
Abstract
The use of viral vectors that can replicate and move systemically through the host plant to deliver bacterial CRISPR components enables genome editing at the whole-plant level and avoids the requirement for labor-intensive stable transformation. However, this approach usually relies on previously transformed plants that stably express a CRISPR-Cas nuclease. Here, we describe successful DNA-free genome editing of Nicotiana benthamiana using two compatible RNA virus vectors derived from tobacco etch virus (TEV; genus Potyvirus) and potato virus X (PVX; genus Potexvirus), which replicate in the same cells. The TEV and PVX vectors respectively express a Cas12a nuclease and the corresponding guide RNA. This novel two-virus vector system improves the toolbox for transformation-free virus-induced genome editing in plants and will advance efforts to breed more nutritious, resistant, and productive crops.
Introduction
Systems derived from bacterial CRISPR and CRISPR-associated (Cas) proteins 1 have revolutionized biotechnology. In plants, CRISPR-Cas holds great promise for unprecedented genome engineering of both model species and crops.2–6 Most common CRISPR-Cas arrangements include a Cas endonuclease, such as Streptococcus pyogenes SpCas9, and a single-guide RNA (sgRNA), which specifically directs the nuclease to a sequence of interest in the genome. As in other taxonomic groups,7–10 virus-derived vectors have been reported as a powerful alternative to express the CRISPR-Cas components at the whole-plant level, avoiding the labor-intensive and time-consuming tissue culture approaches required for stable transformation. These strategies are commonly termed “virus-induced genome editing” (VIGE) and have focused on the delivery of one or more sgRNAs using RNA or DNA virus vectors in transgenic plants that stably express the Cas nuclease.11–19
Expression of a Cas nuclease using a plant virus–derived vector able to move systemically through a plant was long considered unachievable due to cargo constraints. However, the innovative work by Ma et al. 20 demonstrated efficient genome editing by delivering both the sgRNA and SpCas9 at the whole-plant level using a vector derived from sonchus yellow net virus (SYNV; family Rhabdoviridae). Despite this unprecedented achievement, additional virus-based systems for the co-expression of Cas nucleases and sgRNAs at the whole-plant level are still required to improve the current toolbox for crop engineering. Notably, each viral vector has its own unique properties, particularly a specific host range.
To expand the virus-based tools for transformation-free genome editing in plants, we co-expressed the Cas nuclease and the guide RNA using two compatible viral vectors that replicate in the same cells and coordinately move systemically through the whole plant. We chose a potyvirus vector to express the Cas nuclease. Potyviruses (genus Potyvirus) are the largest group of plus-strand RNA viruses, with more than 200 currently known species that collectively can infect a large range of host plants. 21 We also focused on a Cas12a (formerly Cpf1) nuclease, which is a component of a class 2 type V CRISPR system, isolated from Lachnospiraceae bacterium ND2006 (LbCas12a). 22 Genome editing using LbCas12a has been demonstrated in plants,23–26 and the coding sequence corresponding to this nuclease is smaller than that of SpCas9. We also co-expressed the guide RNA using a recently described potato virus X (PVX, genus Potexvirus, family Virgaviridae) vector, which efficiently induces hereditable gene editing in Nicotiana benthamiana plants that stably express SpCas9. 27 Our results demonstrated efficient DNA-free N. benthamiana genome editing using the two compatible RNA virus vectors.
Methods
Viral vectors
Guide RNAs to target N. benthamiana Flowering locus T (NbFT; SolGenomics Niben101Scf01519g10008.1) and Xylosyl transferase 1 (NbXT1; Niben101Scf04205g03008.1) were selected using the CRISPR-P online tool as described by Bernabé-Orts et al. 28 (Supplementary Table S1). Nucleotide (nt) sequences of recombinant viral clones are shown in Supplementary Figures S1 and S2. These clones were built using the primers shown in Supplementary Tables S2 and S3.
Plant inoculation
N. benthamiana wild type and transformed plants expressing tobacco etch virus (TEV, genus Potyvirus) nuclear inclusion b (NIb) protein 29 were grown at 25°C under a 12 h/12 h day/night photoperiod. Plants that were 4–6 weeks old were agro-inoculated, as previously described.27,30 Tissue samples (approximately 100 mg) from the first symptomatic upper non-inoculated leaf were collected at different days post inoculation (dpi), as indicated, for virus progeny and plant genome-editing analyses.
Analysis of Viral Progeny
RNA was purified from leaf samples using silica gel columns. 27 cDNA was synthesized using RevertAid reverse transcriptase (Thermo Fisher Scientific) and primer D179 (Supplementary Table S4). Polymerase chain reaction (PCR) with Thermus thermophilus DNA polymerase (Biotools) was used to amplify the TEV coat protein (CP) cistron (primers D178 and D211; Supplementary Table S4) or a fragment of the LbCas12a open reading frame (ORF; primers D3604 and D3605; Supplementary Table S4). The presence of full-length LbCas12a coding sequence in the viral progeny was assayed by reverse transcription (RT)-PCR using primers D2567 (RT) and D570-D571 (PCR; Supplementary Table S4). PCR products were separated by electrophoresis in 1% agarose gel followed by staining with ethidium bromide.
Analysis of N. benthamiana genome editing
DNA from leaf samples was purified using silica gel columns. 27 N. benthamiana genome fragments were amplified by PCR using high-fidelity Phusion DNA polymerase (Thermo Fisher Scientific; Supplementary Table S5). PCR products were separated by agarose gel electrophoresis, purified from the gel, and subjected to Sanger sequencing (Supplementary Table S5). The presence of sequence modifications was analyzed using the inference of CRISPR edits (ICE) software.
Results
A dual virus–based vector system to co-express Cas nucleases and guide RNAs in plants
Initially, we built a TEV recombinant clone in which the cDNA of a human codon-optimized LbCas12a replaced that of the viral NIb protein (TEVΔNIb::LbCas12a). Our previous work showed that this vector could express large exogenous sequences, such as a whole bacterial metabolic pathway to biosynthesize lycopene or the saffron carotenoid cleavage dioxygenase.29,31 However, since the virus lacks the viral RNA-dependent RNA polymerase NIb, it replicates only in plants that express this protein. 31 In our recombinant clone, the sequence coding for LbCas12a replaced most of the NIb cistron and was flanked by the native nuclear inclusion a protease (NIaPro) cleavage sites that mediate the release of the nuclease from the viral polyprotein (Fig. 1A and Supplementary Fig. S1).
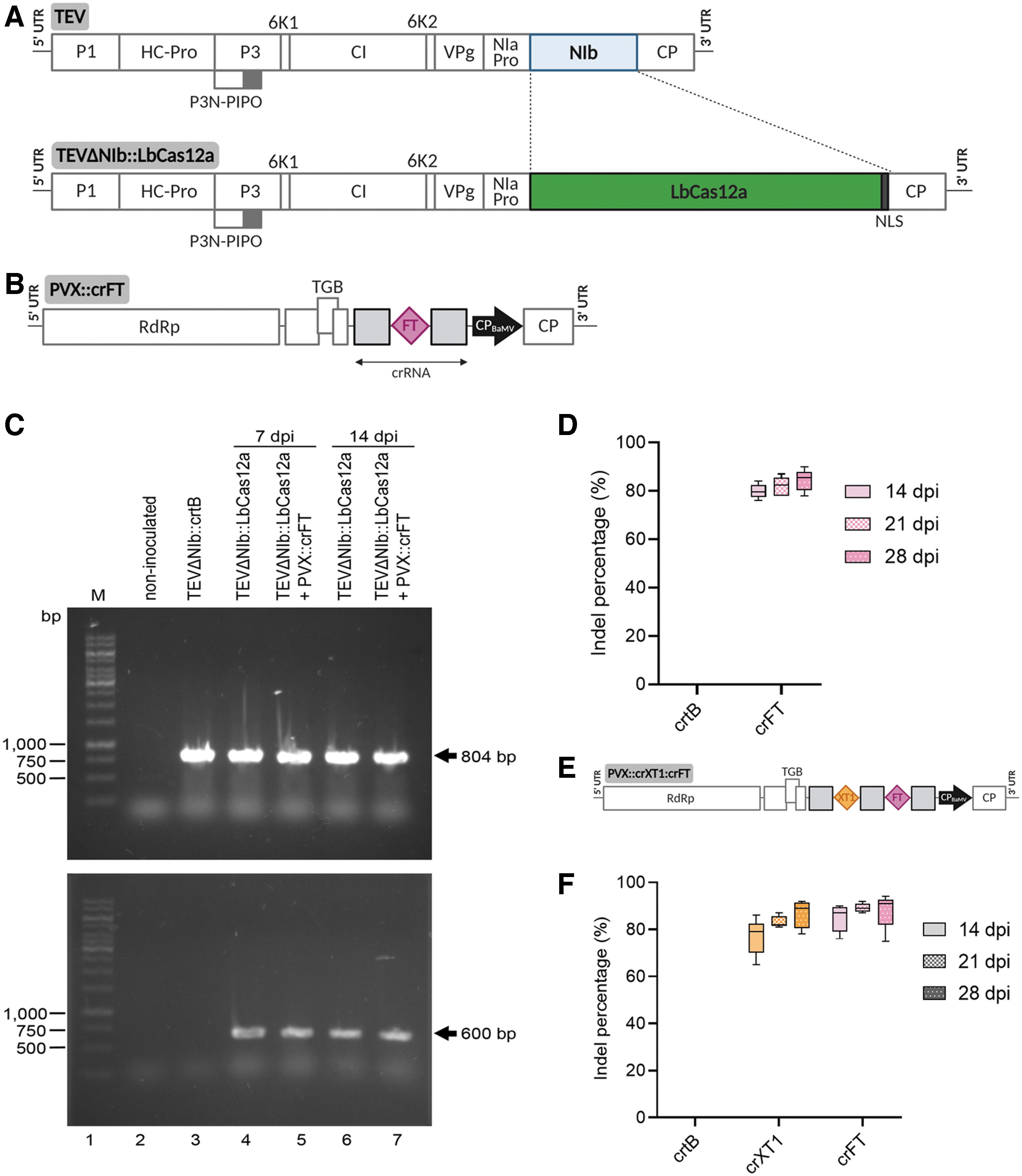
DNA-free gene editing in Nicotiana benthamiana based on virally delivered LbCas12a and crRNA.
Next, we built a recombinant version of PVX in which the LbCas12a CRISPR RNA (crRNA) was expressed under the control of the viral CP promoter, and the 29 initial codons of PVX CP were deleted to improve the stability of the recombinant clone. 32 The viral CP was expressed from a heterologous promoter derived from that of the CP of bamboo mosaic virus (BaMV; genus Potexvirus). Based on previous work by Bernabé-Orts et al. 28 assessing the efficiency of the Cas12a-mediated gene editing of several N. benthamiana loci, NbFT was selected as the target gene (PVX::crFT; Fig. 1B and Supplementary Fig. S2). The 65 nt crRNA was cloned downstream of the PVX CP promoter and consisted of a 23 nt protospacer sequence specific to the target gene flanked on both 5′ and 3′ ends by a conserved 21 nt scaffold, also known as a direct repeat.
N. benthamiana plants constitutively expressing TEV NIb under the control of cauliflower mosaic virus (CaMV) 35S promoter and terminator 29 were co-inoculated with a 1:1 mix of two cultures of Agrobacterium tumefaciens C58C1 transformed with plasmids harboring TEVΔNIb::LbCas12a and PVX::crFT constructs. Controls included inoculation of TEVΔNIb::LbCas12a alone and inoculation of TEVΔNIb::crtB, which allows visual tracking of virus systemic movement due to the yellow pigmentation of infected tissue induced by Pantoea ananatis phytoene synthase (crtB). 31 At 7 dpi, typical symptoms of TEV infection emerged in the upper non-inoculated leaves of all plants. Notably, at 14 dpi, irregular chlorotic spots appeared in the leaves of plants co-inoculated with TEVΔNIb::LbCas12a and PVX::crFT but not in those inoculated with TEVΔNIb::LbCas12a alone (Fig. 2). PVX infection is characterized by the appearance of vein banding, ring spots, and leaf atrophy. 33 Therefore, the observed phenotypic alteration could have been due to LbCas12a-mediated editing of NbFT or an effect of the co-infection. Samples from the first systemically infected upper leaf were collected at 7 and 14 dpi, and TEVΔNIb progeny was studied by RT-PCR analysis (Fig. 1C). An 804 bp specific region of the TEV genome corresponding to the CP cistron was amplified in all virus-inoculated plants, confirming the presence of the virus (Fig. 1C, top panel). However, for viral vectors carrying big cargos, genomes larger than wild type are likely to recombine to smaller sizes, thus triggering the loss of heterologous genes. 34 An additional RT-PCR analysis was performed, and a 600 bp cDNA corresponding to a fragment of the LbCas12a coding sequence was exclusively amplified from plants inoculated with TEVΔNIb::LbCas12a alone or co-inoculated with TEVΔNIb::LbCas12a and PVX::crFT, regardless of sampling time (Fig. 1C, bottom panel). A complementary RT-PCR analysis using primers hybridizing on the flanking borders of the viral vector confirmed the presence of full-length LbCas12a coding sequence in the viral progeny at 14 dpi, although it also indicated that viral progeny partially lost the cargo at this time point (Supplementary Fig. S3). Taken together, these results suggest that the expression of LbCas12a nuclease expands from the onset of viral infection until an undetermined time point in which the viral vector completely loses the heterologous gene.

35S::NIb N. benthamiana plants and representative leaves from these plants inoculated with TEVΔNIb::LbCas12a (left) or co-inoculated with TEVΔNIb::LbCas12a and PVX::crFT (right) at 14 dpi. Chlorotic spots in leaves from co-inoculated plants are indicated with red arrows.
Next, DNA was purified from leaf samples collected at 7 and 14 dpi, and a 550 bp fragment of the NbFT gene covering the LbCas12a target site was amplified by PCR. Sanger sequencing of the PCR products and ICE analysis revealed robust gene editing at 14 dpi in plants co-inoculated with TEVΔNIb::LbCas12a and PVX::crFT, reaching an insertion and deletion (indel) percentage of up to 75% (Fig. 1D and Supplementary Fig. S4). No significant differences were observed at 21 and 28 dpi. The indel distribution was consistent with the deletion-enriched mutagenesis profile characteristic of Cas12a activity, being mainly 5–10 bp deletions. 28 These results indicate that the simultaneous delivery of CRISPR-Cas12a components through two compatible viral vectors (i.e., TEVΔNIb and PVX) allows highly efficient targeted mutagenesis in N. benthamiana.
Multiplex genome editing using the dual virus–based vector system in plants
A key advantage of CRISPR-Cas genome editing is the capacity to target several loci at once by the simultaneous expression of several guide RNAs (i.e., multiplexing). Based on our observations in Cas9-expressing plants, 27 we wondered whether the PVX vector could allow the delivery of multiple, functional crRNAs for Cas12a-mediated editing. To investigate this, we selected NbXT1 as the second target gene. As Cas12a can self-process crRNAs due to its RNase III activity, NbXT1 and NbFT crRNAs were arranged in tandem under the control of the same CP promoter, thus creating the PVX::crXT1:crFT construct (Fig. 1E and Supplementary Fig. S2). N. benthamiana plants constitutively expressing TEV NIb were co-inoculated with a 1:1 mix of two A. tumefaciens cultures carrying TEVΔNIb::LbCas12a and PVX::crXT1:crFT. A. tumefaciens transformed with TEVΔNIb::crtB was used as a control in this assay. The first systemically infected leaf was sampled at 14, 21, and 28 dpi, following extraction of genomic DNA and PCR amplification of the target sites. ICE analysis revealed efficient gene editing on both NbXT1 and NbFT, with average indel percentages ranging from 76% to 88%, which was maintained regardless of the sampling time (Fig. 1F). In addition, the absence of statistically relevant differences in gene editing among NbXT1 and NbFT suggested that LbCas12a can efficiently self-process tandemly arrayed crRNAs. Time-course comparison with the single crRNA construct, in the case of NbFT, also revealed that multiplexing does not affect the editing efficiency, since the mutation rates achieved with both strategies were similar (compare Fig. 1D and F).
Dual vector CRISPR-Cas genome editing in wild-type plants
The fact that TEVΔNIb infectivity depends on the supplementation of viral NIb from a transgene may be perceived as a limitation of this genome-editing system, as the approach is still bound to a previously transformed plant. An alternative strategy for supplying NIb activity consists of the co-inoculation of TEVΔNIb with a recombinant PVX expressing NIb. 30 We wondered whether a single PVX vector could: (1) provide NIb activity for the replication of TEVΔNIb, and (2) deliver the crRNA for LbCas12a-mediated gene editing. Thus, the coding sequence of the TEV NIb cistron plus an additional amino-terminal Met was inserted within the expression cassette in PVX. NbFT-specific crRNA was added downstream of NIb without any linker sequence, so that both NIb and crFT expression were under the control of the PVX CP promoter (PVX::NIb:crFT; Fig. 3A and Supplementary Fig. S2). Wild-type N. benthamiana plants were co-inoculated with a 1:1 mix of two cultures of A. tumefaciens carrying TEVΔNIb::LbCas12a and PVX::NIb:crFT. A. tumefaciens transformed with TEV::crtB was this time used as a control. At 7 dpi, the apical leaves of the co-inoculated N. benthamiana plants became symptomatic (i.e., leaf curling). At 14 dpi, necrotic spotting and interveinal mottling were observed in systemic leaves, which became more noticeable over time (Fig. 4). Samples from the first systemically infected upper leaf were collected at 14 dpi, and the 550 bp fragment of the NbFT gene covering the LbCas12a target site was amplified by PCR (Fig. 3B). ICE analysis of the PCR products exhibited 20% indels in plants co-inoculated with TEVΔNIb::LbCas12a and PVX::NIb:crFT (Fig. 3C). These results indicate that a single PVX vector can supply the viral NIb activity that allows TEVΔNIb to replicate and systemically spread in wild-type N. benthamiana, as well as to perform crRNA delivery for LbCas12a-mediated genome editing.
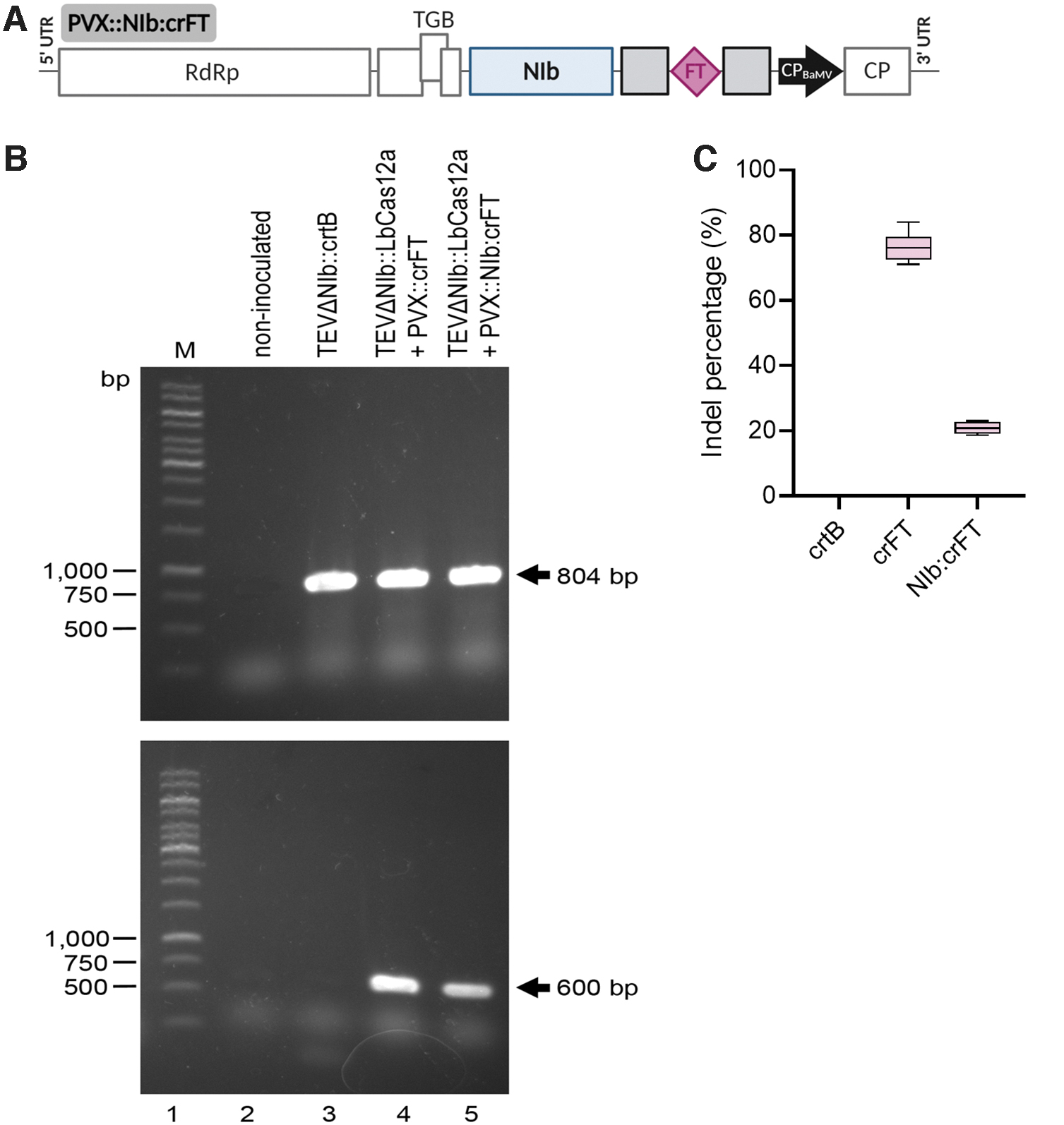
Engineering of a single PVX vector for complementation of defective TEVΔNIb and expression of LbCas12a crRNA.
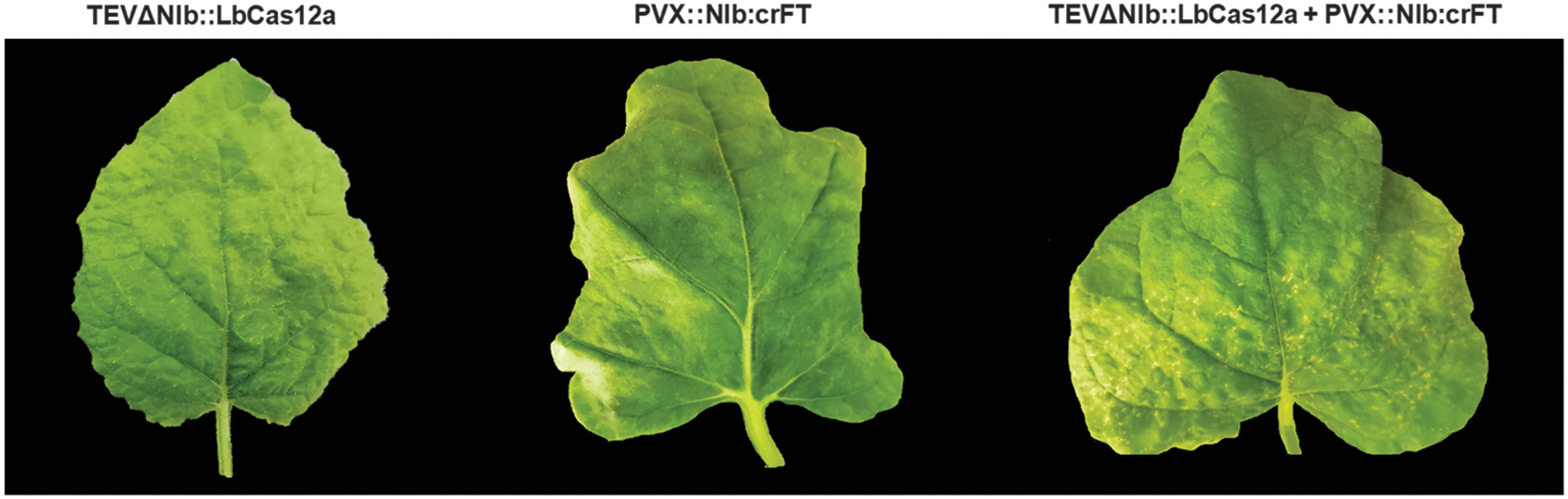
Representative leaves from wild-type N. benthamiana plants inoculated with TEVΔNIb::LbCas12a alone (left), PVX::NIb:crFT alone (middle), or co-inoculated with TEVΔNIb::LbCas12a and PVX::NIb:crFT (right) at 14 dpi.
Discussion
In this study, we describe the engineering of a dual RNA virus system for the delivery of CRISPR-Cas12a components for genome editing in plants. The system consists of two compatible plus-strand RNA viruses, specifically the potyvirus TEV and the potexvirus PVX, to express the Cas nuclease and the guide RNA, respectively. Notably, the TEV vector (TEVΔNIb) contains the deletion of the RNA-dependent RNA polymerase NIb in order to accommodate the large coding sequence of the Cas nuclease. Our previous efforts to express a functional Cas nuclease using a full-length potyvirus vector in plants had been unsuccessful. However, recent works have reported successful Cas9 expression and DNA-free genome editing using single vectors in N. benthamiana.20,35–37 These contrasting results emphasize the diverse properties of vectors derived from viruses belonging to different genera and families, and the necessity for a large spectrum of molecular tools, operating in a wide range of host species, to tackle challenging VIGE goals in crop plants. Notably, SYNV is a minus-strand RNA virus, the inoculation of which entails some complexity, 38 and as with each plant virus, it exhibits a particular host range. 39
In this work, we aimed to express a Cas nuclease using a potyvirus vector for DNA-free plant genome editing. The genus Potyvirus is the largest among the plant RNA viruses and comprises more than 200 species that infect a wide range of host plants from many different botanical families. 21 Therefore, it offers a wealth of genetic resources for VIGE. Although we were unable to express the Cas nuclease successfully using a full-length potyvirus vector, we demonstrated that TEVΔNIb allows the transient expression of LbCas12a and that PVX can perform both single or multiple crRNA delivery as well as providing the NIb activity. Our two-virus delivery system resulted in efficient targeted editing in both NIb-expressing and wild-type N. benthamiana plants, reaching indel percentages of up to 80% and 20%, respectively (Figs. 1 and 3). This dual vector system not only incorporates the enormous genetic resources of potyviruses for VIGE, but also demonstrates that compatible RNA virus vectors can be used to deliver several CRISPR-Cas components simultaneously. This could be useful for more sophisticated arrangements in plant genome editing and gene expression regulation studies. However, caution must be taken when implementing these designs because positive-strand RNA viruses, such as potyviruses and potexviruses, are prone to homologous recombination. 40 Homology must be avoided in flanking sequences of transgenes and heterologous genes, such as NIb in this work, to avoid undesired acquisition by viral vectors (Supplementary Fig. S5). What is also of importance is that our dual RNA virus system is based on two positive-strand viral species that belong to different families and are able to co-infect the same cells. When designing systems with multiple viral vectors, superinfection exclusion must be taken into consideration because phylogenetically related viruses are generally excluded to co-infect the same cells. 41
Recently, we developed a PVX vector to express multiplex SpCas9 sgRNAs efficiently at the whole-plant level. 27 Current results with LbCas12a confirm that PVX can be easily engineered for the simultaneous delivery of guide RNAs regardless of the nature of the Cas nuclease, which highlights its usefulness in a variety of multiplexing approaches. Efficient genome editing in our dual-virus system implies that LbCas12a is able to find and process the crRNA either in the whole PVX genome, in viral subgenomic transcripts or in both. Particularly in the first case, LbCas12a-mediated cleavage will produce replication-incompetent viruses. The observation that LbCas12a expression has no effect on PVX vector infectivity suggests that either the whole viral genome is not the preferred target or cleavage is not that quantitative to abolish infectivity. We obtained similar results when flanked Cas9 guide RNAs with cleavable tRNA sequences in the PVX vector. 27
In contrast to previous reports of DNA-free VIGE that used SpCas9,20,36 we selected Cas12a. This was due to the following unique features of the nuclease 42 : (1) the cleavage of target DNA is directed by a single crRNA shorter than that of SpCas9 sgRNA; (2) the protospacer adjacent motif (PAM) is T-rich (5′-TTTN-3′); (3) DNA cleavage results in cohesive ends with 4 or 5 nt overhangs, which might facilitate homology-directed repair (HDR); (4) it exhibits RNase activity useful to facilitate multiplex gene editing; and (5) it is smaller than SpCas9 (3.8 vs. 4.2 kb), which is important in terms of viral delivery. Moreover, the unique characteristics of the CRISPR-Cas12a system, unlike that of Cas9, such as the recognition of a T-rich PAM and the induction of staggered ends that facilitate homologous recombination, limit the range of target sequences and thus reduce off-target activity. 42 Cas12a orthologues from Francisella novicida U112 (FnCas12a), Acidaminococcus sp. BV3L6 (AsCas12a), and LbCas12a were first experimentally validated in mammalian cells.43–45 In plants, targeted mutagenesis was achieved in rice and tobacco using any of the orthologs.23–26 A DNA-free approach based on the delivery of AsCas12a or LbCas12a loaded with crRNA was also validated in wild tobacco and soybean protoplasts. 46 Here, we focused on LbCas12a, since previous works reported that this nuclease possesses higher efficiency than FnCas12a or AsCas12a.24,28 We showed here that LbCas12a is also effective for plant genome editing when virally delivered.
In conclusion, our dual RNA virus–based system broadens the current toolbox for DNA-free VIGE and will contribute to applications in plant functional genomics and crop improvement.
Footnotes
Acknowledgment
The authors thank Verónica Aragonés for excellent technical assistance.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by grants BIO2017-83184-R and PID2019-108203RB-I00 from the Ministerio de Ciencia e Innovación (Spain) through the Agencia Estatal de Investigación (co-financed by the European Regional Development Fund) and H2020-760331 Newcotiana from the European Commission. M.U. and M.V.-V. are the recipients of fellowships FPU17/05503 from the Ministerio de Ciencia e Innovación (Spain) and APOSTD/2020/096 from the Generalitat Valenciana (Spain), respectively.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.