
Abstract
Promising to provide powerful genetic control tools, gene drives have been constructed in multiple dipteran insects, yeast, and mice for the purposes of population elimination or modification. However, it remains unclear whether these techniques can be applied to lepidopterans. Here, we used endogenous regulatory elements to drive Cas9 and single guide RNA (sgRNA) expression in the diamondback moth (DBM), Plutella xylostella, and test the first split gene drive system in a lepidopteran. The DBM is an economically important global agriculture pest of cruciferous crops and has developed severe resistance to various insecticides, making it a prime candidate for such novel control strategy development. A very high level of somatic editing was observed in Cas9/sgRNA transheterozygotes, although no significant homing was revealed in the subsequent generation. Although heritable Cas9-medated germline cleavage as well as maternal and paternal Cas9 deposition were observed, rates were far lower than for somatic cleavage events, indicating robust somatic but limited germline activity of Cas9/sgRNA under the control of selected regulatory elements. Our results provide valuable experience, paving the way for future construction of gene drives or other Cas9-based genetic control strategies in DBM and other lepidopterans.
Introduction
Gene drives are heritable elements capable of autonomously increasing their frequency within a gene pool.1,2 Traits associated with the gene drive will also spread and could be arranged to include examples beneficial for pest control (e.g., a sex-specific fitness cost), reducing vector-borne virus transmission (e.g., virus-refractory transgenes), or conservation (e.g., resistance to disease/pesticides).3,4
One example of a gene drive technology is the CRISPR-Cas9-based homing drive.4,5 In its simplest form, this system requires a source of Cas9 and a single guide RNA (sgRNA) expression cassette to be integrated into a genome at the precise site specified by the sgRNA. As this integrated “homing element” has disrupted its linked sgRNA site, it is immune to further cutting. However, in a heterozygote, the homologous “wild-type” (WT) chromosome may be targeted for cleavage by Cas9, and in the resulting double-strand break (DSB) repair, the locus harboring the integrated homing element may be used as a template for homology-directed repair (HDR), transforming the heterozygous cell into one homozygous for the homing element (a process known as “homing”). If this process occurs in the germline, it can bias the inheritance of the homing element above the expected 50:50 ratio. 5
The binary nature of the CRISPR-Cas9 system allows variations on this form, for example with one or both components integrated at sites away from the homing element. Such “split-drive” (sgRNA cassette remains in homing element)6–9 or “trans-drive” (neither component remains in homing element) 10 designs are predicted to act in a more “controllable” way once released into a target population because any component not included in the homing element will not benefit from super-Mendelian inheritance and will likely reduce in frequency over time due to associated fitness costs.11,12 As both components are required in an individual for the drive to function, the efficiency of the drive will thus reduce over time as one or more components become limiting. Although potentially less efficient than the simple “all in one” design outlined above, the potential to limit the spread of a gene drive geographically and temporally may prove beneficial to the regulation, perception, and field deployment of such technologies. 13
To date, CRISPR-Cas9 homing drives have been demonstrated in various dipterans (prominent examples include Drosophila melanogaster 14 and the mosquitoes Anopheles gambiae, 15 Anopheles stephensi, 16 and Aedes aegypti 17 ), yeast, 6 and mice. 18 Here, we explore the potential of such technology for the first time in a lepidopteran—the diamondback moth (DBM) Plutella xylostella—focusing on a “split-drive” design. Lack of previous research into a lepidopteran gene drive is surprising, given the extreme and wide-ranging importance of this insect order, for example as major agricultural pests and invasive species, 19 and as producers of silk and components of industrial processes. 20 DBM provides an appropriate model for such work because it is both a globally important pest of brassica crops21,22 and is tractable in terms of genetic engineering technologies, having been the subject of previous genetics-based pest control strategy development.23,24
Methods
Insects
DBM transgenic lines were generated from the Vero Beach WT strain, 25 with rearing conditions described previously.26,27
Identification and expression profiling of germline candidate genes
To drive Cas9 expression in specific tissues and developmental stages, germline-active promoters must be identified and characterized. Referring to previous reports,28–31 amino acid sequences of nine germline candidates—BGCN, Shutdown, SDS3, Meiw68, SIWI, NanosO, NanosP, NanosO, and NanosM (origins listed in Supplementary Table S2)—were downloaded from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/) and blasted against the DBM pacbiov1 genome database (http://ensembl.lepbase.org/Plutella_xylostella_pacbiov1/Info/Index) to identify putative homologs.
Expression patterns of these DBM candidates were compared with reverse transcription polymerase chain reaction (RT-PCR) using the endogenous gene 17S as a loading control. DBM samples collected at different developmental stages (i.e., embryonic, larval, and adult stages) were dissected, frozen in liquid nitrogen, placed in RNAlater (Thermo Fisher Scientific), and stored at −80°C. Total RNA was extracted with the RNeasy Mini Kit (Qiagen), followed by removal of gDNA with DNase I (Thermo Fisher Scientific). Purified RNA was diluted to 20 ng/μL and used to produce cDNA pools with the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). RT-PCR was conducted using the Q5 High-Fidelity 2 × Master Mix (NEB). All primers used in the current study are available in Supplementary Table S1.
Construct design
Adult ovary total RNA was used to amplify the 5′ and 3′ UTR of Pxmeiw68 and PxnanosP using the SMARTer RACE 5′/3′ Kit (Takara). Regulatory sequences 5′ and 3′ were then cloned from fourth instar larval gDNA (NucleoSpin Tissue Column; Macherey-Nagel). PiggyBac constructs AGG1906 (Pxmeiw68-Cas9; GenBank accession number OK145566) and AGG2093 (PxnanosP-Cas9; GenBank accession number OK145567) were developed as for AGG1536 (Pxvasa-Cas9) previously, 26 except for replacing the Pxvasa promoter, Pxvasa 3′ UTR, and SV40 terminator with corresponding regulatory fragments from Pxmeiw68 and PxnanosP as well as the fluorophore marker 3′ UTR with P10. All plasmids were assembled with NEBuilder HiFi DNA Assembly Cloning Kit (NEB; see Fig. 1A).
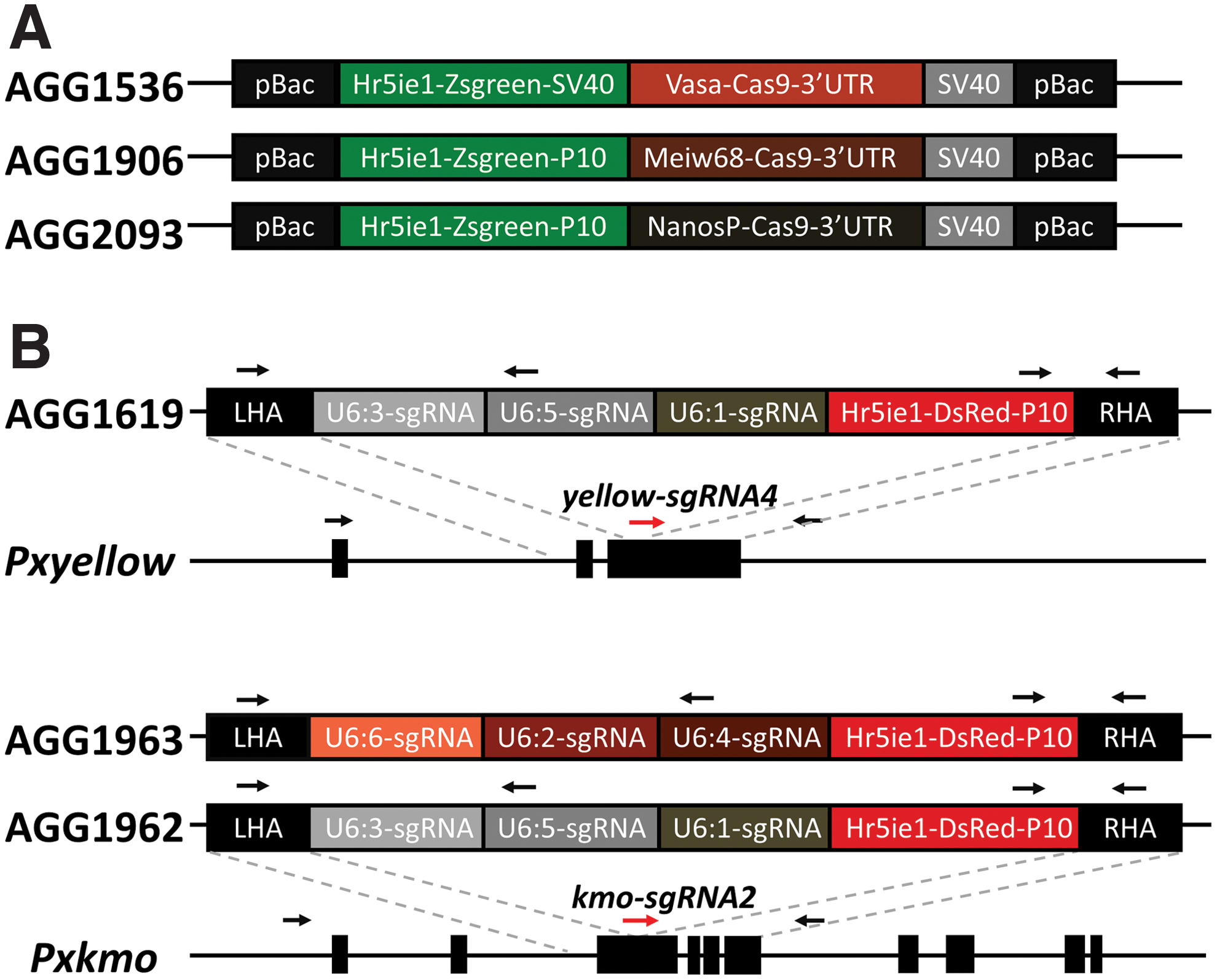
Construction of transgenic lines.
For constructing sgRNA expressing lines (“homing elements”), two pigmentation genes Pxyellow and Pxkmo 27 were selected as targets for CRISPR-based site-specific insertion (“knock-in”) of sgRNA cassettes due to the ease of visually scoring null mutations in these genes. Between 800 and 1,000 bp upstream/downstream regions immediately flanking each sgRNA cleavage site were cloned from gDNA as homology arms for mediating HDR repair. Six RNA polymerase III PxU6 promoters 32 were divided into two sets of three and used to express relevant sgRNAs for each gene (see Fig. 1B). Within each construct, although multiple U6 promoters were used, only a single sgRNA was expressed (all U6 promoters within the same construct expressed the same sgRNA). sgRNAs were designed with CHOPCHOP (https://chopchop.cbu.uib.no/). sgRNA expression cassettes were synthesized and cloned into the homology arm destination plasmid, alongside a Zsgreen-expressing marker (Fig. 1B; Genewiz; GenBank accession numbers AGG1619:OK145568, AGG1962:OK145570, and AGG1963:OK145569).
Assembled constructs were purified with NucleoBond Xtra Midi Prep Kit EF (Macherey-Nagel) before injection.
Development of transgenic lines
A Pxvasa-Cas9 line (1536A) was built previously. 26 To mitigate issues associated with “positional effects” of the transgene insertion site, additional Pxvasa-Cas9 lines were generated here, initially using the same method as previously described. Subsequently, to improve transformation efficiency, piggyBac helper plasmid and codon-optimized mRNA were replaced with a commercially synthesized piggyBac mRNA (TriLink Biotechnologies). Constructs (AGG1906 or AGG2093) were injected at 500 ng/μL alongside 600 ng/μL piggyBac mRNA to generate Pxvasa-Cas9, Pxmeiw68-Cas9, and PxnanosP-Cas9 lines.
For constructing homing elements, sgRNAs were in vitro transcribed (150 ng/μL), complexed with 300 ng/μL Cas9 protein 27 and injected alongside the relevant sgRNA plasmid (800 ng/μL). Injection, transformant, and insertion-site identification were as previously described.25,27
For resolving the 5′ junction between the integrated homology arm and the genomic flanking sequence in 1619P15, the Cas9 Sequencing Kit (Oxford Nanopore Technologies) was used because PCR could not confirm the junction. In brief, high molecular weight (HMW) genomic gDNA was extracted from homozygous 1619P15 pupae using the Monarch HMW DNA extraction kit for tissue (NEB). The Cas9 Sequencing Kit (Oxford Nanopore Technology) was used to prepare a library targeting the insertion. Targets GAACTCGGTGATGACGTTCT
The Pxkmo knockout line was generated previously. 27 The Pxyellow knockout line was generated following previous protocols, 35 except using a newly designed sgRNA (Pxyellow-sgRNA4) targeting the third Pxyellow exon.
Assessment of Cas9 somatic editing and homing efficiency
To assess somatic editing efficiency, reciprocal crosses were conducted using Cas9 and sgRNA lines. In each cross replicate, four Cas9 heterozygote “grandparents” and four sgRNA heterozygote “grandparents” were crossed, with F1 eggs collected and reared until pupation. F1 pupae were screened for fluorescence and body/eye pigmentation patterns based on mutant phenotypes reported previously.27,35
Note that Pxyellow mosaics are only observable when they co-occur in an individual displaying the “stripy” phenotype. This phenotype occurs naturally and consists of two darkly pigmented areas running laterally along the dorsal surface of the pupae. Therefore, the number of Pxyellow mosaics was calculated against the number displaying this phenotype rather than the total number of pupae. The mutagenesis of target sites was confirmed in a subset of somatic mosaics using a T7 endonuclease assay (NEB).
To assess “homing” activity, transheterozygous F1s containing both Cas9 and sgRNA transgenes (parents) were selected from the above cross and themselves crossed to Pxyellow or Pxkmo knockout lines, depending on which sgRNA line was used in the initial cross (Fig. 3). In each cross replicate, a minimum of three transheterozygotes and three knockout individuals were reciprocally crossed, with five replicates conducted for each combination. An exception to the above process was made for the 1906 lines where, due to the large number of lines to assess, the “grandparent” factor was not applied.
In all cases, the third instar larvae (Pxyellow cross) or pupae (Pxkmo cross) of F2 progeny were screened for fluorescence (to assess gene drive activity) and knockout phenotypes (to assess germline/somatic editing efficiency). Control crosses were conducted by reciprocally crossing each sgRNA line as heterozygotes to its respective knockout line and scoring the same details as above.
Statistical analyses
Analyses of sgRNA cassette inheritance was conducted using R v3.6.2 (The R Foundation for Statistical Computing), 36 and figures were generated using ggplot237 and patchwork. 38 The proportion of offspring inheriting the sgRNA cassette was analyzed using a binomial GLM (“logit” link) with control cross outcomes set as the intercept. Approximate 95% confidence intervals were calculated using the Sidak adjustment for multiple comparisons.
Figures of germline cleavage percentages were made using GraphPad software. Differences were analyzed with one-way analysis of variance or an independent t-test using SPSS Statistics 22.0 (IBM Corp).
Western blot
Moth carcass, ovary, and testis samples from 2- to 3-day-old adults were homogenized on ice in 100 μL 1 × passive lysis buffer (PLB) including Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific). After 15 min of incubation at room temperature, samples were centrifuged for 10 min at 10,000 g and 4°C. An additional 50 μL PLB + protease inhibitor was added to the samples and incubated for a further 15 min at room temperature.
Protein concentrations were determined using the DC Protein Assay (Bio-Rad) using a bovine serum albumin standard curve. Absorbance values were obtained using a microplate reader (GloMax) at 750 nm.
For each sample, equal amounts of total protein (35 μg) were combined with Laemmli sample buffer (Bio-Rad) and denatured for 5 min at 95°C.
Samples were loaded onto 4–20% precast polyacrylamide gels (Bio-Rad) and run at 200 V. A chemiluminescent protein ladder (8–260 kDa; LI-COR) was loaded alongside the protein samples to estimate the position of the 158 kDa Cas9 (FLAG-tagged) band. Proteins were transferred onto a nitrocellulose membrane (Bio-Rad). After transfer, unspecific binding sites were blocked overnight at 4°C with 3% milk powder (Marvel) in Tris-buffered saline solution (Bio-Rad) including 1% Tween 20 (TBS-Tween; Sigma–Aldrich). Anti-FLAG-tag antibody (Abcam) was diluted 1:1,000 in 3% milk/TBS-Tween, and the membrane incubated overnight at 4°C. Membranes were developed using Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific), scanned using a Gel Doc imager (Bio-Rad), and analyzed using Image Lab software (Bio-Rad).
Results
Cas9 line generation and characterization
Previously, we showed that Pxvasa was preferentially expressed in adult soma and gonads, and the selected Pxvasa promoter fragment drove Cas9 somatic and gonadal transcription in a single generated line (1536A). 26 Here, three additional 1536 lines were developed (Fig. 1 and Table 1). Moreover, homologs of nine putative germline expressing genes were identified in DBM (Supplementary Table S2). RT-PCR showed that Pxbgcn, Pxsds3, Pxshu1, Pxshu2, Pxpiwi, and PxnanosN were ubiquitously transcribed in embryos, larvae, and adult tissues, whereas PxnanosO and PxnanosM were expressed in female ovaries and early embryos. In contrast, detectable Pxmeiw68 expression was restricted to adult gonads, and PxnanosP was preferentially detected in adults (especially female ovaries; Supplementary Fig. S1A), making them better candidates for further construction of Cas9 lines for homing drive generation. The putative promoters and 5′/3′ regulatory regions of these two genes were cloned to direct Cas9 expression, with seven 1906 (Pxmeiw68-Cas9) and five 2093 (PxnanosP-Cas9) lines subsequently generated (Table 1). Flanking PCR confirmed independent insertion sites of each line (Supplementary Table S3). Paired expression profiling of Cas9 and endogenous genes (Pxmeiw68, Pxvasa, and PxnanosP) showed that Cas9 mRNA was transcribed in both testes and ovaries of all 1536, 2093, and 1906 lines except 1906B (Supplementary Fig. S1B). Unexpectedly, it was also observed that expression levels of Cas9 were much higher in adult carcasses than in gonads—the opposite trend for the respective endogenous genes (Supplementary Fig. S1B).
PiggyBac-based Construction of the Pxmeiw68-Cas9 and PxnanosP-Cas9 Transgenic Lines
sgRNA line generation and characterization
Homozygous null mutations in Pxyellow or Pxkmo cause visible phenotypes in the body or eye pigmentation of DBM, respectively, making them scorable markers for genome editing events.27,35 As such we chose these two genes as targets for building sgRNA expression lines. Although six PxU6 promoters (PxU6:1 to PxU6:6) have been identified and characterized in DBM somatic cells, 32 no information is available on their germ-cell activities. Therefore, we employed a conservative approach, utilizing all six in two separate arrays (PxU6:3,5,1 and PxU6:6,2,4) to express sgRNAs in three knock-in constructs (Fig. 1B). In each construct, the three chosen promoters each expressed an identical sgRNA, which was the sgRNA used to target the construct to its genomic location.
Using CRISPR-mediated site-specific knock-in, two AGG1619 lines (yellow-sgRNA4 expressed by PxU6:3,5,1 promoters), two AGG1962 lines (kmo-sgRNA2 expressed by PxU6:3,5,1 promoters), and three AGG1963 (kmo-sgRNA2 expressed by PxU6:6,2,4 promoters) lines were isolated (Table 1). Interestingly, PCR analysis of the genomic location of these lines revealed a high number of noncanonical repair events (Supplementary Fig. S2). Of the seven insertions, two were deemed “off target,” judging by the inability to generate PCR flanking bands and the lack of expected knockout phenotype in these lines when inbred (1962B, 1963A); two were in the correct locus but showed truncations of the outer transgene/homology arm sequences or genomic flanking regions (1619T18, 1962A); and three were perfectly integrated but displayed no (1619P15), very mild (1963C), or severe (1963B) internal deletions within the transgene sequence.
Note that the 5′ junction of 1619P15 could not be resolved by PCR and required targeted sequencing of the region to identify the repair junction (Cas9 Sequencing Kit; Oxford Nanopore Technology; Supplementary Figures S2 and S3). As 1619P15, 1962A, and 1963C were the most intact, each possessing a different sgRNA expression cassette and at least one perfectly repaired flanking sequence (both junctions perfectly repaired in 1963C and 1619P15), they were maintained for further cross experiments.
Somatic activity of CRISPR-Cas9 system (F1 analysis)
To test the CRISPR-Cas9 system's ability to function when engineered into DBM, we first crossed heterozygous Cas9 and sgRNA F0s and analyzed the phenotypes and genotypes of their F1 progeny with regards to the target locus (Pxyellow or Pxkmo, respectively). Regarding insertion sites, these crosses theoretically resulted in four equally represented genotypes: Cas9-only, targetsgRNA only, Cas9 + targetsgRNA transheterozygotes, and nonfluorescent WT.
Pxyellow analysis
Broadly, Pxyellow somatic mosaics were observed in Cas9 + yellowsgRNA transheterozygotes in all crosses involving 1536 and 2093 lines, while only two out of seven 1906 lines (1906B and 1906E) showed observable mosaicism in F1s (Fig. 2A). Choosing a subset of these lines for finer-scale analysis, we calculated the somatic editing rates of F1 individuals using 1536C, 1906B, 1906E, and 2093A (Table 2). All (100%) of
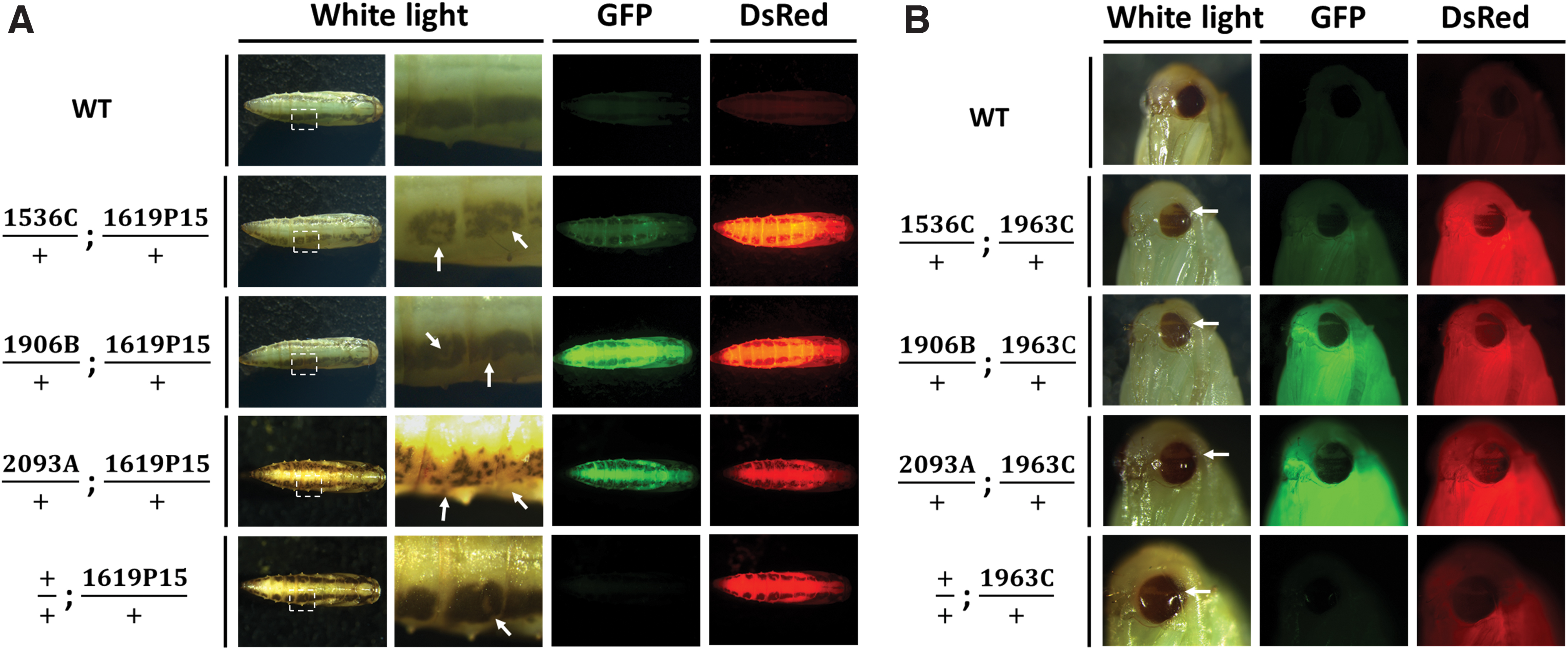
Somatic mosaicism present in F1 transheterozygotes.
Individuals and Percentages of Different Genotypes and Phenotypes Shown in F1s Generated from Pxyellow Homing Crosses
Pxkmo analysis
Regarding crosses involving the 1963C line, Pxkmo mosaicism broadly concurred with Pxyellow results above. At a finer scale, however, the observable mosaic phenotype was generally lower in 1536C (54.4–84.3%), 1906B (4.0–9.7%), and 1906E (0–1.8%) F1 transheterozygotes, although 100% of 2093A transheterozygotes continued to show some form of eye mosaicism (Table 3). Mosaicism of other F1 genotypes was again absent except in crosses involving the 2093A line where it was present only in kmosgRNA individuals. Interestingly, however, here it occurred both where Cas9 had been deposited by the male (14% F1 mosaic) and female (20.4% F1 mosaic) parents.
Individuals and Percentages of Different Genotypes and Phenotypes Shown in F1s Generated from Pxkmo Homing Crosses
A random subset of mosaic transheterozygotes were collected for both 1963C (
Germline activity of CRISPR-Cas9 system (F2 analysis)
F1 transheterozygotes derived from the above experiments were reciprocally mated to relevant knockout lines (Pxyellow–/– for F1s carrying 1619P15 and Pxkmo–/– for F1s carrying 1963C/1962A), and their F2 offspring were then scored for fluorescence and knockout phenotypes (Fig. 3). Under Mendelian inheritance, 50% of these F2 would be expected to carry the relevant sgRNA-expressing transgene, with significant deviation above this taken to be an indication of inheritance bias caused by gene drive activity. For the control crosses, heterozygous Cas9 and sgRNA lines were independently, reciprocally, mated to the two knockout lines, with F2 data collected as above (Supplementary Fig. S5).
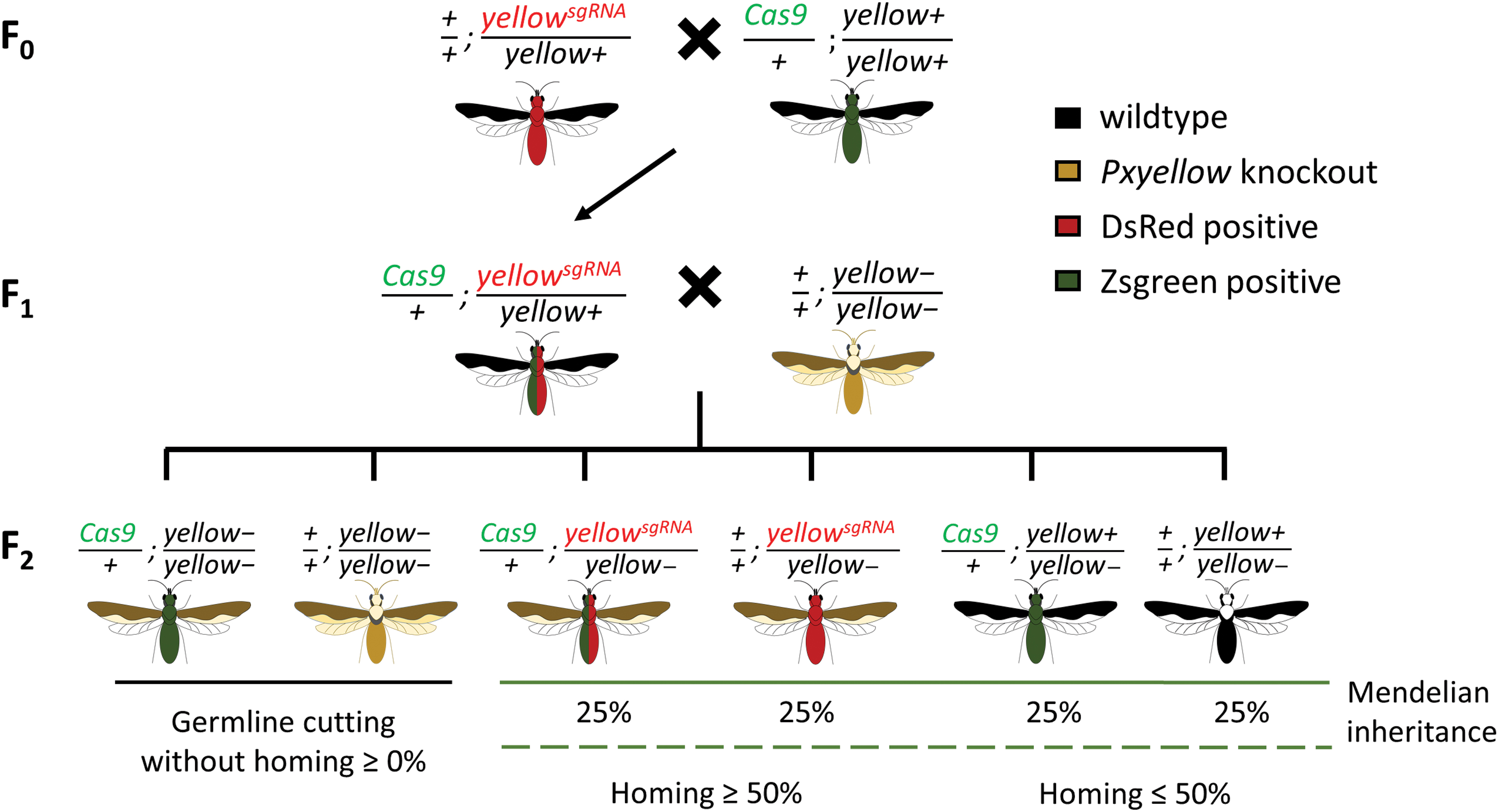
Crossing scheme for the experiment. Taking the Pxyellow cross group for example, based on mendelian inheritance, only two pigmentation phenotypes as well as four genotypes will be observed in the F2 generation:
Results showed no significant deviation from 50% inheritance for any of the three sgRNA-expressing transgenes assessed at a significance threshold of p ≤ 0.05 (Fig. 4A), indicating that any gene drive activity, if present, was too low to detect. Interestingly, however, the observation of some WT and Cas9-only individuals with full Pxkmo or Pxyellow knockout phenotypes in the F2 progeny indicated that germline expression and activity of Cas9 was nonetheless taking place (Fig. 4B), resulting in cleavage and repair through end joining rather than HDR/homing. These full knockout phenotypes could have derived from either cleavage of a parental WT chromosome in the F1 germline or cutting in the F2 following deposition of Cas9 by the F1. Given that these individuals displayed full knockout phenotypes (complete white eyes or yellow bodies) rather than the mosaic phenotypes observed in the F1 analysis (where no such full knockout phenotypes were observed), we believe the former (germline cutting) hypothesis to be the more likely. It cannot be ruled out that very early cutting in the F2 embryo may have contributed to this observation, however. Regarding 1619P15 (Fig. 4B-a), this activity was present in crosses involving the 1536 and 2093 but not 1906 lines.
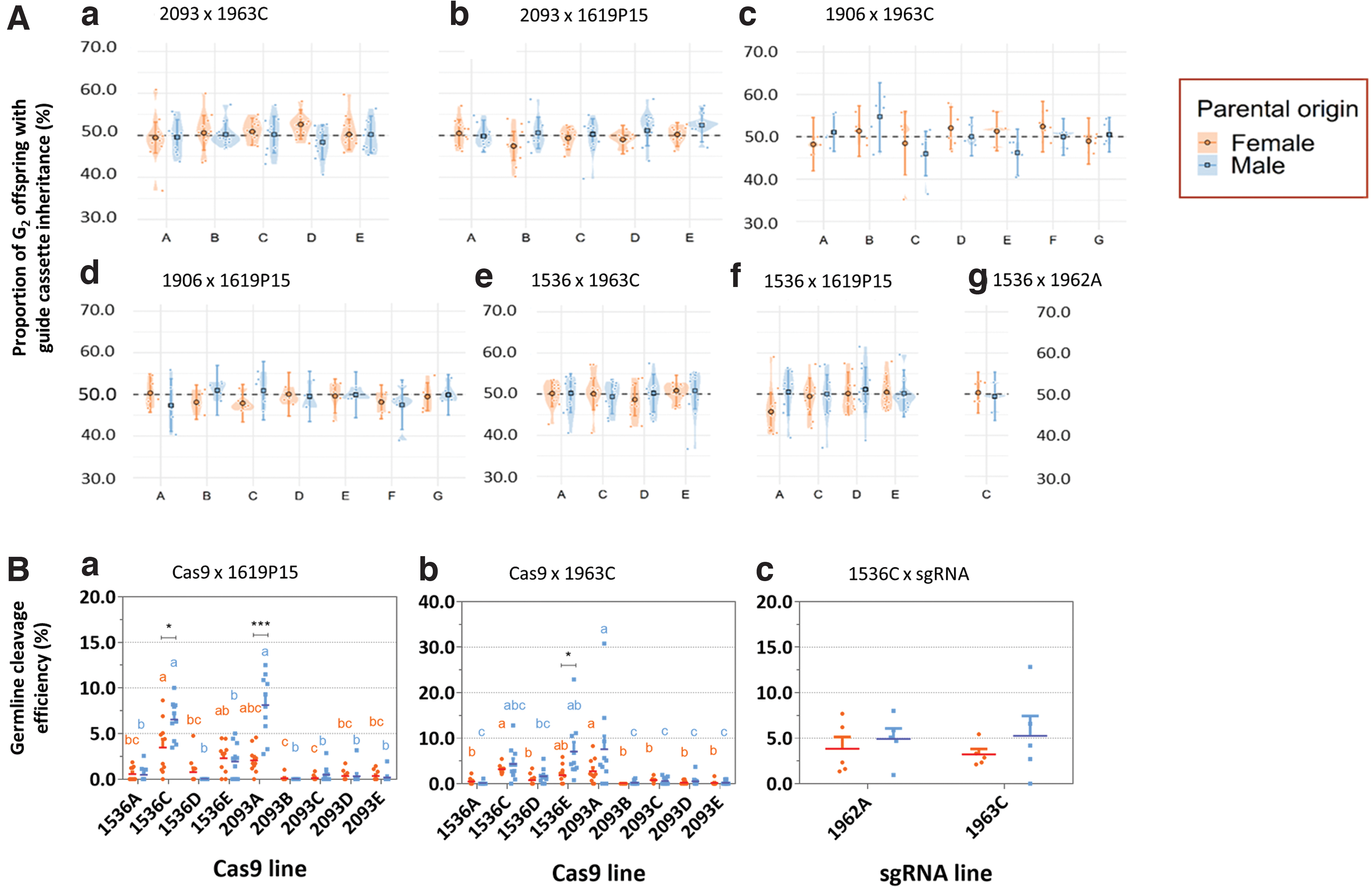
Assessment of the inheritance of sgRNA cassettes and the germline cleavage efficiency.
Comparing putative germline cleavage rates by sex of transheterozygous F1 parent between Cas9 lines, we found that 1536C and 2093A lines showed the highest average cleavage when assessing F1 male parents (6.0% and 7.7%, respectively), whereas 1536C exhibited the highest average germline cutting when assessing F1 female parents (2.9%). When comparing between sexes within each Cas9 line, the germline Cas9 activity in male transheterozygous parents was significantly higher than in female parents for both 1536C and 2093C crosses. Other comparisons were nonsignificant.
Similarly for crosses involving 1963C (Fig. 4B-b), putative germline cutting events were detected exclusively in F1 WT and Cas9-only individuals from 1536 and 2093 crosses. Specifically, 2093A showed the highest germline editing levels, independent of whether assessing male or female F1 transheterozygous parent (male F1 = 7.6%, female F1 = 2.8%). It was also noted that male F1 parents showed higher germline cleavage rates than females in the 1536E crosses. To compare the cleavage efficiency induced by different PxU6 promoters, we also conducted 1536C × 1962A crosses. Although null mutants derived from germline cleavage were also found in F2s, no significant differences in frequency of these events were observed between 1962A and 1963C crosses involving 1536C (Fig. 4B-c).
A random selection of WT F2 knockout mutants were individually sequenced at the relevant target site. Multiple indel alleles were observed separate from those previously established as occurring in each knockout line 27 (Supplementary Fig. S6).
Finally, gonads and carcasses of WT, 1536C, 2093A, and 2093D heterozygotes were dissected for comparing Cas9 protein expression. At a broad scale, it was noted that Cas9 protein levels generally appeared higher in carcasses than gonads given equal amounts of total protein loaded (35 μg) in all three transgenic lines, while no Cas9 was expressed in WT (Supplementary Fig. S7)—a result in line with Cas9 transcription patterns detected by RT-PCR (Supplementary Fig. S1). However, unlike Cas9 mRNA patterns, Cas9 protein was not observed in 2093D gonads. This result concurs with the observation of both somatic and germline Cas9 activity (cutting) in the 1536C and 2093A lines but no germline activity in the 2093D line, strengthening the argument that cutting events detected in the F2 generation resulted from Cas9/sgRNA activity in the F1 germline.
Discussion
Previous studies integrating CRISPR-Cas9 components into lepidopterans are limited to the highly domesticated model species Bombyx mori (silkworm). There, using the IE1 promoter to drive Cas9 resulted in somatic editing of transheterozygotes39–41 while using the B. mori nanosP promoter resulted in both somatic mosaicism of transheterozygotes31,42–53 and some germline activity.54–56 However, these studies provide no rates for these activities, and due to their experimental design, it is often difficult to disentangle the effects of germline edits from somatic mosaicism. Here, we expand the use of integrated CRISPR-Cas9 components to a free-living agricultural pest of global importance using an experimental design that allowed us to quantify and clarify different editing activities and also to assess the possibility of homing-based gene drive. Using three different endogenous promoters, which were putatively germline specific, we developed Cas9 expressing DBM lines. Testing these lines, we were able to demonstrate all three forms of Cas9 nuclease activity—somatic expression, resulting in mosaicism of F1 transheterozygotes; trans-generational deposition, resulting in F1 targetsgRNA-only mosaics; and within germline expression, resulting in F2 nonfluorescent (WT) and Cas9-only target gene full-knockouts. Our results indicate that, in general, Pxvasa-Cas9 and PxnanosP-Cas9 drove higher rates of Cas9 activity than Pxmeiw68-Cas9.
We observed that somatic Cas9 expression from somatic activity of the vasa and nanosP promoters14–16,57 was much enhanced relative to native vasa and nanosP in all Cas9 lines. This may be an interaction with the Hr5 enhancer used to aid expression of the fluorophore marker in our constructs. Although nanosP promoter activity has been reported as more germline restricted than vasa in D. melanogaster,57,58 these two promoters both achieved high levels of somatic Cas9 expression in DBM, resulting in 100% observable mosaicism in multiple lines, similar to reports in Drosophila suzukii when the vasa and nanos promoters were used to drive Cas9. 63
For some gene drive applications in DBM, it may be beneficial to reduce this somatic overexpression, for example by employing alternative regulatory elements without long-distance enhancers to drive marker expression, such as the 3xP3 or OpIE2 promoters, both of which have been characterized in lepidopteran transgenesis. 25 Nonetheless, “leaky” somatic expression of Cas9 may be a significant problem in constructing population-suppression gene drives targeting fertility/viability genes due to the generation of resistant alleles and the high fitness cost in drive heterozygotes.59,60 On the other hand, the extremely high level of somatic editing activity demonstrated here would be of great utility in building alternative forms of genetic control technologies such as “precision-guided SIT” (pgSIT/TI-pgSIT),61–63 HomeR drive, 64 and ClvR/TARE drive systems65,66 by targeting essential fertility/viability genes. However, at least in the latter two technologies, this would need to be paired with significantly increased germline Cas9 activity. Future work could seek to identify suitable target genes for these phenotypes in DBM and utilize the novel tools generated here to arrange such a system for the first time in a lepidopteran.
Both maternal and paternal deposition of Cas9 occurred in PxnanosP-Cas9, although maternal Cas9 was more abundant, judging by mosaicism of targetsgRNA-only F1 progeny. Our results contrast with a similar study in D. melanogaster where maternal but not paternal deposition of Cas9 was evident. 67 Similar deposition, however, was not observed in Pxvasa-Cas9 lines, although such carryover occurs at high levels in vasa-Cas9 A. gambiae 59 and vasa-Cas9 D. melanogaster 67 transgenics, and Cas9 protein was clearly detected in the ovaries and testes of a Pxvasa-Cas9 line here. It is possible that deposition of Cas9 protein or mRNA occurred in Pxvasa-Cas9 lines but not at a level high enough to be observed in the relatively small areas (eyes and dorsal stripes) where mosaicism could be detected.
Germline activity of Cas9 was confirmed by the hypothesized inheritance of null Pxkmo/Pxyellow alleles by F2 non-Cas9 (WT and targetsgRNA) individuals. However, compared with studies in D. melanogaster60,67 where both vasa and nanos promoters have been tested, rates of germline cutting were relatively low, with a maximum mean of 8.1 ± 1.0% of WT chromosomes mutated. This was surprising because the high rate of somatic mosaicism observed suggested the expressed Cas9 protein and Pol III sgRNAs were highly capable of acting at the target site. A possible explanation could be either insufficient Cas9 expression/translation or sgRNA expression from the employed promoters within germline cells. While RT-PCR and Western blot analysis of gonads from Cas9 lines confirmed the presence of Cas9 mRNA/protein, we cannot exclude the possibility that this may have largely represented expression within the somatic cells of gonadal tissues. Similar to studies assessing CRISPR-based split-drive in A. aegypti, 17 we observed that different Cas9 lines generated with a single construct (e.g., 2093A and 2093D) varied in Cas9 expression (RT-PCR and Western blot) and subsequent germline cleavage rates, likely due to the “positional effect” of semi-random piggyBac insertion. Future studies may seek to employ a site-specific approach once suitable “safe-harbor” sites have been identified, or an “integral” gene drive approach where endogenous regulatory elements can be hijacked for canonical Cas9 expression. 68 Interestingly, when comparing between sexes within each cross, significant differences (where they occurred) showed a higher level of cleavage in male rather than female germlines, whereas the opposite trend would have been expected given the endogenous expression patterns of Pxvasa and PxnanosP.
Germline editing and conversion rates can vary according to the locus being targeted by the homing element, likely affected by chromosome/chromatin structure, sgRNA efficiency, or sequence diversity in the population.15,60 As such, we constructed sgRNA expressing lines by site specifically inserting guide cassettes into two loci: Pxkmo and Pxyellow. To our knowledge, this is the first report of HDR-based long fragment knock-in using CRISPR-Cas9 in the Lepidoptera, although site-specific integrations using the more complex microhomology-mediated end joining (MMEJ)-based PITCH technologies and TALEN-based integration have been demonstrated in B. mori.44,69,70 However, knock-in efficiency was relatively low (0.4–0.7%), compared with highly efficient knockout in DBM (57.1%). 27 Integration rates were also lower than comparable reports in dipterans.71–73 Interestingly, our results showed a large number of noncanonical repair events, with only two of the seven generated homing elements lines showing a precise, perfect integration. This runs contrary to experience in other insect species published,17,57,70,73 and also our extensive experience in mosquitos, where off-target integration may occur relatively often, but those constructs integrated at the correct location are almost always integrated perfectly. A possible reason for imperfect integration detected here is that repeat sequences existing either inside the knock-in cassette (e.g., the multiple U6 promoters) or between the cassette and flanking genomic regions may have caused noncanonical repair of DSB (e.g., MMEJ) and the loss of partial knock-in sequences. 75 It is also noted that previous efforts to generate Cas9-mediated insertions using plasmid templates in B. mori have failed, while similar TALEN-based integrations are relatively efficient. 45 Therefore, it may be that some part of the DSB repair process in lepidopterans differs from other insects studied so far, reducing the efficiency of donor single-strand invasion following the blunt ends breaks created by Cas9, but not the overhanging breaks created by TALENs.
Other genome engineering tools (e.g., TALEN,45,69 TAL-PITCH, and CRIS-PITCH 70 ) could be employed to increase the integration efficiency of CRISPR-Cas9 split-drive components in DBM, although for this purpose the integration site mediated by these technologies would need to coincide fortuitously with the chosen sgRNA cleavage site. Additionally, we successfully utilized Cas9-based targeted sequencing of the integration junction to resolve the location of one of these homing element lines (1619P15). To our knowledge, this is the first published use of such a system and may be of benefit to other researchers seeking to resolve transgene integration junctions that are similarly difficult to resolve using conventional PCR-based techniques.
Despite testing three putatively germline-active Pol II promoters and six U6 Pol III promoters at two independent loci, we were unable to demonstrate significant gene drive. One explanation for this could include insufficient expression of Cas9 mRNA or its translation in tissues and at times required for a “homing” reaction to occur. The simultaneous presence of Cas9 protein and sgRNA in a germline-specific time window is critical for homing, especially in early meiosis when the germ cell is in a recombination-orientated state and homologous chromosomes are in close enough proximity to act as HDR templates.67,76
We were able to demonstrate here that Cas9 protein was present in gonadal tissues of both sexes and that it was able to mediate heritable germline DNA breakage events after complexing with sgRNAs. However, the timing of these events may have presented before or after this narrow window, and/or the expression levels might be too low in germ-cells to achieve distinguishable biases in inheritance, as has been similarly reported recently in mice. 77 As an additional hurdle, the imperfect repair of the 5′ homology arm/genome interface in 1962A may have reduced the ability of this integration to act as a repair template. This, however, did not apply to 1619P15 or 1963C, which were perfect integrations. Finally, we are unable to discount the possibility that the six known U6 promoters in DBM do not function efficiently in the germline. We recommend additional germline-specific regulatory components to be tested in DBM to increase germline cleavage and conversion efficiency.
Conclusion
In summary, this study has identified and characterized endogenous genetic components (i.e., germline active Pol II and Pol III promoters) and completed the first CRISPR-mediated site-specific knock-in of large fragments in the Lepidoptera. With these components, we built and tested the first split-drive system in a lepidopteran, achieving a very high level of somatic editing. Although we did not see significant homing, Cas9-mediated germline cleavage as well as maternal and paternal Cas9 deposition was observed. Our results provide valuable experience, paving the way for future construction of gene drives, or other Cas9-based genetic control strategies in the global pest, DBM, and other lepidopterans.
Footnotes
Acknowledgments
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by European Union H2020 Grant nEUROSTRESSPEP (634361). In addition, THS and LA were respectively supported by a Impact Acceleration Account grant (BB/S506680/1), and three core fundings [BBS/E/I/00007033, BBS/E/I/00007038 and BBS/E/I/00007039] from the UK Biotechnology and Biological Sciences Research Council. XX was supported by a CSC Scholarship from the Chinese Government.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.