Qiagen) for in vivo experiments. The sgRNA sequences are listed in the Supplementary Table S1. The pegRNA sequences are listed in the Supplementary Table S2.
Cell culture and electroporation
A HEK293T-derived cell line carrying the TLR-MCV1 reporter was generated in-house and is described elsewhere. 26 Primary mouse embryonic fibroblasts (MEFs) were isolated from the TLR-MCV1 mouse embryos using methods described elsewhere. 31 Primary bone marrow-derived macrophages were prepared from the TLR-MCV1 mouse bones using methods described elsewhere. 32 All cells were cultured in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin under 37°C and 5% CO2.
Electroporation was performed using the Neon electroporation system (ThermoFisher) according to the manufacturer's protocol. Briefly, for Cas9 ribonucleoprotein (RNP) electroporation into bone-marrow-derived macrophages (BMDMs) or MEFs, 10–20 pmol SpyCas9 (UCB QB3-MacroLab) and 30–60 pmol sgRNA (IDT) were first incubated under room temperature for 30 min, then mixed with 100,000–200,000 cells in buffer R, and electroporated in 10 μL Neon tips using the following parameters: 1720 V—10 ms—2 pulses (BMDMs) or 1650 V—20 ms—1 pulse (MEFs).
For HDR experiment, 20 pmol SpyCas9, 50 pmol sgRNA, and 2.2 pmol double-stranded DNA (dsDNA) donor (either triethylene glycol [TEG] or unmodified donor) were electroporated into 200,000 primary MEFs. The dsDNA donor was produced by polymerase chain reaction (PCR) using Q5 mastermix (NEB), the plasmid template containing donor sequence (Supplementary Table S5), and TEG primers (Spacer 9 modification at the 5′-end of primers; IDT) or unmodified primers (IDT). Then donor was purified using PCR cleanup kit (Qiagen).
For prime-editing plasmid electroporation into HEK293T cells or MEFs, 400 ng pegRNA, 1200 ng PE2, with or w/o nicking 133 ng sgRNA, and 100,000–200,000 cells were mixed in buffer R, and then electroporated in 10 μL Neon tips using the following parameters: 1150 V—20 ms—2 pulses (HEK293T cells) or 1650 V—20 ms—1 pulse (MEFs). After electroporation, cells were plated in prewarmed DMEM containing 10% FBS and incubated for 72 h before analysis.
Flow cytometry
Seventy-two hours postelectroporation, the cells were trypsinized, washed in phosphate-buffered saline, and resuspended in phosphate-buffered saline with 2% FBS. Flow cytometry was performed using MACS-Quant VYB (Miltenyi Biotec) according to the manufacture's protocol. Briefly, forward scatter area (FSC-A) versus side scatter area was used to gate for live cells. Then FSC-A versus forward scatter height (FSC-H) was used to gate for singlet cells. mCherry+ cells were gated using the Y2 channel, and GFP+ cells were gated using the B1 channel. Data were analyzed using FlowJo software.
Genomic DNA isolation, amplicon sequencing, and data analysis
The genomic DNA (gDNA) from cell culture was isolated using QuickExtract DNA extraction solution (Lucigen). Mouse gDNA was purified using DNeasy blood and tissue kit (Qiagen).
Amplicon sequencing libraries were prepared following two-step PCR amplification. Briefly, for the first PCR, the primers (Supplementary Table S3) containing genomic sequence and Illumina forward/reverse adapter sequence were used to amplify the TLR-MCV1-edited genomic site. Then, for the second PCR, the Illumina-barcode primers were used to amplify the first PCR product. NEBNext Ultra II Q5 Master Mix was used for both PCR reactions. The second PCR products were pooled together, purified by gel electrophoresis using QIAquick gel extraction kit (Qiagen), and cleaned up again using QIAquick PCR purification kit (Qiagen). The DNA concentration was measured using Qubit dsDNA HS assay kit (Invitrogen). The amplicon library was then sequenced on the Illumina Miniseq according to the manufacturer's protocol.
The amplicon sequencing data was analyzed as described previously. 33 Briefly, sequencing reads were first demultiplexed using bcl2fastq (Illumina); then the demultiplexed fastq files were batch-analyzed using CRISPResso2. 34 The nuclease editing efficiency was determined using NHEJ mode with quantification window size (w) = 10, minimum average quality score (q) = 30, and ignore_substitutions = TRUE. The percentage of indels was calculated as the percentage of the modified reads.
The prime editing efficiency was determined using HDR mode with quantification window size (w) = 10 (for mCherry prime editing [PE] analysis) or 40 (for GFP PE analysis), minimum average quality score (q) = 30, and ignore_substitutions = TRUE. The percentage of correct edits = 100% × (the unmodified reads aligned to PE amplicon)/(the total of unmodified and modified reads aligned to both WT and PE amplicons). The percentage of un-edit = 100% × (the unmodified reads aligned to WT amplicon)/(the total of unmodified and modified reads aligned to both WT and PE amplicons). The percentage of indels was calculated as 100% − (the percentage of correct edits) − (the percentage of un-edited reads).
Pronuclear microinjection and in vivo experiments
All mouse studies were conducted according to the Institutional Animal Care and Use Committee (IACUC) at University of Massachusetts Chan Medical School. The pronuclear microinjection was performed at the Transgenic Animal Modeling Core, UMass Chan Medical School, using methods described elsewhere. 35 Briefly, C57BL/6J (Stock #000664) mouse were obtained from Jackson Laboratory. Superovulated females were mated, and their zygotes were collected at E0.5. The pronuclei were injected with the injection mixtures including SpyCas9 protein (IDT), SpyCas9 messenger RNA (mRNA; TriLink), sgRNA (Synthego) targeting Rosa26 locus, and donor plasmid containing TLR-MCV1 transgene, and Rosa26 homology arms. Finally, zygotes were transferred to pseudo-pregnant recipients and allowed to go to term.
For AAV injection, adenoassociated virus serotype 9 (AAV9) vectors were produced by the Viral Vector Core at Horae Gene Therapy Center, UMass Chan Medical School. The viral titers were determined using gel electrophoresis, silver staining, and droplet digital PCR. For liver and heart analysis, 1 × 1012 gc per AAV was injected intravenously into each mouse. For lung analysis, 4 × 1011 gc per AAV was injected intratracheally. For brain analysis, about 2.5 × 1010 gc per AAV was injected intrastriatally into each side of the mouse brain. For muscle analysis, about 2–4 × 1011 gc per AAV was injected intramuscularly into each mouse. The mice were sacrificed and harvested for the tissues at 4 weeks postinjection for downstream analysis. For plasmid hydrodynamic tail vein injection, 30 μg PE2 and 15 μg pegRNA or sgRNA plasmids were injected into each mouse. The mice were sacrificed, and their tissues were harvested 2 weeks postinjection for downstream analysis.
Immunohistochemistry
The immunohistochemical staining was performed using methods described previously.36,37 Briefly, mouse tissues were fixed with 4% formalin overnight, embedded with paraffin, and sectioned. The tissue sections were dewaxed, rehydrated, and stained accordingly. Antibody used here: anti-RFP (Rockland 600-401-379), 1:300 dilution. Leica DMi8 microscopy was used to image the immunohistochemistry (IHC) slides.
TLR-MCV1 mouse strain availability
The TLR-MCV1 reporter mouse has been deposited with the Jackson Laboratory for distribution under Stock number 038717.
Data availability
Illumina amplicon deep sequencing data will be available at the NCBI Sequencing Read Archive (SRA) database. All other data are available upon request.
Results
Generation and in vitro validation of the TLR-MCV1 reporter mouse
We harnessed CRISPR-Cas9-mediated HDR to achieve the targeted integration of the TLR-MCV1 transgene into the mouse Rosa26 safe harbor locus. The pronuclear microinjection mix include a sgRNA targeting the mouse Rosa26 locus, SpyCas9 protein, and mRNA, as well as a donor plasmid carrying the TLR-MCV1 cassette and Rosa26 homology arms. Successful knock-in of the TLR-MCV1 transgene was confirmed through PCR genotyping analysis and Sanger sequencing (Supplementary Fig. S1 and Supplementary Table S4).
We then validated the functionality of the integrated TLR-MCV1 transgene by in vitro experiments. Initially, primary MEFs were isolated and cultured. Subsequently, the sgRNA-SpyCas9 RNP complex was introduced into the primary MEFs via electroporation. The resulting mCherry expression after gene editing was confirmed and assessed using flow cytometry (Fig. 1C, D). Likewise, following electroporation of the RNP complex, mCherry expression was also observed in primary BMDMs from the mouse (Fig. 1E, F). In both MEFs and BMDMs, we observed an occurrence of 20–30% mCherry+ cells (Fig. 1D, F), indicating the functional success of the TLR-MCV1 transgene in vitro. The actual extent of editing would be higher, since this editing percentage reflects the fraction of indels that resulted in correction of the mCherry reading frame.
Furthermore, we aimed to validate the expression of GFP following the repair of its broken region by HDR-based gene editing. In this experiment, we utilized a double-stranded DNA donor featuring a TEG group on both 5′-ends, which have previously demonstrated its capacity to enhance HDR efficacy in mammalian cells. 35 After electroporating both the RNP and the donor into primary MEFs derived from the TLR-MCV1 mouse, we observed successful GFP expression (Fig. 2A, B). Consistent with previous results, the TEG donor exhibited higher HDR editing when compared to the unmodified donor (Fig. 2A, B). As anticipated, the RNP alone or the donor alone yielded only background-level signals (Fig. 2A, B). Taken together, we have successfully confirmed the ability of cells derived from the TLR-MCV1 mouse to report on both HDR outcomes (by GFP repair) and end-joining outcomes (by frameshifts leading to mCherry expression).
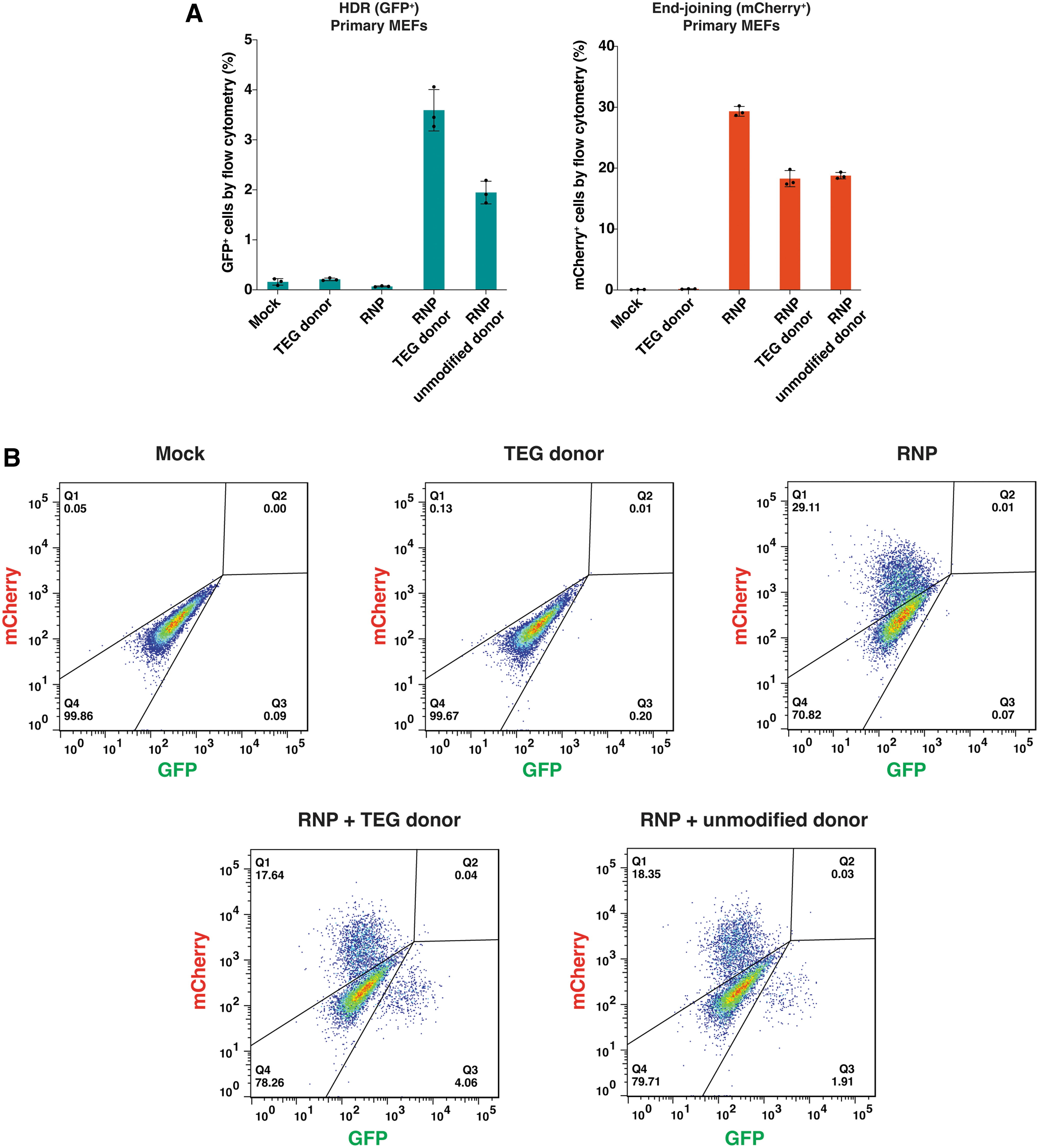
HDR-based editing in primary cells from the TLR-MCV1 reporter mouse.
In vivo nuclease editing of the TLR-MCV1 reporter mouse
To further illustrate the utility of the TLR-MCV1 mouse, we used the AAV9 to deliver the sgRNA and either SpyCas9 or Nme2Cas9 for nuclease editing across various tissue and organ types. We investigated the liver and heart after intravenous injection of AAVs. Following systemic delivery of SpyCas9+sgMCV1 via dual-AAV or Nme2Cas9+sgMCV1 via single-AAV, mCherry expression was observed through IHC staining (Fig. 3A), and indels were confirmed via amplicon sequencing (Fig. 3B, C) in the liver and heart. Interestingly, the abundance of mCherry+ cells surpassed that of indels in the heart. One plausible explanation is the multinucleation and nuclear polyploidization phenomenon in cardiomyocytes, 38 leading to the lower number of indels observed, but resulting in a higher proportion of mCherry-expressed cells, as even a single reading-frame-corrected copy is theoretically sufficient for detection by IHC staining. Moreover, we detected mCherry expression and indels in the lung (Fig. 3A, B) after intratracheal injection of AAVs.
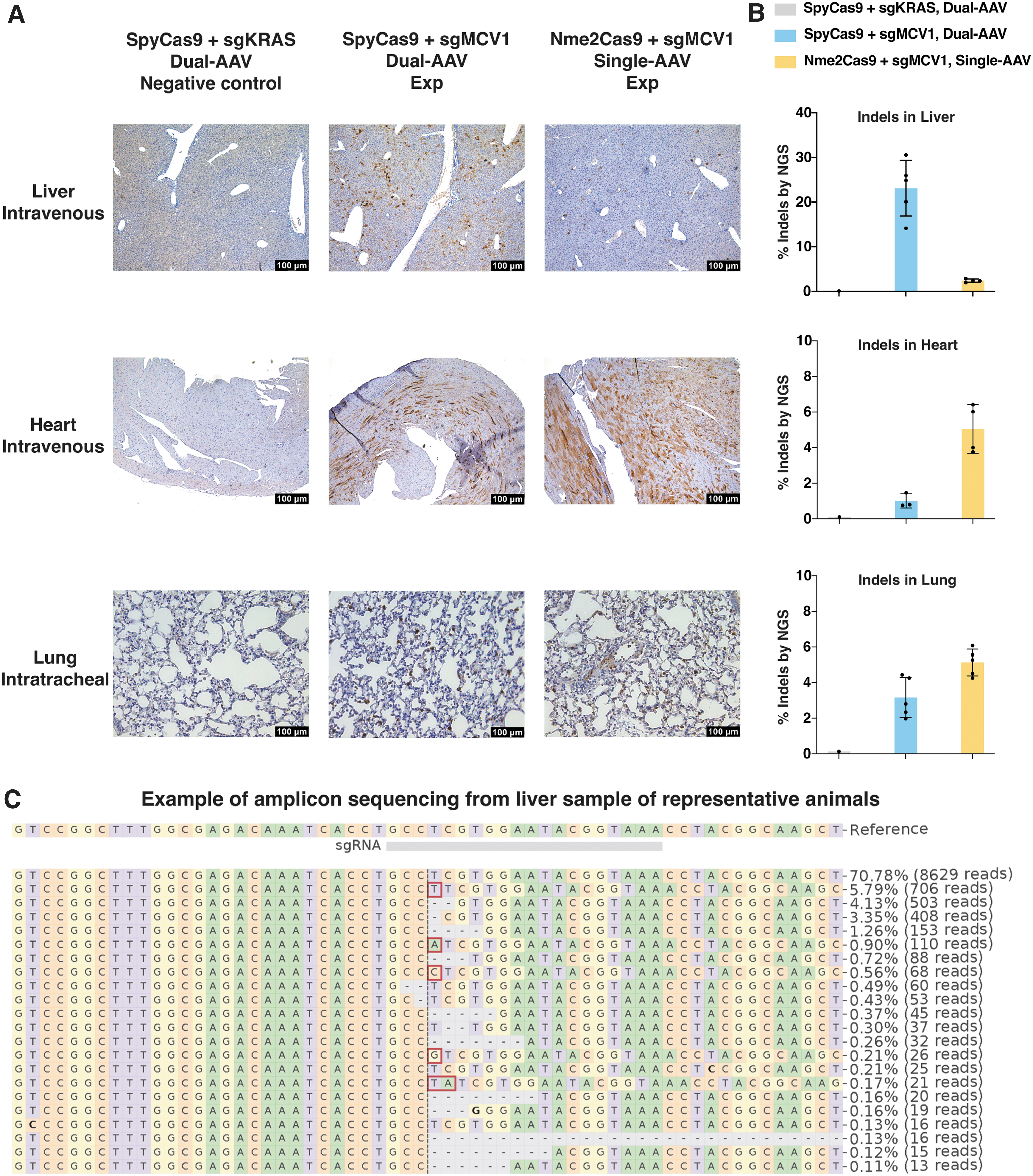
In vivo nuclease editing at the TLR-MCV1 reporter using SpyCas9 or Nme2Cas9 delivered by AAV9.
Notably, SpyCas9 exhibited greater nuclease editing efficiency in the liver, whereas Nme2Cas9 displayed higher editing efficiency in the heart and lung. This observation likely reflects the variations of in vivo delivery among different organs, their interplay with the intrinsic editing capacities of distinct effectors, and the impact of both on the ultimate in vivo editing outcomes. SpyCas9 and sgRNA were delivered using dual-AAV due to the larger combined size, while Nme2Cas9 and sgRNA were delivered using single-AAV owing to their smaller size. When in the liver, a much less challenging organ for in vivo delivery, SpyCas9 could likely maintain its inherent higher editing despite potential delivery limitations associated with dual-AAV. Conversely, in the case of difficult-to-deliver organs like the heart and lung, single-AAV delivery could prove advantageous for overcoming the inherent activity disparities between Nme2Cas9 and SpyCas9, and facilitating higher in vivo editing efficiency with the former.
Additionally, intramuscular and intrastriatal injection of AAVs enabled mCherry expression in muscle and brain, respectively, showing evidence of gene editing after local delivery to these tissues (Supplementary Fig. S2). Taken together, our findings demonstrate the successful in vivo nuclease editing of the TLR-MCV1 reporter mouse using different editing effectors in diverse tissues and organs.
Prime editing for restoring mCherry expression at the TLR-MCV1 reporter in vitro
We next explored the potential of the TLR-MCV1 reporter for prime editing applications due to the growing momentum of this editing strategy. Our initial focus was to correct the mCherry frameshift. Indels with 1 ± 3n bp can fix the frameshift and restore mCherry expression (Fig. 1A). Consequently, we devised four distinct prime editing strategies, namely 1 bp insertion, 1 bp insertion+PAM mutation, 4 bp insertion, or 4 bp insertion+PAM mutation (Fig. 4A). The inclusion of PAM mutation aims to enhance prime editing efficiency, as previous research has indicated that it minimizes the interference from the mismatch repair pathway. 39 To avoid potential complications arising from precise indels generated through prime editing versus stochastic indels caused by double-stranded breaks, we excluded the use of a nicking sgRNA (as used in PE3 approach) 11 for mCherry frameshift correction via prime editing.
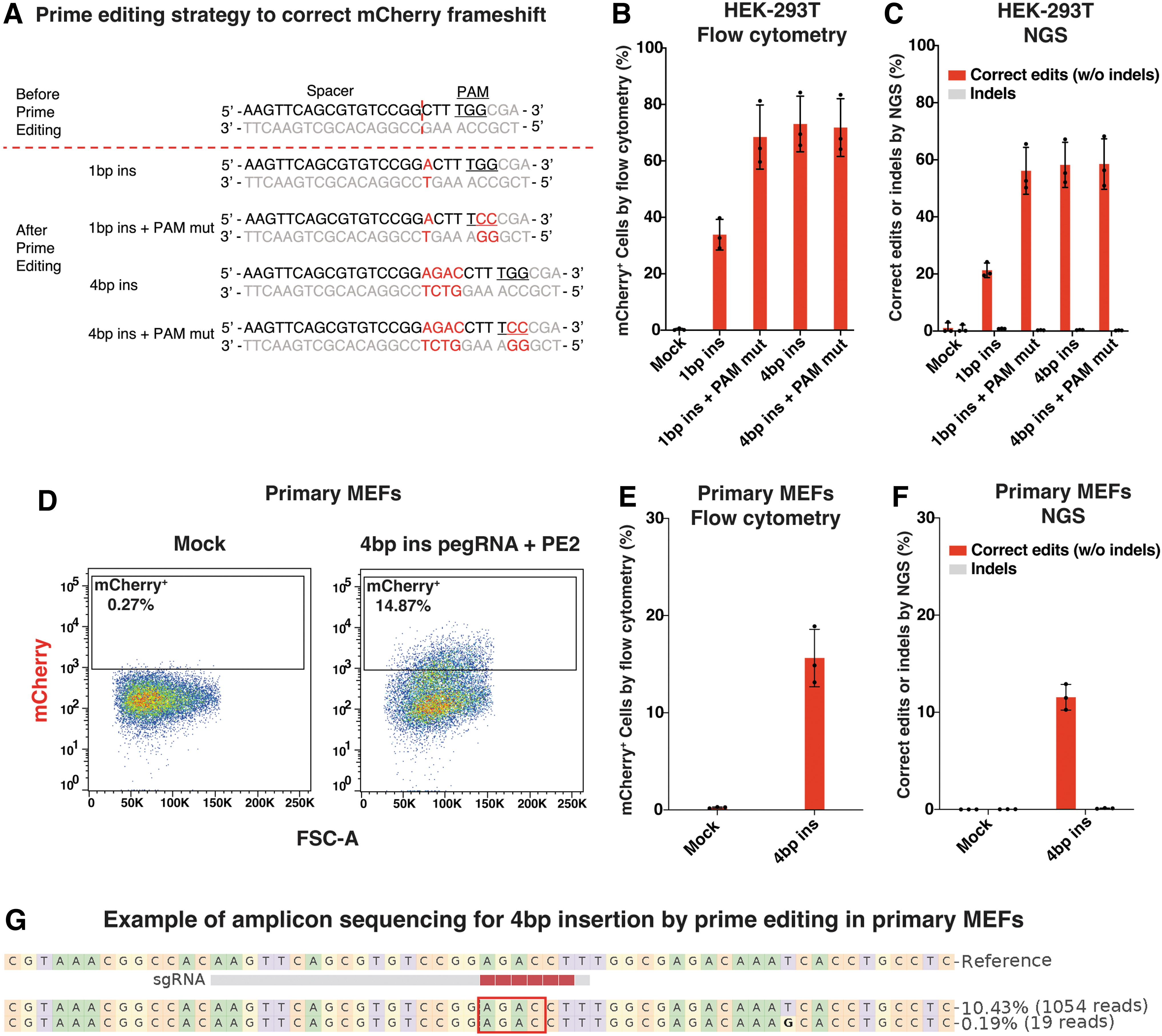
Prime editing for restoring mCherry expression at the TLR-MCV1 reporter in vitro.
Initially, we introduced the pegRNA and prime editor effector plasmids into HEK293T cells harboring the TLR-MCV1 reporter via electroporation. The expression of mCherry was assessed using flow cytometry (Fig. 4B), and the precision of editing was confirmed through amplicon sequencing (Fig. 4C). Notably, the combination of 1 bp insertion and PAM mutation exhibited enhanced editing compared to 1 bp insertion alone, as expected. However, the 4 bp insertion and PAM mutation did not yield further improvements over 4 bp insertion alone, likely due to reaching a plateau of editing efficiency.
Subsequently, we selected the 4 bp insertion prime editing strategy and evaluated it in primary MEFs obtained from the TLR-MCV1 mouse. Similarly, following electroporation of the plasmids, mCherry expression was observed (Fig. 4D, E), and the editing precision was confirmed (Fig. 4F, G). Because primary MEFs are hard to edit, the prime editing efficiency in primary MEFs was lower compared to that observed in HEK293T cells. Collectively, our results show successful prime editing for the restoration of mCherry expression at the TLR-MCV1 reporter in vitro.
Prime editing for correcting GFP expression at the TLR-MCV1 reporter in vitro
We next proceeded with the investigation of correcting GFP expression at the TLR-MCV1 reporter using prime editing methodology. The successful implementation of prime editing would repair the broken region of GFP and rescue its expression (Fig. 5A). We designed three pegRNAs with different primer binding site (PBS) lengths and included a nicking sgRNA for initial evaluation. In addition to employing amplicon sequencing to discriminate between correct edits and indels, the presence of mCherry+ cells can also serve as the indicator of indels, while the abundance of GFP+ cells can provide a measure of correct edits.
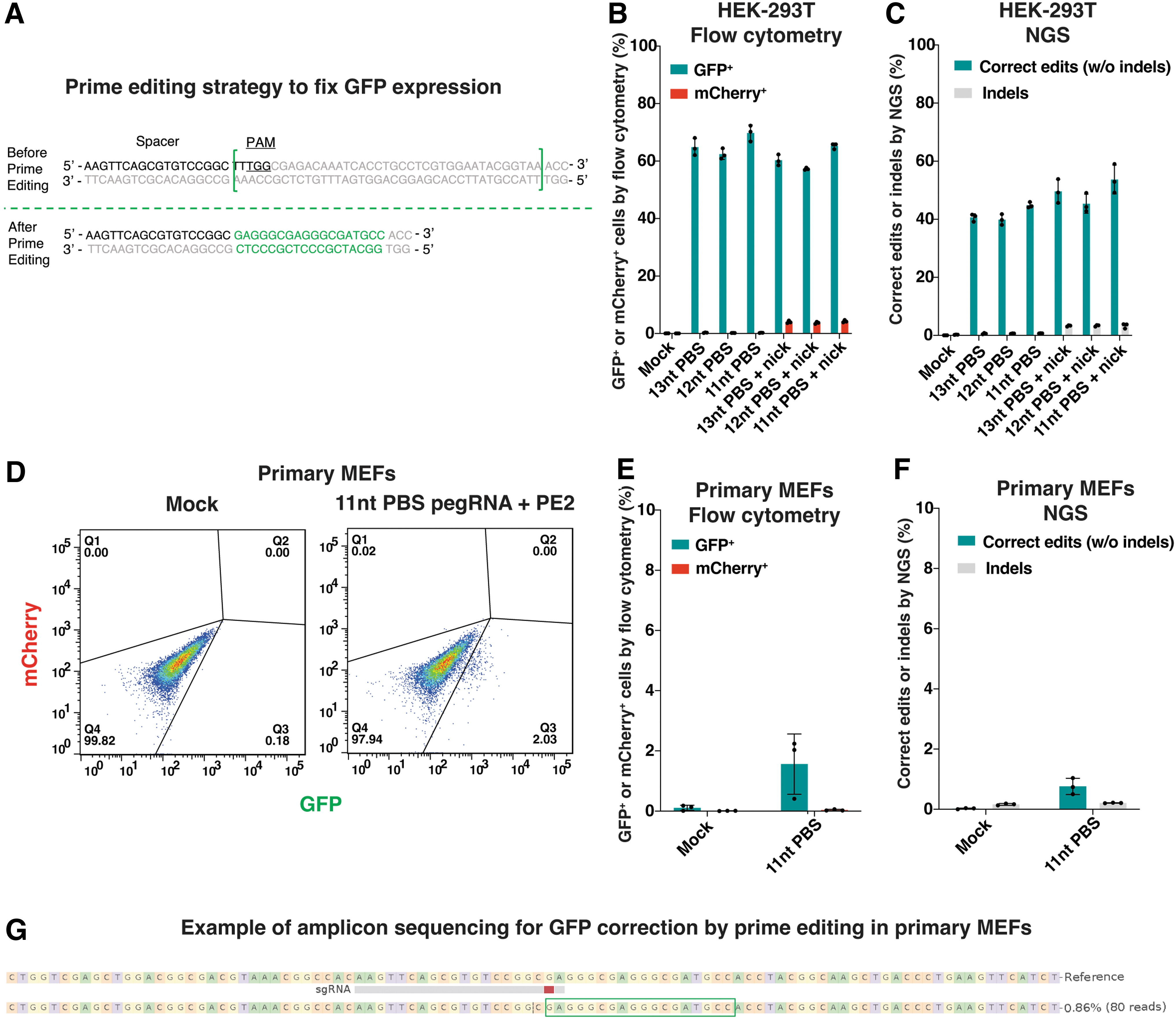
Prime editing for correcting GFP expression at the TLR-MCV1 reporter in vitro.
At first, HEK293T cells harboring the TLR-MCV1 reporter were electroporated with the pegRNA and prime editor effector plasmids, with or without the nicking sgRNA. Flow cytometry was used to assess the presence of GFP+ and mCherry+ cells (Fig. 5B), while the precision of edits was confirmed through amplicon sequencing (Fig. 5C). Notably, comparable editing efficiencies were observed among pegRNAs with different PBS lengths. The inclusion of nicking sgRNA did not lead to improved prime editing but rather introduced additional by-products, as evidenced by an increase in mCherry+ cells and indels.
Later, we chose the 11nt-PBS-length pegRNA and evaluated its performance without nicking sgRNA in primary MEFs obtained from the TLR-MCV1 mouse. Similarly, after electroporation of the plasmids, GFP expression was observed (Fig. 5D, E), and the correct edits were confirmed (Fig. 5F, G). Although prime editing is much more challenging in primary MEFs, we successfully demonstrate the capability of prime editing to restore GFP expression at the TLR-MCV1 reporter in vitro.
In vivo prime editing in the TLR-MCV1 mouse
We subsequently aimed to demonstrate the in vivo functionality of the TLR-MCV1 mouse as a platform for prime editing. Considering the relative ease of achieving mCherry frameshift correction compared to repairing the broken GFP region using prime editing, we selected and evaluated the 4 bp insertion prime editing strategy in the TLR-MCV1 mouse for the purpose of activating mCherry expression. Through hydrodynamic tail vein injection of prime editor and pegRNA plasmids (Fig. 6A), we successfully observed precise 4 bp insertions in the mouse liver, as confirmed by amplicon sequencing (Fig. 6B, C). Furthermore, mCherry expression was detected via IHC staining (Fig. 6D). Overall, our findings substantiate the suitability of the TLR-MCV1 reporter mouse as a valuable tool for in vivo assessment of prime editing.
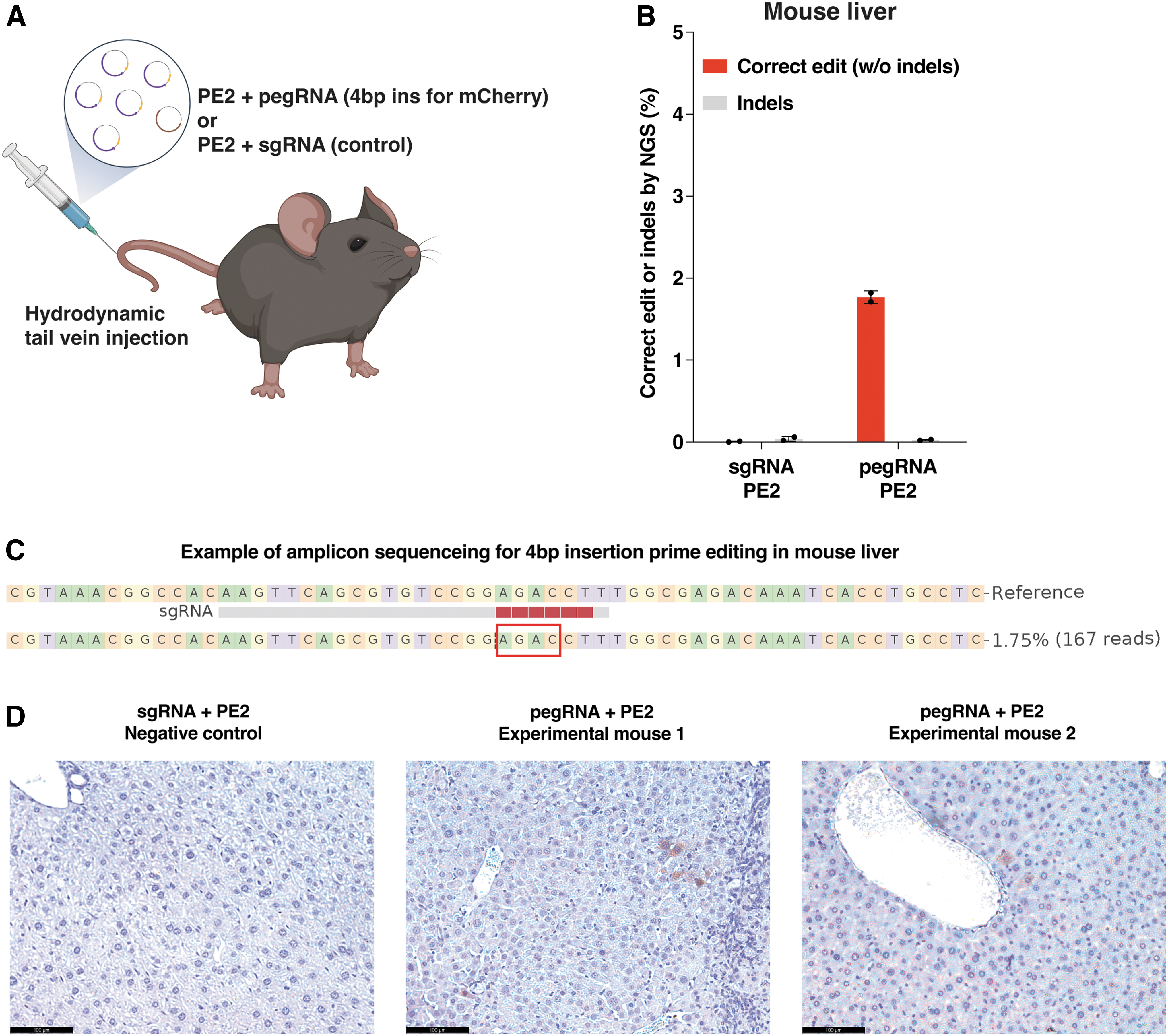
In vivo prime editing for restoring mCherry expression at the TLR-MCV1 reporter.
Discussion
In this study, we have established a fluorescent reporter mouse model that serves as a versatile and powerful tool for the evaluation of genome editing in vivo. PAM requirements for a broad range of effectors (including SpyCas9, Nme1Cas9, Nme2Cas9, SauCas9, CjeCas9, Geo1Cas9, Geo2Cas9, AspCas12a, LbaCas12a, FnoCas12a, and most engineered or evolved derivatives thereof) were included during the initial reporter design, 26 and additional editors that have emerged since SauriCas9 40 and ScCas9 41 are also accommodated. The TLR-MCV1 reporter mouse has been successfully validated for its utility with nuclease editing both in vitro and in vivo, enabling the assessment of in vivo delivery and editing efficiency in various tissues and organs.
Furthermore, this reporter mouse is useful for prime editing applications, including both mCherry frameshift correction and repair of the broken region of GFP. While most of the in vivo work on prime editing has focused on endogenous loci, the availability of a fluorescent reporter mouse that is well suited for prime editing outcomes should facilitate studies of increasing PE delivery and efficacy in new tissues and cell types in vivo. Overall, this reporter mouse will enable further development and comparative analysis of diverse nuclease and prime editors in vivo, paving the way for advancements in gene editing research and therapeutic applications.
Footnotes
Acknowledgments
We thank Judith Gallant at the UMCMS Transgenic Animal Modeling Core for pronuclear microinjection, the UMCMS Viral Vector Core for AAV production, and the UMCMS Morphology Core for tissue sectioning. We thank Mona Motwani from Kate Fitzgerald lab for the help with the isolation of BMDMs. We are grateful to all the lab members for their valuable discussions, advice, and helpful feedback.
Authors' Contributions
Z.C., S.Y.K., W.X., E.J.S., and J.K.W. designed the experiments. Z.C. and S.Y.K. conducted molecular biology and cellular experiments. Z.C. and S.Y.K. built, genotyped, and maintained the mouse line with help from A.M., K.S.G., K.K., and Y.C. Z.C. and S.Y.K. performed the mouse experiments with help from M.H., M.S., S.-Q.L., K.K., and N.G. Z.C. and S.Y.K. conducted high-throughput sequencing and bioinformatic analyses. W.X., E.J.S., and J.K.W. supervised the research. Z.C., S.Y.K., W.X., E.J.S., and J.K.W. wrote the manuscript with input and comments from all other authors.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This work was funded by the National Institutes of Health (Somatic Cell Genome Editing Consortium, UG3/UH3 TR002668 to E.J.S. and J.K.W., and UG3/UH3 HL147367, P01 HL158506 to W.X.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.