
Abstract

Gene therapy in a mouse model for amyotrophic lateral sclerosis (ALS) illustrates the rapid deployment of base editing in therapeutic modeling of neurodegenerative disease.
In the United States, baseball is the national sport. ALS is better known as Lou Gehrig's disease after the legendary first baseman of the New York Yankees who succumbed to it in 1941. However, it was Jean-Martin Charcot (1825–1893) who first defined the distinctive pathology and degeneration of motor neurons in ALS.1,2 Typically, the disease starts with a mild motor abnormality that then spreads, leading to complete paralysis and death within a few years. As with almost all neurodegenerative disorders, there is no effective treatment.
While the majority of ALS is “sporadic”, without known family history, about 10% is familial and usually inherited in an autosomal dominant manner. Many associated genes and mutations have been discovered, 3 providing a route toward understanding ALS, particularly using transgenic overexpression. 4 However, the genetic diversity of ALS is also problematic; with numerous targets, it is a challenge to identify key nodes for therapeutic intervention.
Mutation in the gene encoding superoxide dismutase 1 (SOD1) causes roughly 15% of familial and 1% of sporadic ALS. 5 SOD1 was the first ALS-linked gene identified, 6 and research has revealed that mutations destabilize the mature protein, leading to a dominant toxic gain of function. 4 Motor neurons are most susceptible to the cytotoxic effects of disordered and aggregated SOD1, either cell autonomously or indirectly via other cell types.7,8 Importantly, under conditions where unfolding and aggregation are promoted, the wild-type SOD1 protein can also adopt a cytotoxic conformation.9,10 Although the precise nature of the toxic SOD1 species remains elusive, new RNA- and DNA-targeted technologies aimed at reducing SOD1 levels offer hope for treatment.
Antisense oligonucleotides (ASOs) targeting both mutant and wild-type alleles were developed in a mutant human SOD1G93A overexpressing model. 11 ASO treatment has shown promising results, and a current Phase III clinical trial (NCT02623699) will inform whether this will be the first effective treatment for SOD1-ALS. For largely sporadic diseases such as ALS, ASOs are moving toward broader targets. For example, a trial targeting Ataxin 2 (ATXN2) 12 in sporadic ALS is imminent. Other “generic” ALS targets, such as Stathmin 2 (STMN2), should follow.13,14 The RNA targeting effect of ASOs may be transient, requiring lifelong administration. Effective central nervous system (CNS) delivery after peripheral administration 15 would make treatment more practical, but there is likely to be future competition from new gene therapy approaches.
The Next Step
The enduring effects of DNA-targeted therapies offer the next step toward more precise treatment for genetic ALS. Gene editing using CRISPR-Cas9 relies on double-strand break repair.16–18 Although CRISPR-Cas9 gene therapy trials are underway, 19 there are risks to this approach due to the unpredictability of insertions and/or deletion (indel) mutagenesis (e.g., large deletions, unintended protein species). This is where base editing offers some potential advantages.
CRISPR single-base editors are a genome artist's equivalent of an eraser and pencil; hybrid proteins that harness sequence-specific targeting of CRISPR-Cas9 but also draw upon a nucleotide-modifying capacity. 20 Nucleotide modifiers act via base excision and DNA-mismatch repair and can generate modifications in both actively dividing and nondividing cells such as neurons. 21
A recent paper in Molecular Therapy from the laboratory of Thomas Gaj at the University of Illinois addressed the feasibility of in vivo base editing in an ALS model using an adeno-associated virus 9 (AAV9) vector to deliver a Streptococcus pyogenes (Sp)Cas9 cytidine base editor (CBE) to the CNS. 22 This report builds upon a previous study targeting indels to Exon 2 of SOD1 in neonatal SOD1G93A mice with Staphylococcus aureus (Sa)Cas9. 23
To test the efficacy of CBEs, Colin Lim, Thomas Gaj, and colleagues used an elegant dual split-intein strategy (Figure 1) to deliver the much larger APOBEC1-SpCas9n CBE to the mouse spinal cord. The utilization of a more clinically relevant study design (adult, pre-symptomatic mice with delivery to the cerebrospinal fluid, premature stop codon rather than indels), but differences in the promoter, exon, and main cell type that was targeted, makes it difficult to compare the overall outcome of the two approaches.
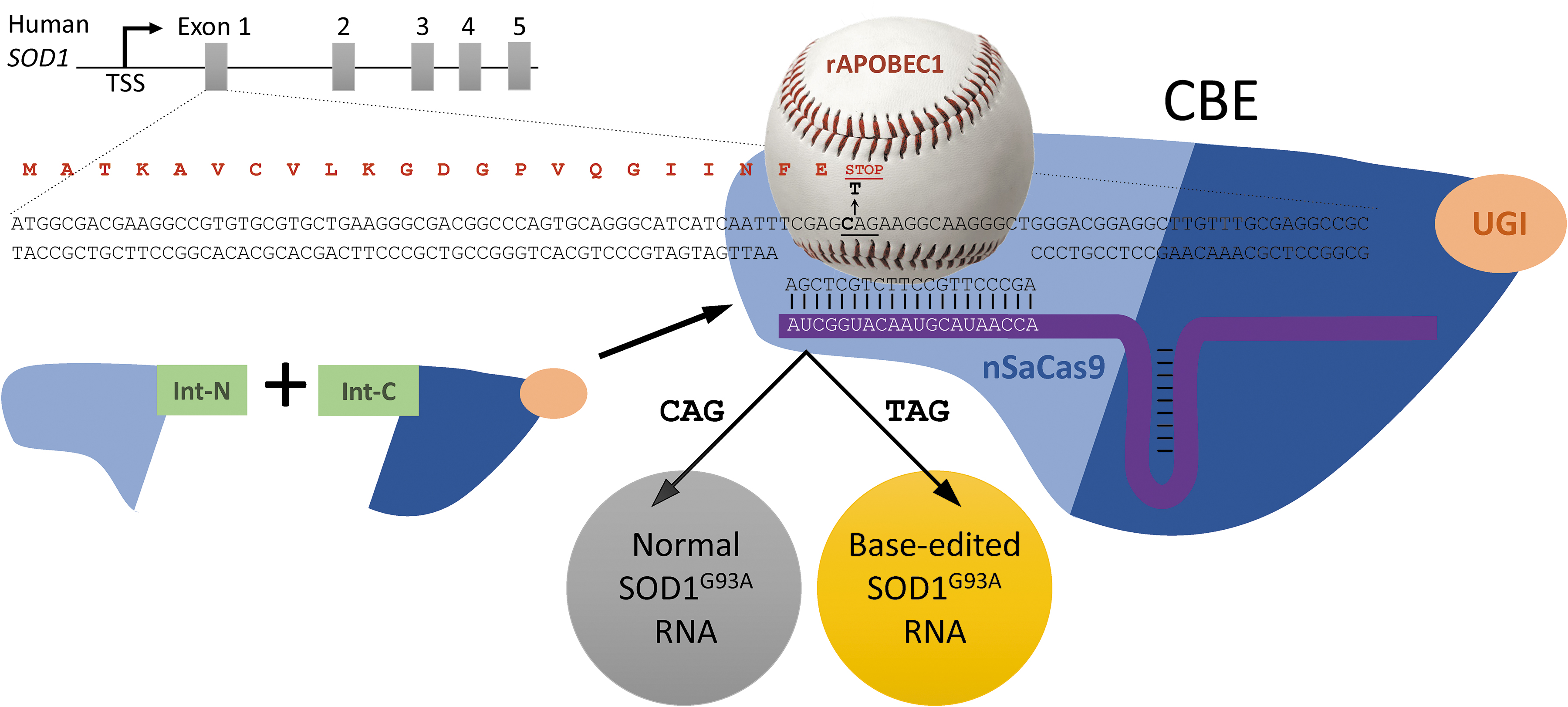
Base editing strategy for SOD1. Schematic representation of the human SOD1 gene with part of the Exon 1 sequence shown below. A cytidine base editor (CBE) consisting of the Cas9 D10A nickase variant from Staphylococcus aureus (nSaCas9; blue) fused with the rat rAPOBEC1 cytidine deaminase (baseball) and the uracil glycosylase inhibitor protein (UGI; orange). The CBE was expressed as two separate split-intein constructs (bottom left). When nSaCas9 is complexed with a sgRNA (purple line) targeting Exon 1 of SOD1, the target site loses base contacts, allowing rAPOBEC1 to access the site for deamination of cytidine bases. UGI prevents base excision repair. A CAG to TAG codon change results in inclusion of a premature stop codon in the base-edited SOD1G93A RNA, which is likely to be subject to non-sense mediated decay (not investigated) and reduction in SOD1G93A protein level (adapted from Lim et al. 22 ).
However, using CBE, Lim et al. reported editing 1.2% of human SOD1 transcripts in 6.5% of spinal cord cells that were dually transduced (mainly astrocytes), leading the authors to propose an “effective editing rate” of 20%. Disease progression was delayed, and several motor symptoms were improved, with very few off-target events. Lim et al. quantified SOD1 aggregates at end stage using an epitope that is hidden in SOD1 aggregates in vivo. 24 So, it remains unclear to what degree CBE reduced disease-associated aggregate load. However, even though the efficiencies of cell-type delivery and editing may be improved, the degree of efficacy achieved in vivo is promising.
Major League
Genetic disorders will benefit from precise genome editing, but will in vivo base editing make it to the major league for ALS treatment? The technological advance of delivering large, functional, multipartite proteins to the CNS of adult animals is an important step. As the authors acknowledge, improving and refining delivery and expression systems will be required to translate this approach for patient benefit. Development of single-cell sequencing will also help the interpretation of editing effects in neurons and other cell types, which is a central problem in ALS.
The SOD1G93A model 4 is widely used for translational studies, 25 and with some 25 copies of the human transgene, it provides a useful test-bed for in vivo base editing. However, switching the field of play could help accelerate the rate of translation of new gene therapy approaches to clinical trials. Overexpression models do not allow quantification of the degree of knockdown that may be beneficial in humans. The recent discovery of neuromuscular symptoms in children carrying a homozygous loss of function mutation in SOD126,27 further highlights the importance of addressing the consequences of SOD1 knockout at the cellular level. 28 A fully humanized SOD1 knock-in mouse would facilitate the testing of gene therapy in a more natural genomic context and under physiological expression levels. 29
Finally, from a patient perspective, it is crucial that ethical and regulatory considerations keep pace with the astounding rate of technological development in this field. The recent successes of ASO trials may have struck out some of the demand for unregulated gene and stem cell therapy clinics, providing ALS patients and their families with real hope of effective therapies. Time will tell if base editing can emerge from the minor leagues to challenge the big hitting of ASOs.