
Abstract
Cas9 targets genomic loci with high specificity. For knockin with double-strand break repair, however, Cas9 often leads to unintended on-target knockout rather than intended edits. This imprecision is a barrier for direct
Introduction
CRISPR technologies have enabled the development of agents to edit the genome in naive host cells and whole organisms. The development of high-performance
A key challenge for genome editing
The Cas9 toolkit offers a variety of editing modalities, including double-strand break (DSB) repair-based editing, base editing, and reverse transcriptase-based (Prime) editing. 4 DSB repair-based approaches are maximally versatile for knockin, allowing insertions from DNA donors that enable knocking in larger domains, such as fluorescent proteins. However, such approaches are also the most imprecise, often leading to unintended disruptions in gene function. 5 Current DSB repair-based knockin methods produce the desired editing outcome the minority of the time, an order-of-magnitude less than unintended on-target insertions or deletions (indels). 3
In this study, we present a high-throughput workflow to quantify editing outcomes for creating and identifying editing agents with increased performance and their optimal combinations for knockin applications. We established editing
Methods
Plasmid design and construction
Mammalian expression plasmids and knockin donor template plasmids were constructed with a combination of standard cloning techniques. For gRNA expression constructs, oligos (Integrated DNA Technologies) were annealed and cloned into a custom hU6 backbone using Golden Gate Assembly (GGA). 6 Cas9 expression constructs were assembled by a modified mMoClo system. 7
Briefly, individual parts were cloned, BsaI adapters added, and internal sites removed by polymerase chain reaction (PCR) using KAPA HiFi HotStart DNA Polymerase with 2X Master Mix (Roche), or synthesized (Integrated DNA Technologies). Parts were subsequently assembled into expression constructs using NEB GGA Kit (BsaI-HF v2) according to the manufacturer's recommendations. Homology arms for donor templates were PCR amplified from CD1 mouse genomic DNA with adapters for GGA. gRNA binding site (GRBS) parts were generated by oligo annealing. GRBS and homology arms were assembled with knockin sequences using GGA. See Supplementary Table S1 for sequences.
Cell line culture and transfection
BFP knockin HEK293 cells (HEL:BFP) were developed in the Corn Lab and were the kind gift of Chris Richardson. 8 Cells were maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) media plus GlutaMax (ThermoFisher Scientific) supplemented with 10% (v/v) fetal bovine serum (FBS). Typically, 20,000–22,500 cells/cm2 were seeded onto 24-well plates the day before transfection. Cells were transiently transfected at 70–80% confluence using Polyethylenimine, Linear, MW 25000 (“PEI,” Polysciences) resuspended to 1 mg/mL in H2O at a 3:1 (v/w) PEI:DNA ratio with 250 ng DNA per plasmid (750 ng total DNA) diluted in Opti-MEM (ThermoFisher Scientific) and added dropwise to cells.
Flow cytometry
Cells were trypsinized, pelleted, and resuspended in Dulbecco's phosphate-buffered saline (PBS) containing 0.1% FBS. At least 20,000 live cells (typically 80,000+) were analyzed using an LSRII cell analyzer with HTS (BD Biosciences). Blue fluorescent protein (BFP) and mTagBFP were measured with a 407 nm laser and a 450/50 emission filter. Green fluorescent protein (GFP) and mNeonGreen were measured with a 488 nm laser, a 505 LP mirror, and a 530/30 emission filter. mCherry was measured with a 561 nm laser, a 600 LP mirror, and a 615/25 emission filter. Data were analyzed with FlowJo v10.6.2 (Flowjo LLC). Live cells were gated by size and granularity using forward scatter (FSC-A) versus side scatter (SSC-A). Singlets were gated using SSC-A versus SSC-H (Supplementary Fig. S1). At least three biological replicates were run with internal technical duplicates or triplicates.
Sequence analysis of knockin products
Transiently transfected HEK:BFP cells were sorted using a FACSAria II sorter (BD Biosciences). Genomic DNA was extracted from sorted cells using the Genomic DNA Clean & Concentrator kit (Zymo Research). PCR fragments were amplified using KAPA HiFi HotStart DNA Polymerase with 2X Master Mix (Roche), gel extracted with Zymoclean Gel DNA Recovery kit (Zymo Research), and submitted for Sanger sequencing (Genewiz). See Supplementary Table S1 for primer sequences. Alignment of sequencing results was performed using Benchling. Analysis of editing outcomes by decomposition of Sanger sequencing data was performed using the inference of CRISPR edits (ICE) Analysis tool v2 (Synthego) as previously described. 9
Animals
All animal experimental protocols were approved by the University of Maryland Baltimore Institutional Animal Care and Use Committee and complied with all relevant ethical regulations regarding animal research. Experiments were performed on outbred strain CD1 mouse pups (Charles River Laboratories). Analyses are thought to include animals of both sexes at approximately equal proportions, as no sex determination was attempted. No statistical method was used to predetermine sample size.
Primary cell culture and cuvette electroporation
CD1 mouse pups were euthanized by decapitation on postnatal day 0. The skin was sterilized and removed from the pup's back using sterile surgical tools. Skin was placed dermis-side down on cold 0.25% Trypsin with ethylenediaminetetraacetic acid (Invitrogen) and incubated at 4°C overnight. The epidermis was separated from the dermis in a sterile hood. Dermis was minced with a razor blade and triturated in warm 10% FBS 1 × GlutaMAX DMEM using a glass pipette 10–20 times to separate individual cells.
The suspension was then transferred to a 50 mL conical tube and centrifuged at 150 g. The cell pellet was resupsended in 10% FBS 1 × Glutamax DMEM and filtered through a 100 μm cell strainer (BD Biosciences). Cells were counted using a hemocytometer and cell viability was estimated using Trypan Blue (Sigma). Approximately 4–5 × 10 6 cells were used for each electroporation. Cells were centrifuged at 150 g and resuspended in 100 μL AMAXA nucleofection solution (Lonza) at the proper concentration and combined with 1–3 μg of desired DNA mixture in a cuvette.
The cuvette was electroporated with the AMAXA biosystems Nucelofector II (Lonza) using the manufacturer's settings for Mouse Embryonic Fibroblasts. After electroporation, the solution was immediately transferred to 12-well glass bottom plates (#1.5H; Cellvis), which were pretreated with poly-L-lysine (Sigma Aldrich) diluted 1:12 in sterile PBS the night before, containing prewarmed sterile-filtered DMEM (ThermoFisher) supplemented with 10% FBS (Gibco) and 1 × GlutaMAX (Gibco) at the desired density and incubated at 37°C/5% CO2. Half volume fresh medium was exchanged the next day.
In utero electroporation
Electroporations of plasmid DNA were performed
A midline incision was made to expose the uterine horns. Using pulled (Narishige PC-100) and beveled (Narishige EG-45) glass micropipettes connected to a pneumatic aspirator, DNA solution was injected into one lateral brain ventricle for cerebral cortex electroporation on E14.5, or in the fourth ventricle for cerebellar Pukinje cell electroporation on E11.5. Around 4 × 50 ms square pulses of 35 V (NEPA21 electrokinetic platinum Tweezertrodes connected to a BTX ECM-830 electroporator) were applied to target the nascent sensorimotor areas of the cortical plate. When using the triple electrode, 6 × 50 ms square pulses at 35V was performed on E14.5 and 25V on E11.5.
Typically, four to six pups were electroporated per dame. Uterine horns were placed back inside the abdominal cavity, and monofilament nylon sutures (AngioTech) were used to close muscle and skin incisions. After term birth, electroporated mouse pups were noninvasively screened for unilateral cortical or cerebellar fluorescence using a fluorescence stereoscope (Leica MZ10f with X-Cite FIRE LED light source) and returned to their dame until postnatal day 7 (P7) or P14. When possible, to minimize interdame variation, control and experimental electroporations were performed in littermate pups from the same dame.
Histology and immunolabeling
Tissue was prepared by intracardial perfusion with PBS and 4% paraformaldehyde. Brains were cut to 80 μm coronal sections on a vibrating microtome (Leica VT1000). Sections were immunolabeled in blocking solution consisting of 5% bovine serum albumin and 0.2% Triton X-100 in PBS for 30 min, and then incubated overnight at 4°C with primary antibodies diluted in blocking solution. Sections were washed in PBS, incubated for 3–4 h at room temperature with secondary antibodies diluted 1:400–1:1000 in blocking solution. Following PBS washes, sections were mounted on slides with Fluoromount-G Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI) (ThermoFisher Scientific). For antibodies used, see
Microscopy and image analysis
Fluorescence images were acquired using a Nikon Ti2-E inverted microscope fitted with an automated registered linear motor stage (HLD117; Pior Scientific), a Spectra-X 7 channel LED light engine (Lumencor), and standard filter sets for DAPI, FITC, TRITC, and Cy5. Images were stitched and analyzed with NIS-Elements (Nikon) using an automated script to identify and count electroporated cells in brain sections. Knockin-positive neurons were counted manually using ImageJ (NIH) and independently by at least two blinded investigators. Five 80 μm sections, centered at the middle of the anteroposterior axis of the electroporation field and taken every other section, were analyzed per brain, with counts aggregated across sections from the same brain.
Statistical analysis
All statistical values are presented as mean ± standard error of the mean. For experiments containing more than two conditionals or groups, statistical significance was calculated using a one-way analysis of variance (ANOVA) with Tukey's multiple comparison test, with a single pooled variance. For experiments containing two conditionals or groups, statistical significance was calculated using a two-tailed Student's
Citation diversity statement
Toward awareness and mitigation of citation biases, we used cleanBib to assess the predicted gender and predicted racial/ethnic category of the first and last author of our cited references. The gender breakdown of our references is 11.05% woman (first)/woman (last), 9.28% man/woman, 22.18% woman/man, and 57.49% man/man. The racial/ethnic breakdown of our references is 24.39% author of color (first)/author of color (last), 13.8% white author/author of color, 31.36% author of color/white author, and 30.45% white author/white author. These analyses exclude self-citations and are subject to the caveats and limitations outlined in the cleanBib documentation.
Results
Quantifying editing efficiency and precision through BFP-to-GFP conversion
To quantify and compare
Fluorescence was used as a surrogate for editing outcomes following transfection of these cells with Cas9 or Cas9 variants, gRNA targeting the
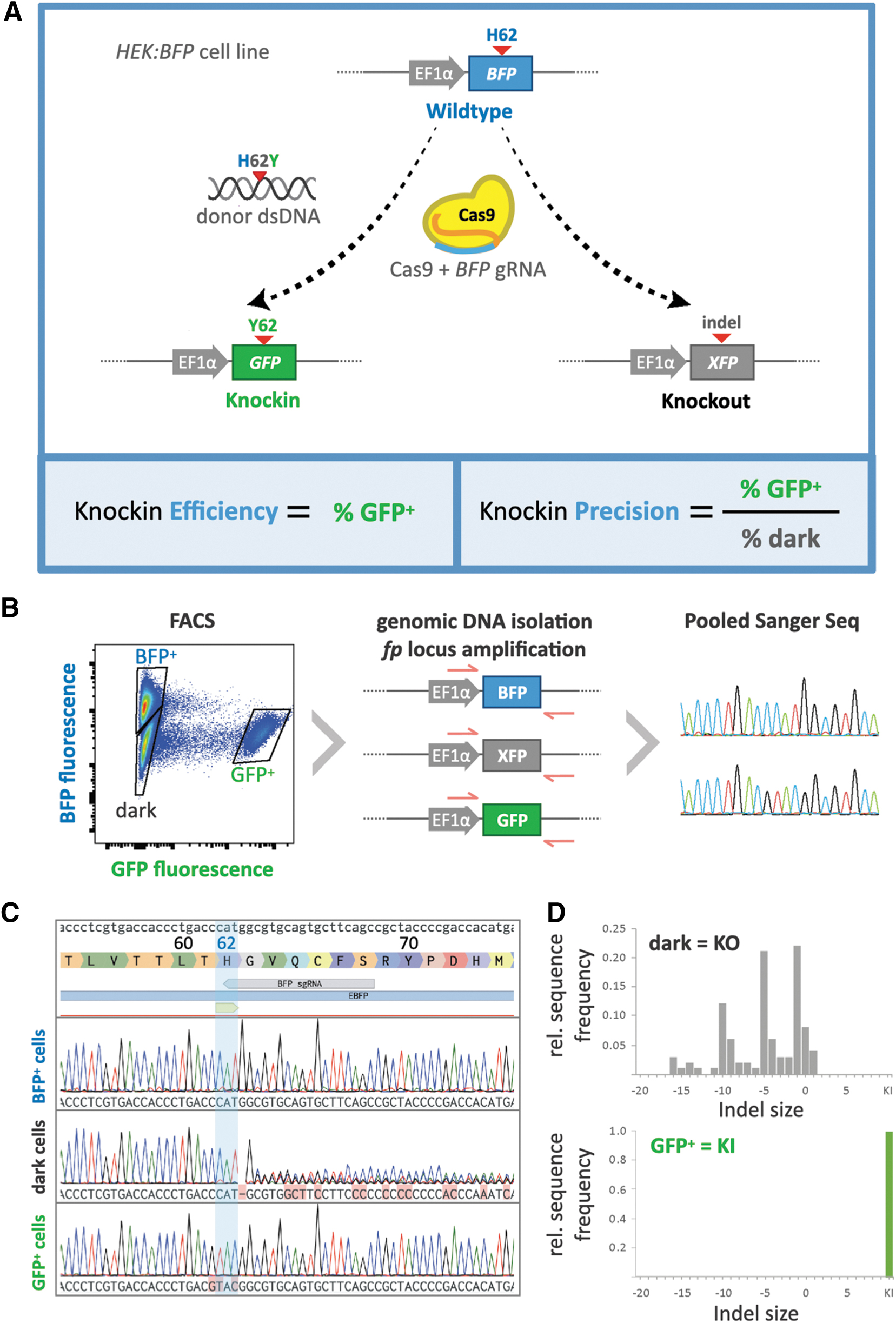
BFP-to-GFP editing as a platform to quantify knockin efficiency and precision.
To validate our workflow and confirm that fluorescence readouts correspond to predicted genotypes in HEK:BFP cells, we used fluorescent activated cell sorting (FACS) to sort and collect the three phenotypic cell populations (BFP+, GFP+, and dark) that emerge following treatment with BFP/H26Y editing agents. Genomic DNA was extracted from sorted cell populations and used as template in PCR with primers flanking the region targeted by the BFP gRNA within the
As expected, the genotype of the BFP+ population matched that of the WT
Combinatorial screening of editing agents for enhanced performance
We sought to identify elements of DSB repair knockin agents that in combination offer improved efficiency and precision of editing. We investigated a matrix of combinations of three factors, which have individually been shown to enhance knockin performance: (1) WT
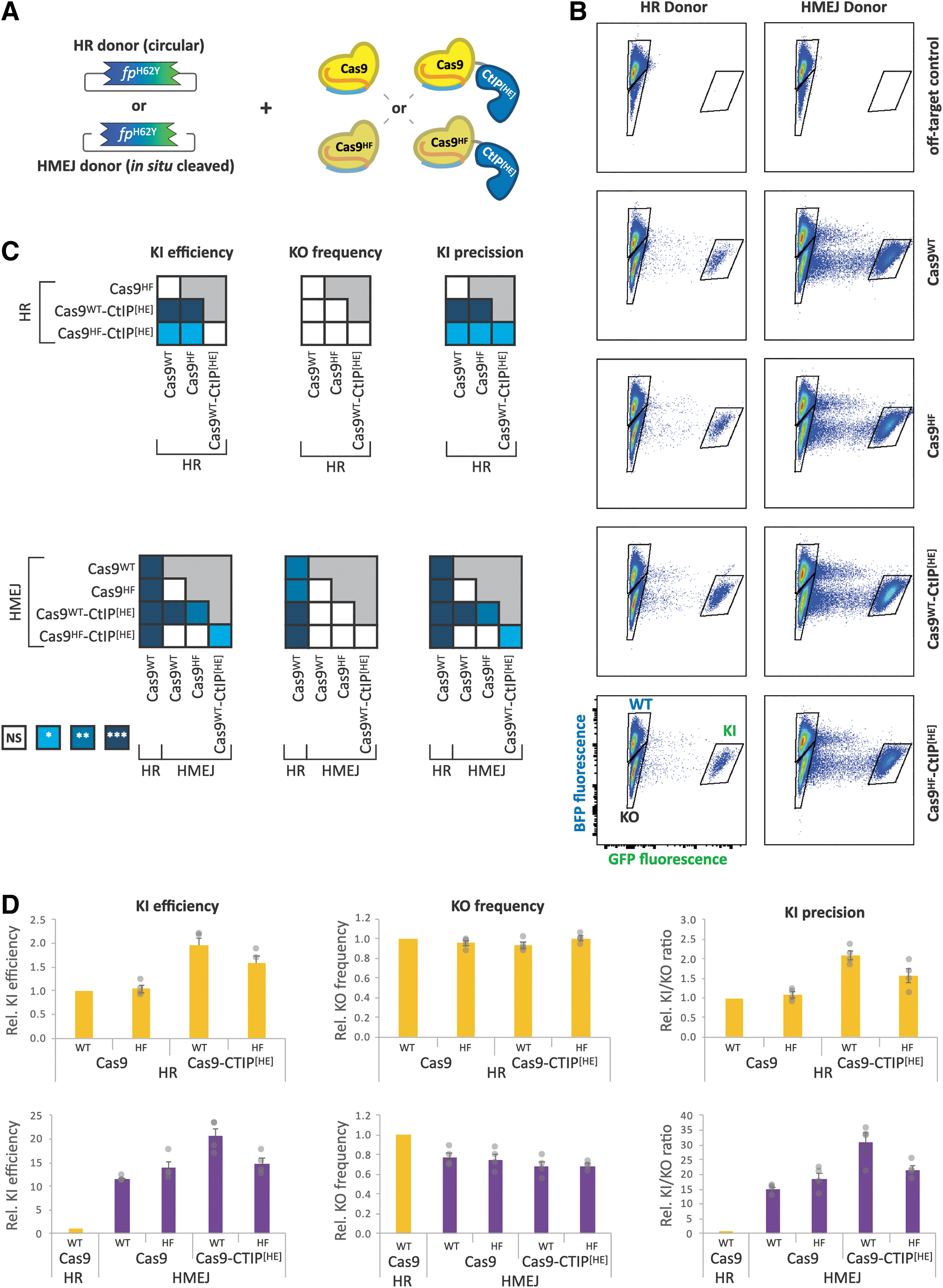
Cas9-CtIP[HE] fusion and HMEJ donors additively improve knockin precision.
Both knockin donors were provided on plasmids with the knockin sequence flanked by ∼800 bp homology arms, the length of which did not significantly affect results within a range of 500–1500 bp (data not shown). The HMEJ donor differed from the HR donor by the insertion of GRBS flanking the homology arms, which are cleaved by Cas9 to create linear dsDNA donors in cells. The orientation of the GRBSs did not have significant effects on editing performance (Supplementary Fig. S2).
Transfection of HEK:BFP cells with Cas9 variant, donor template, and the
In contrast to the Cas9 variants, donor architecture impacted knockin efficiency as well as knockout rates. Across all four Cas9 variants, the HMEJ donor showed a 9- to 13-fold increase in knockin efficiency compared to the corresponding HR combination (Supplementary Fig. S4). Of the combinations of Cas9 and donors tested, Cas9WT-CtIP[HE] in conjunction with the HMEJ donor resulted in the highest rate of knockin, 24-fold higher than Cas9WT with the HR donor.
Regardless of Cas9 variant, the HMEJ donor showed a 22–30% reduction in gene disruption, which, together with the improved knockin efficiency, enhanced the precision by about 15-fold relative to the HR donor template permutations (Supplementary Fig. S2). Interestingly, these data suggest the independent and additive contributions of both CtIP[HE] fusion (twofold) and the
Compound fusions on Cas9 improve editing performance
Several groups have independently demonstrated that modulation of DNA repair pathways is an effective way to improve knockin efficiency and precision.19,23,24 To build on the results of the 3-factor screening, highlighting WT Cas9, CtIP[HE] fusion, and HMEJ donor as the best performing combination, we iterated the BFP-to-GFP screening platform with constant HMEJ donor and evaluated the impact of five candidate DNA repair protein domains (dn53BP1,24–26 TIP60,26,27 RNF169, 25 Rad52,23–30 and eRad18 27 ) on editing efficacy and precision when fused N-terminally to Cas9 or Cas9-CtIP[HE] (Fig. 3A).
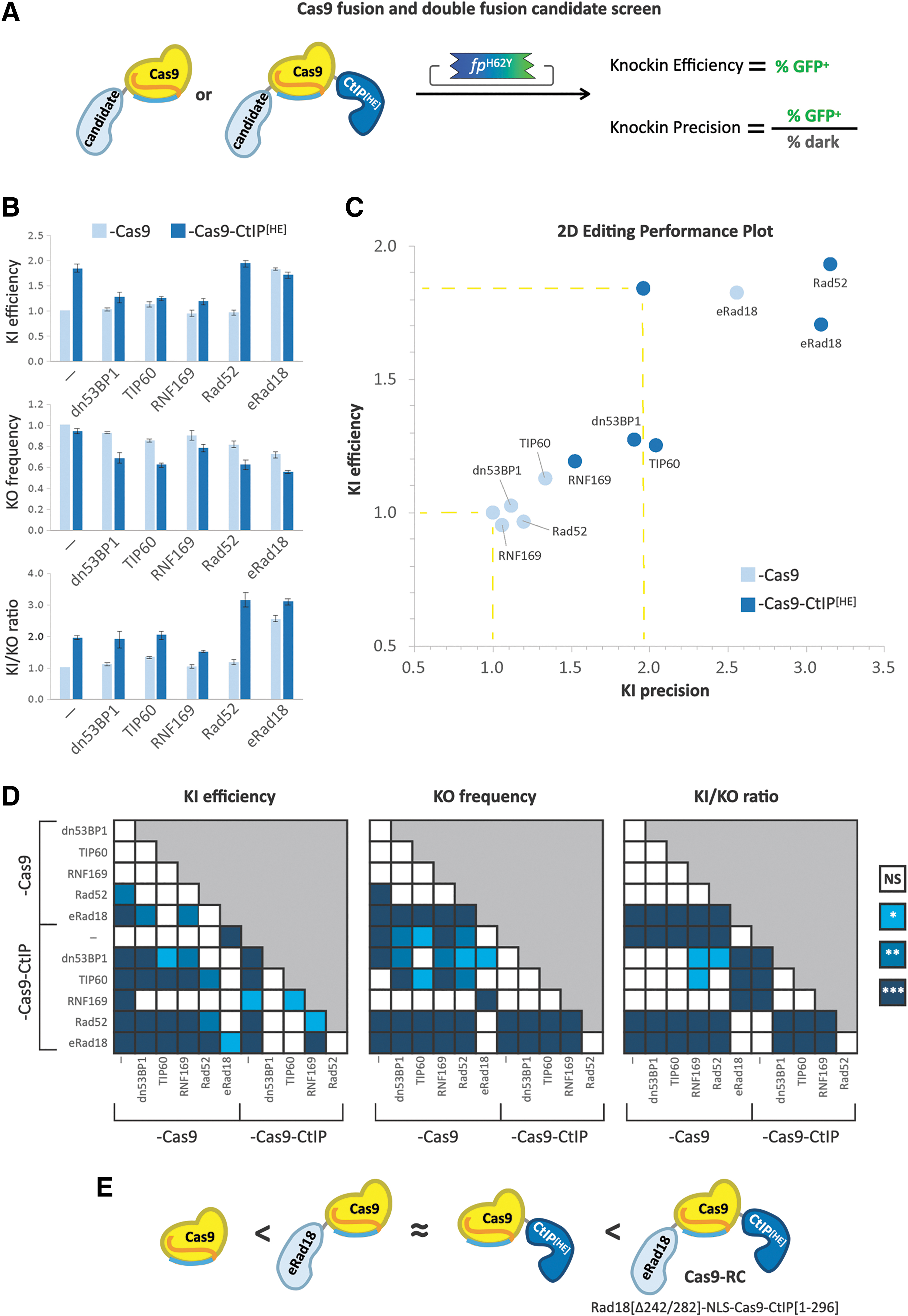
Iterative screening of novel Cas9 fusions and compound fusions with DNA repair domains for increased editing performance.
In the absence of CtIP[HE], only eRad18 fusion to Cas9 significantly improved knockin efficacy, increasing it by 1.8-fold above Cas9 alone, similar to Cas9-CtIP[HE]. With the compound fusions, while addition of dn53BP1, TIP60, or RNF169 to Cas9-CtIP[HE] appeared to abrogate the effect of CtIP[HE] on knockin efficacy, fusion of Rad52 or eRad18 did not have a detrimental impact on efficiency (Fig. 3B, D).
Regarding unintended on-target knockout, dn53BP1-, TIP60-, and RNF169-fused Cas9 did not significantly differ from the Cas9-only control. Rad52 and eRad18 fusion, however, showed 18% and 28% reductions in knockout frequency, respectively (Fig. 3B, D). Interestingly, compound fusion of each of the five DNA repair proteins with CtIP[HE] led to significant reductions in the knockout rate, with eRad18, Rad52, and TIP60 showing the most pronounced decreases (45%, 38%, and 38%, respectively). Despite these reductions in knockout rates, only Rad52 and eRad18 led to significant improvements in overall knockin precision. Without CtIP[HE], eRad18 demonstrated a 2.5-fold increase in the knockin-to-knockout ratio, while combination of either Rad52 or eRad18 with CtIP[HE] led to a 3.1-fold increase in precision relative to Cas9 alone (Fig. 3B, D). These results show that specific combinations of DNA repair domains fused to flank Cas9 can function together to improve both the efficiency and precision of editing.
We elected to move forward with the smallest of these compound fusions, Cas9WT flanked by eRad18 and CtIP[HE], which we call “Cas9-RC.” In addition to C-terminal fusion of truncated human CtIP[HE], Cas9-RC harbors an N-terminal fusion of a deletion variant of human Rad18 protein, eRad18, which lacks the SAP domain between residues 242 and 282 (Δ242/282), for a total of 2,172 residues. eRad18 was shown to enhance homology-dependent repair (HDR) when independently co-expressed with Cas9 by suppressing imprecise end-joining repair pathways. 27
Cas9-RC works with both HMEJ and HDR donor templates. However, because eRad18 suppresses recruitment of 53BP1, which is required for both nonhomologous end joining and microhomology-mediated end joining (MMEJ), 31 we did not test Cas9-RC with MMEJ donors, which would be predicted to perform poorly. Cas9-RC expression plasmids and test knockin plasmids have been made available through Addgene (Cas9-RC with GFP #207404 or mScarlet #207405; test KI-HMEJ #207407, 207408; Supplementary Table S1).
Cas9-RC increases knockin in vivo
The combinatorial screening and iterative optimizations of Cas9 agents yielded Cas9-RC, which showed increases in knockin performance in cultured cell lines. Aiming to develop precision knockin agents for direct editing
To test the knockin efficiency of Cas9-RC
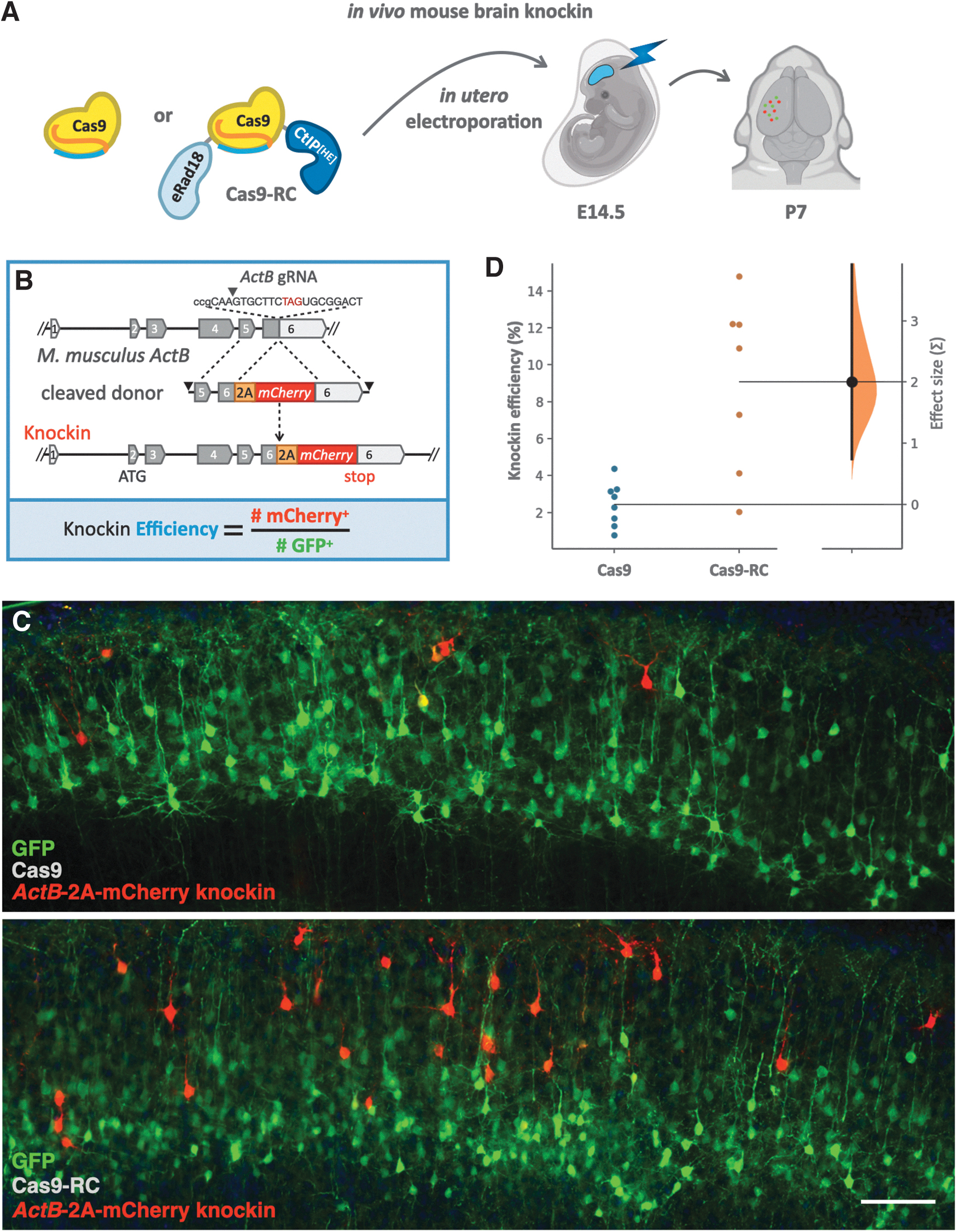
Cas9-RC enhances knockin efficiency
Fluorescent protein knockin applications with Cas9-RC
We went on to use Cas9-RC plasmids and HMEJ donors in knockin applications of three different fluorescent proteins onto three different loci in three different cell types.
We extracted primary mouse fibroblasts from P0 mice and
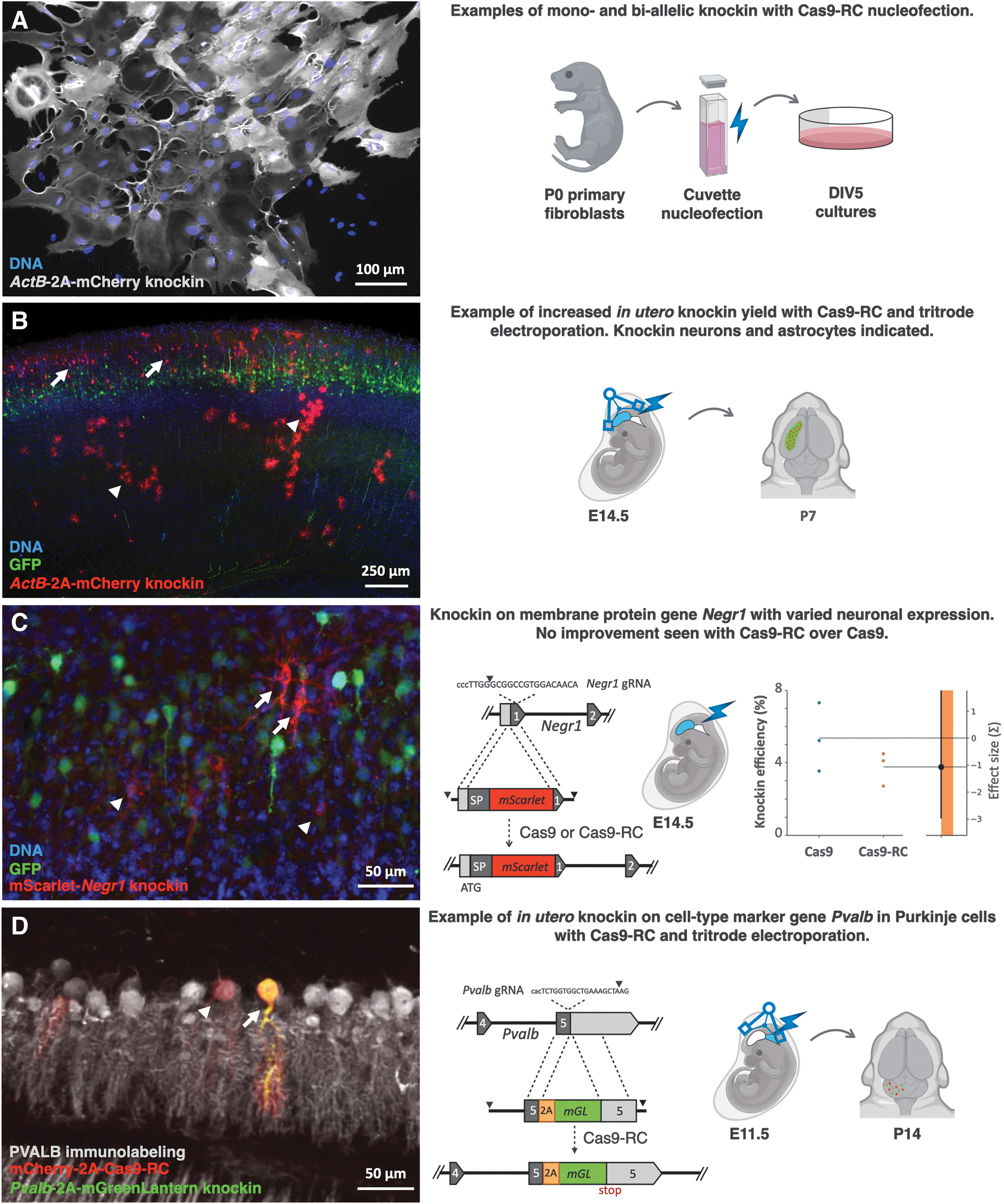
Knockin applications with Cas9-RC.
One of the limitations of Cas9-RC efficiency
We used the same knockin approach to fuse the fluorescent protein mScarlet onto the N-terminus of the GPI-linked membrane protein neuronal growth regulator 1 (Negr1), a protein with variable expression in the mouse brain.
33
We went on to quantify mScarlet fluorescence compared to GFP electroporation marker to assess Cas9-RC knockin efficiency on the
Finally, we used Cas9-RC with tritrode electroporation and supercoiled constructs to target Purkinje cells in the mouse embryonic cerebellum, as a case to examine a difficult to transduce cell type that we were not able to knock in with Cas9. We constructed HMEJ donor and gRNA constructs to knock in the fluorescent protein mGreenLantern downstream of the
Discussion
In this study, we sought to develop high-performance knockin tools by exploring combinations of DNA donor templates, variants of Cas9, and fusion of DNA repair protein domains. We identified novel Cas9 fusions and donor combinations that resulted in knockin with significantly improved metrics of editing efficiency and precision,
Our work builds upon an established reporter cell line as a standardized pipeline to optimize tools for precision genome editing. The BFP-to-GFP screening platform
8
in HEK cells provides a high-throughput quantitative readout of the efficiency of correctly edited cells, while also reporting on the frequency of incorrectly edited cells. By simultaneously evaluating knockin and knockout rates, we identified combinations that optimized both efficiency (overall knockin rate) and precision (knockin rate vs. knockout rate). Knockin of a fluorescence cassette into the highly expressed β-Actin locus similarly enabled direct quantification of efficiency for large inserts at an endogenous gene locus
Quantification at the variably expressed locus
We propose editing
Efficiency and precision can be used as readouts when assessing individual components for holistic performance optimization, as we have done in this study. They can additionally be useful as common metrics to compare performance across distinct editing modalities, for example, DSB repair versus Prime editing. By simultaneously assessing efficiency and precision across a variety of knockin tools, optimal agents can be selected based on the experimental need. For example, with
Using this dual metric performance assessment, our study developed the high-performance DSB repair editor Cas9-RC (Supplementary Discussion). When paired with HMEJ donor templates, Cas9-RC outperformed Cas9 by over 30-fold in human cells and showed potential for threefold increases in the mouse brain, although not at all loci tested. As DSB repair through Cas9-RC enables high performance for large genomic edits, such as fluorescent protein knockin, it complements parallel developments in base editors and Prime editing, which offer high performance, but are limited to smaller edits. Ultimately, a diverse toolkit of precision editors will be useful to broaden the scope of
Conclusion
Fusion of Cas9 to DNA repair protein domains can produce synergistic enhancements of knockin performance. Iterative high-throughput screening based on fluorescent protein conversion is an effective platform to assess knockin efficiency and precision for developing new editing agents. Cas9-RC is a new DSB repair genome editor demonstrating enhanced knockin performance
Footnotes
Acknowledgments
We are grateful for the technical and instrumentation support of the University of Maryland School of Medicine Center for Innovative Biomedical Resources through the Confocal Imaging Facility, the Flow Cytometry Facility, the Genomics Facility, and the Biostatistics Shared Service.
Authors' Contributions
R.R.R.: Conceptualization, methodology, validation, formal analysis, investigation, writing—original draft, visualization, and supervision. M.S.: Methodology, validation, formal analysis, investigation, data curation, writing—review and editing, and visualization. S.N.K.: Validation, formal analysis, investigation, data curation, writing—review and editing, and visualization. G.W.C.: Validation, formal analysis, investigation, data curation, writing—review and editing, and visualization. C.B.: Conceptualization, methodology, investigation, resources, writing—review and editing, visualization, and funding acquisition. C.D.R.: Methodology, validation, formal analysis, investigation, data curation, writing—review and editing, and visualization. A.J.R.: Methodology, validation, formal analysis, validation, investigation, data curation, writing—review and editing, and visualization. J.I.: Formal analysis and investigation. B.A.: Methodology, writing—review and editing, visualization, and supervision. A.P.: Conceptualization, methodology, validation, formal analysis, resources, writing—original draft, visualization, supervision, project administration, and funding acquisition.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the High-Risk, High-Reward Research Program of the National Institutes of Health Common Fund under award no. DP2MH122398 (A.P.) and by the Autism Research Institute through grant no. 30030461 (C.B.). R.R.R. and C.B. were supported through the Cancer Biology T32 Training Program at the University of Maryland School of Medicine funded by the National Cancer Institute under award no. T32CA154274. C.B. was additionally supported by the Schizophrenia & Psychosis-Related Disorders T32 Training Grant of the Maryland Psychiatric Research Center under award no. T32MH067533. J.I. and S.N.K. were supported by the University of Maryland STAR-PREP Science Training for Advancing Biomedical Research Postbaccalaureate Program funded by the National Institutes of Health under award no. R25GM113262.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.