
Abstract
Abstract
Background:
Germline mutations in succinate dehydrogenase (SDH) are associated with multifocal cancers: pituitary gland tumors, pheochromocytomas, paragangliomas, gastrointestinal stromal tumors, and renal-cell carcinomas (RCCs). SDH-deficient renal-cell carcinoma (SDH-RCC) was first identified in 2004 as an inherited kidney cancer with mutations in the SDH gene. SDH consists of A, B, C, and D units. Mutation in the SDHB gene is the most common mutation in SDH-deficient RCCs.
Case Presentation:
We report a case of a 51-year-old healthy man diagnosed with SDHB germline mutation and RCCs. Positron emission tomography/computed tomography (PET/CT) showed a 12 cm tumor in the upper pole of the left kidney. A hand-assisted laparoscopic partial nephrectomy was performed and the histopathology of the tumor showed SDH-deficient RCC with clear surgical margins. Six months after the initial presentation, the patient had a slightly impaired renal function and was disease-free on PET/CT.
Conclusion:
Patients with SDH-deficient RCC are at risk of multifocal kidney tumors and should be offered lifelong follow-up. To preserve the renal function, nephron-sparing surgery is the choice of treatment when feasible, regardless of tumor size.
Introduction and Background
Succinate dehydrogenase (SDH) is a heterotetrameric protein consisting of A, B, C, and D subunits located on the inner mitochondrial membrane. SDH has an important role in cellular energy metabolism in the Krebs cycle and as part of the respiratory chain. 1
Vanharanta et al. first identified SDH-deficient renal-cell carcinoma (SDH-RCC) in 2004 as an inherited kidney cancer with mutations in the SDH gene. 1 The mutation was also shown to be associated with pituitary gland tumors, pheochromocytomas (PCCs), paragangliomas (PGLs), and gastrointestinal stromal tumors (GISTs).2,3 Ricketts et al. described in 2012 that SDH-RCC can develop regardless of mutation history. 2 In 2013, SDH-RCC was integrated into the International Society of Urological Pathology Vancouver Classification. 4
Presentation of Case
We report a case of a healthy 51-year-old man with known SDH deficiency in the family. The patient's nephew was diagnosed with PGL and a gene analysis identified the SDHB gene mutation. Hence, DNA analysis was performed on the rest of the family members.
The patient's DNA analysis showed SDHB deficiency and in February 2017, as part of the primary assessment, a positron emission tomography/computed tomography (PET/CT) scan was performed because of the high risk of multifocal disease. The scan demonstrated an 11.8 × 10.4 cm tumor in the upper pole of the left kidney (Fig. 1). However, the scan showed no sign of metastatic disease. The renal tumor anatomical classification according to the PADUA score was estimated to 12. The patient had a history of back pain for many years prior for diagnosis and explained that he had recently started to feel pain in the left hypochondria. The patient had not experienced hematuria.
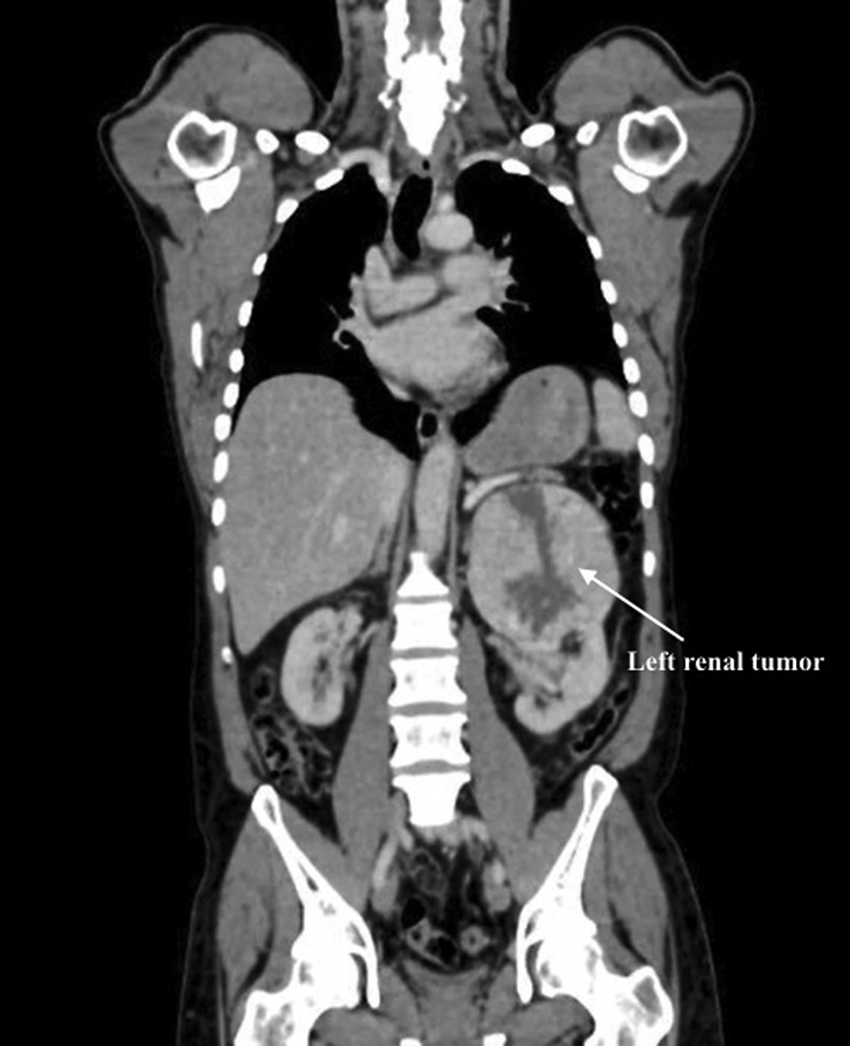
Twelve centimeter tumor on the upper pole of left kidney.
A hand-assisted laparoscopic partial nephrectomy was performed on the 20th of March 2017 with off-clamping technique to preserve the renal function. The estimated blood loss was 600 mL with a surgical time of 143 minutes. The surgery was performed by a senior consultant who had previously performed >130 partial nephrectomies. There were no complications reported within the first 30 postoperative days. Regarding the renal function, the estimate glomerular filtration rate was 90 mL/min 1.73 m2 preoperatively, at 1 month postoperative it was 79 mL/min 1.73 m2, and at 7 months postoperative it was 62 mL/min 1.73 m2.
The histopathology of the tumor revealed SDH-RCC with clear margins, no sarcomatoid dedifferentiation, no necrosis, and no pleomorphic tumor cells (Fuhrman grade 1; Fig. 2). A 6-month follow-up with fluorodeoxyglucose (FDG) PET/CT was planned.
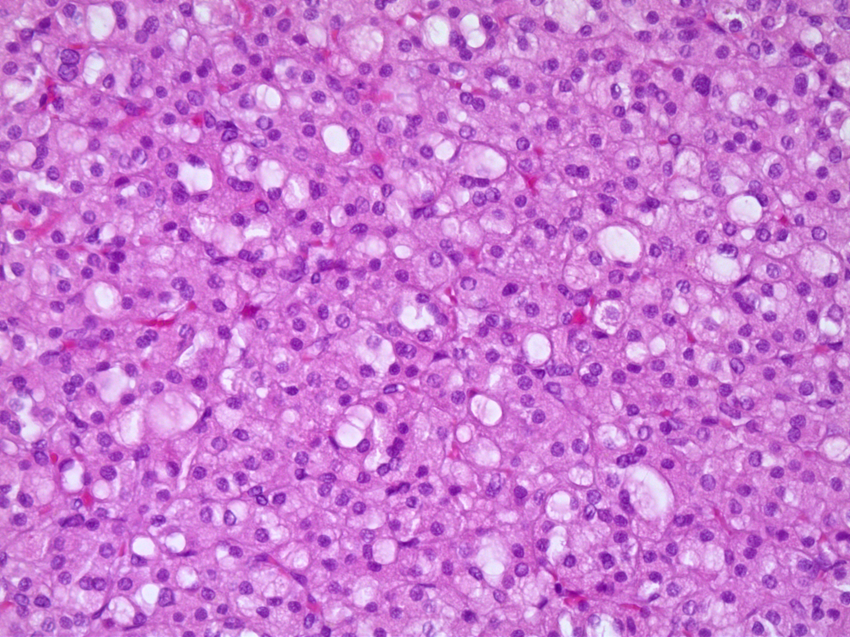
Succinate dehydrogenase-deficient renal-cell carcinoma. 20 × enlarged. Hematoxylin-eosin.
Four months after the surgery, the patient contacted the hospital with pain in the left hypochondria throughout 5 days and described it like the initial pain. Despite the pain, the 6-month planned FDG PET/CT was performed in August 2017. The scan revealed activity in the lungs and in mediastinal lymph nodes. A biopsy of the lungs showed no malignity. The patient also complained of alternating stools, and subsequently a colonoscopy was performed—again with no abnormal findings. Further follow-up control was changed because of the activity in the mediastinal lymph nodes. Scans were planned for every 3 months during the first year and afterward every 6 months during the subsequent 3 years and annually after that.
Discussion and Literature Review
The frequency of SDH-RCC in all RCCs is estimated to be between 0.05% and 0.2%. 4 In SDH-RCC, the male:female ratio is 1.8:1 and the average mean age of debut is reported to be 33, 38, and 37 years, respectively.2–4 Gill et al. reported an average tumor size of SDH-RCC of 51 mm (range 7–90 mm). 4 The most common SDH mutation in SDH-RCC is SDHB deficiency followed by SDHC and SDHD deficiency.1,2 Owing to these findings, patients with early onset (age <45) RCC, with bilateral or multifocal renal tumors, should be considered for SDH germline mutation testing. 2 Furthermore, patients with SDH deficiency tumors of the pituitary gland, PGL, PCC, or GIST, should be considered for testing.2,3 Everyone with an SDH germline mutation in the family should be offered a DNA analysis. Our patient underwent genetic evaluation at an age of 51 years because of a family history of mutation in the SDHB gene and was subsequently found to have a 12 cm tumor in the left kidney. Previous studies have recommended nephron-sparing surgery because of the high risk of bilateral tumors and the lifelong risk of recurring renal tumors.2,4 SDH deficiency patients are young at time of diagnosis and are at risk of multifocal tumors and oncologic treatments, thus depending on as much renal function as possible supporting nephron-sparing surgery.2–4 Despite the large tumor size localized to the upper pole of the left kidney, we, therefore, chose to perform a partial nephrectomy with off-clamping technique, which was carried out effectively.
In 2014, Gill et al. observed metastatic disease in 9 out of 27 patients of which 2 patients experienced a late recurrence in, respectively, 5.5 and 30 years after initial presentation. The risk of metastatic disease increased with high grade nuclei and coagulative necrosis, whereas metastatic disease was rare in conjunction with low-grade tumors. 4 Therefore, extended—if not lifelong—surveillance is recommended.2–4 In our case, the 6 months' control PET/CT scan showed no sign of metastatic disease.
Conclusion
Patients with early onset RCC, those with a family history of genetic mutation, should be considered for gene testing and life-long follow-up in case of an SDH germline mutation. Further evidence is needed to establish the appropriate management for SDH-RCC. At our department, we are advocating nephron-sparing surgery whenever feasible, regardless of tumor size, because of the possibility of bilateral renal tumors, multifocality, and low metastatic risk, as illustrated in our case. Our case also shows that treatment by partial nephrectomy in such patients is feasible in expert hands even in large tumors. Furthermore, we were able to perform the surgery with an off-clamp technique, thereby protecting the remaining tissue from ischemia, and reducing the loss of renal function.
Footnotes
Disclosure Statement
No competing financial interests exist.